Article Text

Abstract
Glioblastoma is the the most common primary brain tumor in adults. Onset of disease is followed by a uniformly lethal prognosis and dismal overall survival. While immunotherapies have revolutionized treatment in other difficult-to-treat cancers, these have failed to demonstrate significant clinical benefit in patients with glioblastoma. Obstacles to success include the heterogeneous tumor microenvironment (TME), the immune-privileged intracranial space, the blood–brain barrier (BBB) and local and systemic immunosuppressions. Monoclonal antibody-based therapies have failed at least in part due to their inability to access the intracranial compartment. Bispecific T-cell engagers are promising antibody fragment-based therapies which can bring T cells close to their target and capture them with a high binding affinity. They can redirect the entire repertoire of T cells against tumor, independent of T-cell receptor specificity. However, the multiple challenges posed by the TME, immune privilege and the BBB suggest that a single agent approach may be insufficient to yield durable, long-lasting antitumor efficacy. In this review, we discuss the mechanism of action of T-cell engagers, their preclinical and clinical developments to date. We also draw comparisons with other classes of multispecific antibodies and potential combinations using these antibody fragment therapies.
- immunotherapy
- central nervous system neoplasms
- T lymphocytes
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Patients with glioblastoma have a poor prognosis with a median survival of approximately 16 months.1–3 Advances in survival have been minimal since the mid-2000s, despite improvements in surgical techniques, radiation therapy and the advent of therapies such as tumor-treating fields.2 Immunotherapy has been evaluated as one potential solution. Immune checkpoint inhibition (ICI) therapies targeting programmed death-1 (PD-1) and its ligand, programmed death ligand-1 (PD-L1), have improved outcomes in malignancies such as melanoma even when it has metastasized to the brain.4 However, similar outcomes have been elusive in glioblastoma, reflecting the complex mechanisms of immune suppression and evasion that it possesses.5 6
Currently, systemically delivered antibody-based immunotherapies approved for patients with cancer falls broadly into two categories. ICIs are monoclonal antibodies (mAbs) which inhibit immune checkpoint signaling. Bispecific antibodies tether tumor cells to T lymphocytes (cytotoxic T lymphocytes (CTLs)) to induce cytolysis, as well as activate innate immune pathways via non-specific binding to the tail region of the antibody (fragment crystallizable region, Fc).7 8 To exert their therapeutic effect in glioblastoma, these therapies must transit the blood–brain barrier (BBB) before reinvigorating immune cells that may have been rendered inert by the tumor microenvironment (TME). While some systemically administered antibodies may be able to penetrate the BBB, the concentrations necessary to produce effects in the brain TME are unknown.9 This intracranial bioavailability may therefore only reflect a small fraction of the total administered dose.
One approach to bypass the BBB involves the direct administration of immunotoxins via convection-enhanced delivery (CED).10–12 These are fusion proteins which consist of an antibody fragment that binds the target cell and a protein toxin fragment which induces cytolysis.13 14 However, this approach is invasive and can be hampered by unequal drug distribution.15 16 A newer approach involves the use of a fusion protein that can be delivered systemically—bispecific T-cell engagers. These consist of two antigen-binding variable fragments that tether the tumor cells to CTLs but differ from their antibody parents in that they do not possess the constant (Fc). As they are smaller in size than traditional mAbs, they may more easily penetrate the BBB.17 18 This small size also allows T cells to closely bind their target, resulting in a high-affinity immune synapse.19 Bispecific T-cell engagers also are highly potent, exerting a therapeutic effect at nanomolar concentrations.20 Bispecific T-cell engagers can therefore potentially access this immune privileged compartment more readily while also exerting a highly potent effect even at low concentrations. This combination makes it an ideal candidate for an immunotherapy-based approach in glioblastoma. However, glioblastoma is uniquely shielded from the immune system due to its location within the central nervous system (CNS). While this privilege is not absolute, a significant proportion of tumors have been noted to be devoid of any tumor-infiltrating lymphocytes (TILs) that can be redirected by bispecific T-cell engagers.21 22 In those tumors that do demonstrate invasion by TILs, they are often induced to be dysfunctional and anergic by the suppressive TME.23 Isocitrate dehydrogenase (IDH) wild-type gliomas also lack a universally expressed tumor-specific antigen which may result in antigen escape and tumor regrowth, making targeting of precisely engineered therapies difficult.24–26
Heterogeneity and local immune suppression have also frustrated the use of bispecific T-cell engagers in other solid malignancies, and to date, these agents have only been approved by the US Food and Drug Administration (FDA) for the treatment of acute lymphocytic leukemia (blinatumomab, Amgen).27 28 In this review, we will discuss the current landscape for bispecific T-cell engagers in glioblastoma, as well as the challenges they face, and describe potential approaches to overcome these.
