Article Text

Abstract
Background γ9δ2 T cells hold great promise as cancer therapeutics because of their unique capability of reacting to metabolic changes with tumor cells. However, it has proven very difficult to translate this promise into clinical success.
Methods In order to better utilize the tumor reactivity of γ9δ2T cells and combine this with the great potential of T cell engager molecules, we developed a novel bispecific molecule by linking the extracellular domains of tumor-reactive γ9δ2TCRs to a CD3-binding moiety, creating gamma delta TCR anti-CD3 bispecific molecules (GABs). GABs were tested in vitro and in vivo for ability to redirect T lymphocytes to a variety of tumor cell lines and primary patient material.
Results GABs utilizing naturally occurring high affinity γ9δ2TCRs efficiently induced αβT cell mediated phosphoantigen-dependent recognition of tumor cells. Reactivity was substantially modulated by variations in the Vδ2 CDR3-region and the BTN2A1-binding HV4-region between CDR2 and CDR3 of the γ-chain was crucial for functionality. GABs redirected αβT cells against a broad range of hematopoietic and solid tumor cell lines and primary acute myeloid leukemia. Furthermore, they enhanced infiltration of immune cells in a 3D bone marrow niche and left healthy tissues intact, while eradicating primary multiple myeloma cells. Lastly, GABs constructed from natural high affinity γ9δ2TCR sequences significantly reduced tumor growth in vivo in a subcutaneous myeloma xenograft model.
Conclusions We conclude that GABs allow for the introduction of metabolic targeting of cancer cells to the field of T cell engagers.
- immunotherapy
- lymphocyte activation
- receptors
- antigen
- T-lymphocytes
- translational medical research
Data availability statement
Data sharing not applicable as no datasets generated and/or analyzed for this study.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Among all immunological subtypes, γδT cells stand out in an unbiased computational analysis for their association with improved overall survival of patients with many different tumor types.1 γδT cells are innate like T cells that are present in both blood and tissue and are known to be important for recognition of foreign pathogens, stress signatures of infected cells, and cancer cells.2 In vitro, γδT cells display very potent and broad tumor recognition; they can target and lyse cancer cells of both hematological and solid origin.3 4 In contrast to αβT cells, γδT cells do not rely on HLA for target cell recognition.5 γ9δ2T cells, a γδ subset mainly present in the blood, are known to recognize an increase in intracellular phosphoantigens (pAg), which can be caused by microbial infections but are also found in many cancers.6 Recognition of intracellular pAg levels by γ9δ2TCRs relies on an inside out mechanism involving RhoB, BTN3A1, and BTN2A1.7–11 The metabolic targeting of tumor cells by γ9δ2T cells paves the way for novel tumor antigens for immunotherapy.12 Unfortunately, the adoptive transfer of ex vivo expanded polyclonal γδT cells associates so far with few clinical responses,13 most likely because of a significantly underestimated diversity, and many mechanisms of tolerance in advanced cancer patients that act against this particular immune subset.12 14 Most recently, restoring the αβ/γ9δ2T cell balance by BTN3A1 blocking antibodies has been suggested to hold great therapeutic promise as a new checkpoint inhibitor15; but only a fraction of tumors is infiltrated by γ9δ2T cells.1 T cells engineered to express a defined γδTCR (TEGs) have been proposed as an alternative strategy11 16–24 in line with the development of chimeric antigen receptor transduced T cells (CAR-T).25 26 However, advanced therapy medicinal products (ATMPs) such as genetically engineered T cells are delivered to patients with a substantial price tag,27 and production processes, as well as clinical implementation are cumbersome.28
To avoid the practical and economic challenges of ATMPs while still using the immune system to attack cancer, an alternative strategy is currently employed for classical antigens like CD19. Bispecific antibodies (bsAb) have been developed, fusing a tumor-targeting domain to a T cell binding domain, to recruit cytotoxic T cells to tumors. Such a bispecific T cell engager (BiTE) combining an anti-CD19 and anti-CD3 domain is now used in daily clinical practice,29 and many other bsAb for cancer immunotherapy are in various phases of clinical development.30 The selection of suitable tumor-associated target antigens for these novel therapies, however, remains very challenging, currently limiting the broad application of CAR-T and bsAb therapy.31
An alternative T cell engager strategy arose by linking the extracellular domain of an αβTCR as a tumor antigen binding domain to a single chain variable fragment (scFv) of a CD3 antibody.32 These αβTCR-bispecifics recognize intracellular peptides presented by MHC molecules, creating the possibility of targeting novel tumor-specific antigens that are not expressed at the cell surface. HLA restriction, however, also limits the use of such αβTCR-bispecifics to tumors with high mutational loads and defined HLA-types. Furthermore, downregulation of HLA is observed as an immune-escape mechanism in approximately 40%–90% of all human tumors,33 thereby greatly limiting the applicability of therapies based on αβTCR mediated tumor recognition.
To overcome these limitations and to combine the tumor specificity and therapeutic potential of γδT cells with the recent success of T cell engagers, we fused the extracellular domain of a γ9δ2TCR to an anti-CD3 scFv. We demonstrate that these Gamma delta TCR Anti