Design and mechanism of action
Bispecific T-cell engagers consist of two linked antigen-binding variable fragments devoid of the constant domain of their parent antibody. These fragments are linked by short flexible linker regions29 resulting in a small construct (approximately 55 kDa), which can bring CTLs into close proximity to the target cell, resulting in a high binding affinity.18 30 CD8+ CTLs, like all T cells, express variable T-cell receptors (TCRs) associated with invariable CD3 subunits. Bispecific T-cell engagers typically link tumor-associated antigens (TAAs) with the CD3ϵ unit of the TCR complex, thereby engaging T cells to form a synapse on the surface of the tumor cell. The T cell is activated, triggering cell death signaling pathways with the subsequent release of granzymes and perforins.31 Given that bispecific T-cell engagers engage the CD3ϵ unit, this means that they are not limited by TCR specificity and can potentially redirect the entire repertoire of T cells. This may also involve T cells that have reactivity against other tumor antigens, leading to epitope spreading. T-cell activation also results in the expansion of the CD8+ T-cell compartment, driven in the context of T-cell engagers by an increase in cytotoxic CD8+ T effector memory cells.32 Importantly, this occurs in a TCR–peptide–major histocompatibility complex (MHC) independent manner, which avoids the potential for immunotherapy driven downregulation of MHC-I and immune escape.20 Furthermore, brain bispecific T-cell engager (BRiTE) has been shown to redirect local regulatory T cell (Treg) to kill glioblastoma tumor in vitro via the granzyme–perforin pathway, potentially overcoming a key element of the immunosuppressive microenvironment.33 34
Bispecific T-cell engagers offer immunotherapy in a manufacturing format which is both scalable and standardizable. In contrast to chimeric antigen receptor (CAR) T cells, T-cell engagers do not require initial lymphodepletion and ex vivo expansion of autologous cells which require transduction (that may potentially lead to variable yields).35 36 Owing to their relatively simple structure of a single-chain polypeptide, bispecific T-cell engagers can be batch manufactured in large quantities using well-established commercial processes such as expression via Chinese hamster ovary cells (eg, as used for single-chain bispecific agents targeting CD19).37 38 While this requires generating a construct that can be readily expressed, these T-cell engagers can be processed into a format without dosing variability, offering an ‘off-the-shelf’ form of immunotherapy.
Further, bispecific T-cell engagers have been shown to drive T cell-mediated cell kill both in vivo and in vitro at very low concentrations (10–100 pg/mL) and at low effector to target cell ratios (E:Ts) in hematological malignancies (<1:90).39 40 Single-chain bispecific antibody fragments have also been demonstrated in vitro to exhibit 100 000-fold superior antitumor cytotoxicity compared with conventional mAbs.19 41 However, it is important to note that in vitro data may not accurately reflect the characteristics of a therapeutic in vivo as retrospective work comparing potency between the two settings for antibodies have reported large discrepancies in binding behavior.42 Many in vitro potency assays are unable to fully account for interactions with the target in the steady state and therefore fully evaluate ligand–target kinetics.43 Nevertheless, the potential for a highly potent immunotherapy that can redirect the entire host repertoire of T cells and be manufactured in a consistent and scalable fashion is a highly attractive prospect. However, significant challenges remain which we will discuss in the following section.
Development and challenges facing T-cell engagers in intracranial malignancy
Choosing the right target: is one enough?
Developing a potent and effective T-cell engager therapy for intracranial malignancy faces many challenges (summarized in figure 1). The ideal CTL-based approach requires the identification of a universally expressed and specific tumor antigen, but this is an unrealistic expectation for glioblastoma. While some subsets of glioma share clonal neoepitopes (IDH1-R132H) and have been targeted by vaccination in humans, this is presented in an MHC class II-restricted manner and does not elicit a CD8+ CTL response.44

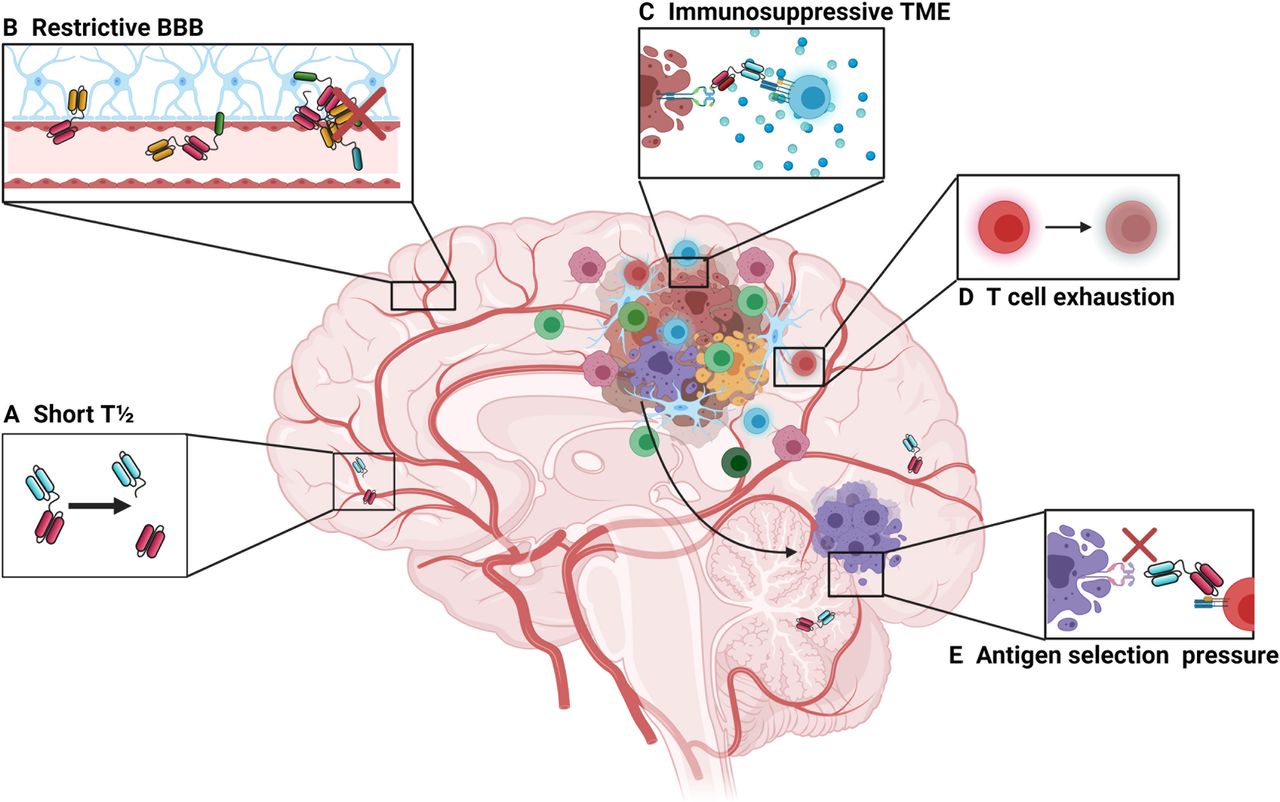
Barriers to bispecific T-cell engager therapies in the intracranial environment. (A) First-generation bispecific T-cell engagers are small in size (approximately 55 kDa), which makes them prone to rapid clearance, giving them a short plasma half-life of approximately 2.5 hours.63 This limits their time to exert their therapeutic effect and can pose a challenge for clinical administration, necessitating continuous infusion strategies. Newer designs involve the use of additional Fc regions, full size antibody constructs or the addition of albumin-binding domains. However, these also increase the size of the construct and may affect trafficking dynamics at the BBB. (B) The BBB prevents the passive movement of cells and molecules that could potentially damage the central nervous system. While the small size of bispecific T-cell engagers makes them theoretically more able to cross the BBB compared with larger, full-sized monoclonal antibodies, the trafficking of effector T cells may be restricted by the BBB. (C) High levels of immunosuppression surrounding tumor may prevent or limit T-cell activation following T-cell engagement. (D) Tumor-infiltrating lymphocytes may become exhausted and anergic, which is driven in part by tumor cells expressing programmed death ligand-1 along with myeloid-derived suppressor cells. (E) Glioblastoma lacks a uniformly expressed major histocompatibility complex-independent tumor-specific antigen which limits bispecific therapy as there may be selection pressure on those tumor cells expressing the target antigen, leading to outgrowth of antigen-negative cells. BBB, blood–brain barrier; TME, tumor microenvironment.
One attractive target is epidermal growth factor receptor variant III (EGFRvIII), which is specific to glioblastoma and is not expressed in non-tumor tissue. Epidermal growth factor receptors (EGFRs) are involved in deregulated cancer signaling pathways, leading to atypical proliferation and growth of tumor cells.45 EGFRvIII is the most common variant which is not presented in an MHC-dependent manner, but is present in only 20%–30% of patients and is not expressed in all tumor cells.46 Scott et al demonstrated that systemically delivered radiolabeled antibodies specific to EGFRvIII were taken up in high levels by tumors in patients with glioblastomas, indicating their ability to accumulate intracranially.47 However, it is notable that this effect was only seen in one of eight patients studied. This may reflect penetration of a radiolabeled antibody through the diseased BBB. However, disruption of the BBB is not uniform in glioblastoma, and there may be regions of immune privileged tumor shielded by intact portions of barrier.48 Further work to determine optimal delivery of systemic bispecific T-cell therapy across intact and disrupted BBB is required.
First in-human trials of EGFRvIII-specific CAR T cells found that disease regression could be induced in a specific manner, with no off-target effects on wild-type EGFR.49 However, O’Rourke et al demonstrated antigen loss and a lack of persistent effector T-cell activity in patients treated with EGFRvIII CAR T cells.24 Brown et al similarly reported achieving efficacy in reducing disease burden when targeting the IL13Rα2 cell surface receptor but described antigen loss in post-treatment tumor samples taken from patients who had experienced recurrence.26 50 While the experience of using bispecific T-cell engagers in clinical glioblastoma is limited, this effect has also been observed with hematological therapies where CD19-negative clones have developed following treatment with blinatumomab or anti-CD19 CAR T cells.51–53 Tandem approaches targeting multiple TAAs are one potential strategy to overcome this obstacle. A tandem CAR targeting HER2 and IL13Rα2 has been shown to enhance survival and mitigate antigen escape in murine models of glioblastoma.54 However, targeting two or more TAAs may ultimately fail if even a small part of the tumor does not express this combination, and such an approach may also significantly increase the risk for off-target toxicity.
Another approach to address heterogeneity may be by inducing partial kill of a tumor, thereby driving antigen shedding by dying tumor cells (epitope spreading).55 56 Concurrent local cytokine production/administration has been shown in vitro and in vivo to drive bystander cell killing, even if those cells in the vicinity are antigen negative.57 58 However, Krenciute et al described antigen escape still occurring in murine models of glioblastoma when IL13Rα2 CAR T cells were induced to express costimulatory interleukin (IL)-15.59 Choi et al reported efficacy in heterogenous murine glioblastoma when using CAR-T cells specific for EGFRvIII but which are also designed to express a bispecific T-cell engagers targeting EGFR wild type. This intracranially administered drug could induce local cytotoxicity, with no EGFR bispecific T-cell engagers detected in the periphery.60 Further, bispecific T-cell engagement of CD3 to the target antigen results in an immune synapse more akin to the natural TCR–MHC peptide complex, resulting in secretion of cytokines and promoting differentiation of naïve T cells to lyse tumor cells, thereby driving a more diverse and efficient immune response.20 61 62
Potent but brief killer
Ensuring persistence of bispecific T-cell engagers to drive ongoing killing at the tumor site is another significant challenge. While the small size of bispecific T-cell engagers allows for them to bring CTLs into close proximity with the target cell, they tend to have a short half-life due to rapid renal clearance (approximately 2.5 hours63). This rapid clearance can limit drug accumulation, particularly in difficult to access compartments such as the brain. A half-life of just 2.5 hours requires dosing regimens that rely on continuous infusion, often requiring patients to have venous access port systems installed which carry their own associated risks.64 Furthermore, the small size can lead to drug stability and aggregation issues.65
Approaches to extend the half-life of bispecific T-cell engagers involve giving the construct a higher molecular weight, which would extend the elimination half-life and make this therapy deliverable via serial infusions while maintaining serum levels.66 These can involve constructions that add a constant domain to the bispecific structure (as per AMG160 targeting PSMA for prostate cancer), or indeed reverting to full size bispecific antibodies such as approaches targeting ENPP3 in renal cell carcinoma (RCC), prostate cancer (via prostate specific membrane antigen (PSMA)) or a mixed valency 2+1 format bispecific antibody targeting claudin-6 in ovarian cancer.67–70
Half-life extended bispecific T-cell engagers are under investigation in a first-in-human phase I study involving patients with B-cell malignancies to evaluate safety and preliminary efficacy (NCT03571828).71 However, bispecific T-cell engagers may transit the BBB more readily due to their small size. Half-life extension modules which increase the molecular weight by adding an Fc domain, or indeed full-size antibody constructs may hinder migration across the BBB.65 Other approaches use the addition of a variable fragment of a humanized albumin-binding antibody.72 Interestingly, the addition of human serum albumin (HSA) may also aid the transiting of small therapeutics across the BBB, and its use in a combination format with bispecific therapies targeting intracranial malignancies may have a dual benefit.73
A hostile microenvironment
Glioblastoma is surrounded by a highly immunosuppressive stroma, in which regulatory T cells (CD4 +CD25+FOXP3), tumor-associated macrophages (TAMs) and myeloid derived suppressor cells are present.56 74 This environment can drive T-cell anergy and apoptosis as well as blunting the impact of innate natural killer (NK) cells. Glioblastoma expresses HLA-G, which inhibits activated NK cells and therefore downregulates their response.75 IDH mutant gliomas also demonstrate resistance to NK activity by epigenetically silencing activating receptor ligands.76 While NK cells can destroy glioma stem cells, phase III studies using NKs or lymphokine-activated killer cells have failed to improve immune response against immunologically ‘cold’ solid tumors.77 78
More recent attention has turned to addressing causes of T-cell failure in the TME. As mentioned previously, regulatory T cells are adept at inducing secondary T-cell failure via immunosuppressive molecular factors such as PD-L1 and CTLA-4, LAG-3, TIM-3 and others.79 80 However, it is notable that ICI as a monotherapy has failed to confer benefit in patients with glioblastoma.81 This may reflect multiple overlapping mechanisms of immune suppression that provide redundancy. Regulatory T cells inhibit the secretion of T-cell cytokines and proliferation by also exerting a downregulatory effect on the production of IL-2 and interferon-γ.82 Overproduction of IDO-1 by glioma not only recruits regulatory T cells but also has a metabolic impact on T-cell activity by reducing the amount of tryptophan available in the microenvironment.83 While there have been several studies evaluating the use of IDO-1 inhibitors in glioblastoma, including combination approaches against PD-1 and IDO-1 (NCT03707457), phase III evaluations of IDO inhibition in other solid malignancies in the CNS (metastatic melanoma) have failed to demonstrate survival benefit.84 However, more recent mechanistic studies suggest that this failure may be due to incomplete blockade of protumorigenic metabolic pathways. Enzymes such as IL-4-induced-1 have also been associated with downstream receptors activated by tryptophan catabolites and whose activity is undisturbed by IDO-1 inhibition.85 86
Stromal cells in the microenvironment also produce highly immunosuppressive cytokines such as transforming growth factor beta (TGF-β) and IL-10.87 88 Preferential production of lactate by tumors via anaerobic metabolism (known as the Warburg effect) can decrease CTL activity as well as migration potential.89 The tumor itself can drive T-cell dysfunction by producing hypoxia-inducing factor-1 alpha (HIF-1a) to promote angiogenesis and proliferation.90 91 Overexpression of HIF-1a can also reduce the migration of CTLs via downregulation of CD62L, resulting in their failure to migrate to the tumor site.92 Taken together, the aforementioned mechanisms contribute to an ‘immune desert’ landscape, characterized by few, if any, infiltrating lymphocytes which bispecific T-cell engagers can redeploy against tumor cells.93
Novel preclinical approaches include combinatorial inhibition of recognized drivers of T-cell exhaustion such as CTLA-4, LAG-3, TIM-3, or IDO with bispecific T-cell engagers, or triggering costimulatory receptors such as 4-1BB, OX40, CD40, or CD27.94 More controllable constructs have also been demonstrated preclinically, with the use of switch receptor constructs targeting PD-1 expression, whereby adding CD28 domains can transform a negative signal into a stimulatory one.95 Work is also under way exploring potential combinatorial cytokine modulation approaches, such as those described in ‘armored’ CAR T cells which are combined with IL-12, IL-15 or IL-18 for enhanced effect.96–98 Should such approaches show promise, these could be translated to the T-cell engager format either as part of the construct or as an adjunct delivered via catheter directly to the tumor. Frewert et al described enhanced CTL activity when infusing IL-1β or interferon-γ intratumorally, making this a logical combination with bispecific T cell engagers.99 Antibody mediated blockade of lactate transporters may also aid in combating T cell dysfunction in the TME, as well as the use of constructs targeting fibroblast activation proteins or expressing heparinase to disrupt immunosuppressive stromal elements.89 100 101
Preclinical development of T-cell engagers for glioblastoma
Murine bispecific T-cell engagers targeting EGFRvIII and CD3ϵ were first described by Choi et al.102 When systemically administered, this construct was found to activate T cells to generate potent antigen-specific lysis of EGFRvIII expression gliomas in vitro (p<0.001) at very low concentrations (10 ng/mL) and at low E:Ts (1:2.5). While this ratio is lower than the previously mentioned <1:90 for CD19 agents, it is notable that bispecific T-cell engagers may benefit from lower E:T ratios in solid tumors due to the process of additive toxicity, as described by Weigelin et al in murine models of melanoma.103 Cytolysis of solid tumor cells may be induced by sequential ‘sublethal’ interactions between CTLs and tumor cells (such as granzyme B-mediated damage to the nuclear envelope and the creation of double-stranded breaks in DNA).103 104 Bispecific T-cell engagers may help to promote this effect on solid tumors by acting as a stabilizing contact which can increase these sublethal CTL interactions at the tumor site.35 103 Choi et al also reported that bispecific T-cell engager therapy could redirect Tregs in vitro to express granzymes and perforins, which may serve to induce further tumor cytolysis.34 Accordingly, the use of bispecific T-cell engagers in murine models of intracranial glioma has been shown to achieve durable and complete cures in up to 75% of mice (p<0.05).102
Following this, a fully human T-cell engager was described by Gedeon et al.105 This fully human construct would avoid potential murine antibody-associated complications such as cytokine release syndrome, unpredictable dose–response relationships, and the formation of human anti-mouse antibodies, leading to rapid clearance of the bispecific T-cell engager from the serum.106–109 This fully human bispecific T-cell engager again exhibited specific binding, cytokine release, T-cell activation and proliferation, and in vitro and in vivo tumor cell lysis in murine models of orthotopically implanted glioma.63 Schaller et al subsequently conducted preclinical studies to determine the minimum anticipated biological effect level and thus to establish a safe dose for first in-human trials (notably 1000-fold lower than prior in vivo dosages).110 This study determined that the theoretical human receptor occupancy was 0.17%, far below industry standard levels.111–113 An extended single-dose toxicity study in vivo using mice demonstrated no evidence of pathological findings related to the bispecific T-cell engager and no neurological toxicity was exhibited.114 Detailed pharmacokinetic analysis demonstrated a relatively short half-life in keeping with other T-cell engagers with a half-life of 8 min and a terminal half-life of ∼2.5 hours.63
Clinical development of EGFR T-cell engagers for glioblastoma
Currently, there are two EGFRvIII targeting T-cell engagers entering phase I trials for glioblastoma. AMG596 is a bispecific T-cell engager (trademarked as BiTE) by AMGEN, which conducted a phase I open-label sequential dose-escalation and dose-expansion trial in humans (NCT03296696). The study evaluated safety, tolerability and pharmacokinetics and pharmacodynamics of AMG596 in patients with both newly diagnosed and recurrent EGFRvIII-positive glioblastoma.115 Like blinatumomab, AMG596 is administered via a continuous intravenous infusion. This T-cell engager was to be trialed as both a monotherapy and in combination with AMG404, a proprietary mAb which blocks binding of the immune checkpoint programmed cell death protein-1 (PD-1).116 However, while this study began enrollment in April 2018, it is unclear if ongoing development may progress due to portfolio prioritization.117 hEGFRvIII-CD3 bi-scFv (BRiTE) is another EGFRvIII bispecific construct which is entering phase I clinical trials (NCT04903795). This consists of anti-human mAb clones 139 (anti-EGFRvIII) and 28F11 (anti-CD3), both of which have been used safely in the clinical environment previously.118–120 This will be trialed as both a monotherapy and in combination with peripheral T-cell infusion. As described, late-stage tumors frequently evade immune response due to inducing T-cell anergy and apoptosis. Increased numbers of intratumoral CD8+ CTLs have also been associated with favorable outcomes in patients with glioblastoma.121–123 Concomitant administration of stimulated CTLs may therefore synergistically enhance the efficacy of this treatment.124 The migration of T-cell engagers across the BBB may also be facilitated by activated T cells which adhere to the brain microvascular endothelium and subsequently cross by diapedesis.125 Concurrent administration of activated T cells could therefore enhance the trafficking of bispecific T-cell engagers into the intracranial compartment, increasing their density at the tumor site and thus the therapeutic effect. However, this approach requires careful monitoring of toxicity, as the release of inflammatory cytokines in the CNS has been associated with immune effector cell-associated neurotoxicity syndromes (ICANS). This condition manifests as a spectrum of symptoms ranging from lethargy and confusion to seizure and coma.126 ICANS has been observed as a potential side effect for bispecific T-cell engagers even without a brain-specific target. Blinatumomab (specific for CD19) has been found to systemically activate T cells which then subsequently cross the BBB in a non-specific manner. It is theorized that these T cells may then encounter sporadic CD19 expressing target cells in the CNS and then release inflammatory cytokines such as IL-6 and IL-1β, which can disrupt the BBB further, allowing for greater ingress of proinflammatory cytokines.125 Importantly, this toxicity can be abrogated by the administration of agents such as natalizumab, which prevents T-cell migration across the BBB.125
EGFR biarmed activated T cells (EGFR BATs) is a separate CD3 bispecific approach under investigation targeting EGFRwt. EGFR BATs are T cells that have been coated with cetuximab (a bispecific antibody targeting EGFRwt) and treated with OKT3 to stimulate them.127 This approach is currently undergoing phase I clinical trials for safety and toxicity in patients with newly diagnosed glioblastoma alongside standard of care treatment (NCT03344250). Despite targeting EGFRwt, which is expressed at several sites around the body, preliminary data report no dose-limiting toxicity of the four patients treated in the first-dose tier.128 A summary of these approaches is shown in table 1.
EGFR targeting T cell engagers for glioblastoma in clinical trials
Future approaches
Given the plethora of targets, agents and obstacles for bispecific T-cell engagers in glioblastoma, it is understandable that new approaches are already under preclinical development (overview shown in figure 2). These consist of both an expansion of the bispecific T-cell engager construct, with more potential antigen targets incorporated, or by combining bispecific T-cell engager therapy with checkpoint inhibitors simultaneously. Given the early stage of preclinical development for many of these approaches, we will discuss the initial findings from multiple malignancies, which may offer insights for future directions in glioblastoma.

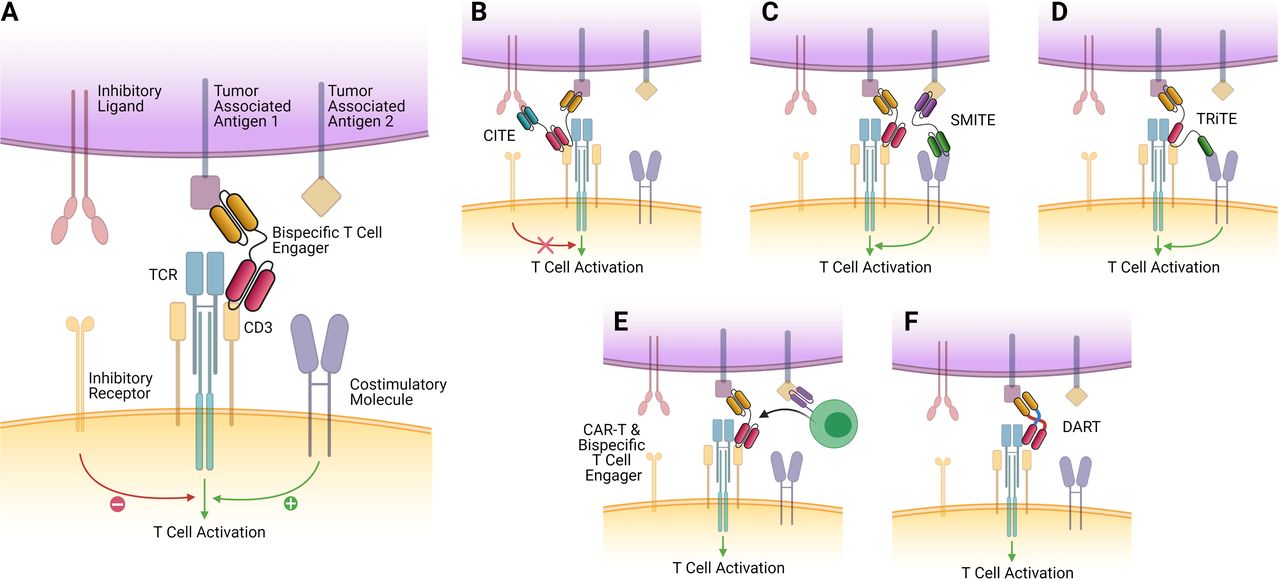
Bispecific T-cell engager constructs and future directions. The ‘classical’ T-cell engager structure, consisting of two antigen-binding variable fragments, devoid of the Fc domain of their parent antibody, linked by a short flexible linker region. It tethers the epsilon subunit of the T cell to EGFRvIII expressing tumor cells, activating the T cell, which then releases granzymes and perforins, resulting in tumor cell death. This approach is currently being evaluated in phase I clinical trials (NCT04903795 and NCT03296696) (B) CiTEs include the extracellular domain of a checkpoint inhibitor (such as PD-1) fused to a traditional T-cell engager scaffold. This allows for synergistic checkpoint blockade alongside T-cell tethering and activation and has demonstrated enhanced efficacy in vitro and in vivo in acute myeloid leukemia (AML).129 (C) SMiTEs are constructed from two separate classical T-cell engagers which target separate antigens. These can offer costimulation of tethered T cells and provide regional checkpoint blockade while also offering the traditional lytic effects formed by the tumor–T cell synapse. (D) TRiTEs can tether T cells to the tumor while also delivering costimulatory signals (eg, via interaction with CD28). (E) CAR T cells which can be engineered to secrete T-cell engagers have been described by Choi et al, who developed a bicistronic construct that resulted in expression of a CAR specific for EGFRvIII, which could also secrete T-cell engagers specific for EGFR wild type, which would only have effect in the local tumor environment. These demonstrated promising efficacy against heterogeneous mouse models of glioblastoma.60 (F) DART proteins are a novel take on the bispecific construct, where two variable fragment chains are linked by disulfide bonds and non-covalent forces, which may result in greater stability and enhanced cytotoxicity.142 CAR, chimeric antigen receptor; CiTE, checkpoint inhibitory T-cell engager; DART, dual-affinity retargeting; EGFR, epidermal growth factor receptor; SMiTE, simultaneous multiple interaction T-cell engager; TCR, T-cell receptor; TRiTE, trispecific T-cell engager.
Checkpoint inhibitory T-cell engagers (CiTEs) and simultaneous multiple interaction T-cell engagers (SMiTEs)
CiTEs offer immune checkpoint blockade in the area of interest only by tethering immune checkpoint domains to classical bispecific T-cell engager construct, reducing the chance for on-target, off-tumor effects.129 SMiTEs consist of two separate bispecific T-cell engagers targeting separate antigens. These have been used to target both CD3, to induce the lytic synapse as described previously, and CD28, to induce a costimulatory signal for engaged T cells. Such constructs have been designed using a CD3-TAA×anti-PD-L1-CD28 format to further enhance their activation and overcome potential anergy in a local fashion.130 Although CD28 stimulation can have deleterious off-target effects, its combination with a bispecific T-cell engager specific to the tumor site may help to ensure specificity and prevent systemic toxicity.131
Trispecific T-cell engagers (TRiTEs)
Other similar approaches involving CD28 include TRiTEs, which have been found to suppress myeloma growth in a humanized mouse model while also stimulating memory/effector T-cell proliferation and reducing Treg cell numbers in primates.132 Bispecific T-cell engagers have also been engineered to include cytokines such as IL-12 to enhance their activity. Do et al described a nanoparticle-based assembly and screening approach before using a modular platform to incorporate the cytokine of interest.133 They reported that the optimally lytic architectures favor high αCD3 to αTAA ratios, and these are improved linearly by increasing IL-12. Given that many current structures offer a 1:1 binding ratio of CD3 to TAA, it may be that combinatorial structures can increase the binding avidity and enhance effect. The wide variety of potential targets, both in terms of ICI and selecting for other TAAs or cytokine inclusion, may also make simultaneous engagement an attractive proposition for addressing glioblastoma heterogeneity and overcoming immune escape. Another TRiTE-like approach is the trispecific T-cell activating construct (TriTAC) platform, which contains three domains that target a TAA, CD3, and HSA. The authors demonstrated that TriTACs have good solid tumor penetrance due to their small size yet have an extended half-life due to HSA binding activity.134
Novel delivery approaches
As discussed previously, treatment penetrance into solid malignancies remains a particular challenge. Oncolytic viruses which deliver therapeutic transgenes can induce local expression of bispecific T-cell engagers and therefore stimulate tumor-resident T cells.135 136 Scott et al developed both a bispecific and trispecific T-cell engager expressing adenovirus and demonstrated they could also be used to deplete immunosuppressive TAMs in vitro.137 This was further developed resulting in an oncolytic virus format which simultaneously produced IL-12, an anti-PD-L1 antibody and a bispecific T-cell engager. In combination with CAR T-cell therapy, this format was able to kill multiple cancer cell lines expressing target antigen while inducing more durable responses against orthotopically implanted tumors.138 Oncolytic viruses such as adenovirus can also kill directly by oncolysis, which may further result in neoantigen release from within lysed cells. Their subsequent presentation by antigen-presenting cells could act as an in situ vaccine, enhancing the specific immune response further.139
Examples of combination therapies of CAR T cell and bispecific T-cell engagers are currently under preclinical evaluation in EGFRvIII expressing glioblastoma. Choi et al developed a bicistronic gene construct that enables coexpression of an EGFRvIII-directed CAR, which also secreted a bispecific T-cell engager targeting wild-type EGFR. These CAR T cells secreted bispecific T-cell engagers which successfully generated T-cell responses against EGFR wild type within the local tumor environment only in addition to EGFRvIII targeted tumor lysis by CARs.49 This resulted in successful clearance of heterogeneous glioblastoma in vivo, and the EGFR-targeting bispecific T-cell engager was only expressed in the local tumor environment, with no systemic detection.60
Of note, this effect was only seen when CAR-T therapy was delivered either intracerebrally or intraventricularly but lost if peripherally given. Similar intracranial approaches are also described by Gardell et al, who directly delivered a retrovirally modified macrophage which could secrete a bispecific T-cell engager specific for EGFRvIII to murine models of glioma. These macrophages could persist in the solid tumor, secreting both the bispecific T-cell engager therapy and IL-12, enhancing the T-cell response.140 However, as stated previously, while these approaches using CED bypass the BBB, these generally require an invasive procedure and can be hampered by uneven drug distribution/coverage at the tumor site.15 16
Targeting other TCRs
Other experimental approaches involve modifying the structure of bispecific T-cell engagers to target other receptors. γ-δ T-cell engagers have been demonstrated to bind to a homogenous effector T-cell population with low levels of immune checkpoint molecule expression.141 Several of these constructs targeting non-CD3 TCRs, in combination with immune checkpoint blockade, are under evaluation (tumorous PD-L1 (NCT03917381), FAP (EudraCT 2017-000292-83), 4-1BB, LAG-3 (NCT04140500), and TIM-3 (NCT03752177)). Another modified approach includes that of dual-affinity retargeting (DART) proteins. These consist of bispecific diabodies with two variable fragment chains assembled by means of disulfide bonding and non-covalent forces. This results in a construct which can provide dual antigen-binding sites but may offer greater stability and enhanced redirection of T-cell killing towards tumor cells.142 Indeed, enhanced cytotoxicity when compared with traditional bispecific T-cell engagers has been observed in preclinical models, and DARTs are capable of redirecting NK cells and T cells in preclinical models of both hematological malignancies (anti-CD16-CD32B DART or anti-CD19-CD3 DART duvortuxizumab (MGD011) and solid tumors (glycoprotein A33-expressing gastrointestinal cancer cells (gpA33;MGD007)).142–145
In glioblastoma, alongside bispecific T-cell engagers, bifunctional antibodies targeting EGFR and TGF-β are undergoing evaluation (BCA101). This seeks to abrogate the potential for TGF-β to induce regulatory T cells in the context of EGFR-driven malignancies (including glioblastoma). BCA101 is currently under evaluation in a phase I trial in advanced solid tumors refractory to standard of care as either a monotherapy or in combination with pembrolizumab, an FDA-approved mAb targeting PD-1 (NCT04429542).146 CDX-527 is another bispecific antibody under investigation for safety, tolerability and activity in multiple solid tumors (NCT04440943). This approach seeks to block the binding of PD-L1 while also including an agonist anti-CD27 domain. CD27 is a member of the tumor necrosis factor receptor family, and its blockade can enhance the immune response while reducing the number of Tregs in the local TME.147 148 This synergistic effect results in increased CD8+ T-cell expansion and effector function.149 150 These novel approaches currently under evaluation in glioblastoma are shown in figure 3.

{kind=link}
{kind=link}
{kind=link}
Novel bispecific approaches currently in phase I trials for glioblastoma. (A) EGFRxTGF-β is a bifunctional antibody, which tethers to tumor cells via the EGFRvIII receptor but blocks the induction of regulatory T cells by binding of TGF-β, therefore preserving local effector T-cell function. This approach is currently being evaluated in phase I trials in multiple advanced solid tumors as either a monotherapy or combined with anti-PD1 agents (NCT04429542). (B) PD-L1xCD-27 is a bispecific antibody which blockades PD-L1 while also including an agonist anti-CD27 domain. Blockade of CD27 (a member of the Tumor Necrosis Factor family) can enhance the immune response while reducing the number of regulatory T cells in the local tumor microenvironment. A phase I trial investigating this approach in multiple solid tumors including glioblastoma is currently under way (NCT04440943). PD-L1, programmed death ligand-1; TGF-β, transforming growth factor beta.
Conclusions
T-cell engagers are a specific and potent antitumor therapy which can overcome the barriers faced by traditional immunotherapy constructs when accessing immune privileged compartments such as the brain. However, significant challenges remain, such as the outgrowth of antigen-negative cells and the profoundly immunosuppressive microenvironment which negates T-cell function. For bispecific T-cell engagers to succeed, combination approaches will be required, possibly with ICI or cytokines, which may reinvigorate the immune response. As development of these molecules continues, it may be possible to merge these constructs into a single agent, although this may also enlarge its size and hamper its ability to cross the BBB. Although the short half-life of bispecific T-cell engagers does not pose an insurmountable clinical challenge, the need for a continuous infusion system makes this a complex therapy to administer at present, and more invasive administration systems will always carry a higher risk of morbidity. Other approaches to extending the half-life of bispecific T-cell engagers such as the addition of large stabilizing proteins may also be of value, but their effect on BBB migration dynamics must be carefully considered. While there are potential mechanisms to enhance trafficking across the BBB, our understanding of the exact mechanism and the degree of carriage into the CNS is not yet fully understood. Work to identify novel MHC independent antigens which are more universally expressed or whether partial killing can drive epitope spreading may also offer a way to overcome tumorous heterogeneity. Combinatorial approaches that can penetrate tumors, negate exhaustion, and drive the presentation of neoantigens to local T cells are likely to have the best chance of inducing efficacious and durable antitumor responses. Combination immunotherapies, however, bring their own challenges in determining the degree of attribution of individual components to efficacy and the potential for additive or synergistic toxicities. As greater numbers of T-cell engaging therapies in varying formats enter clinical trials in glioblastoma, the precise strategy and utility of these promising therapies will become more apparent.
Importance of the review
We summarize the development of bispecific Tcell engagers in glioblastoma, including preclinical and clinical development to date. We explore future designs for Tcell engagers that may help to improve their efficacy and address unique aspects of Tcell engagers, such as their short half-life. Research using similar approaches with bispecific or multispecific antibodies is also elucidated. The unique challenges faced when using immunotherapy within the brain are contextualized with how Tcell engagers may specifically address the hurdles facing development of effective immunotherapy in glioblastoma.
Ethics statements
Patient consent for publication
References
Footnotes
Twitter @TheMohanAditya, @mkhasraw
Contributors MK, JHS, and KS conceived of and designed the work. KS performed the literature search and initially drafted and subsequently revised the manuscript. MK, JHS, KMH, JLR, and AAM developed the manuscript. KMH and AAM developed the figures, which were revised with input from KS and MK. All authors approved the documents for submission.
Funding The authors are supported by the following grants to Duke University: P50-CA190991 (SPORE) (JHS, MK), P01-CA225622 (JHS, MK), U01-NS090284 (JHS), R01-NS099463 (JHS), R01-CA175517 (JHS), and R01-CA235612 (JHS, MK).
Competing interests KS, KLH, JMR, and AAM declare no conflicts of interest. JHS has an equity interest in Annias Immunotherapeutics, which has licensed intellectual property from Duke related to the use of the pepCMV vaccine in the treatment of glioblastoma multiforme. JHS has an equity interest in Istari Oncology, which has licensed intellectual property from Duke related to the use of poliovirus and D2C7 in the treatment of glioblastoma. JHS is an inventor on patents related to PEP-CMV DC vaccine with tetanus, as well as poliovirus vaccine and D2C7 in the treatment of glioblastoma. MK reports personal fees from Jackson Laboratory for Genomic Medicine and research funding from AbbVie and Bristol-Myers Squibb were paid to his institution for glioblastoma research.
Provenance and peer review Commissioned; externally peer reviewed.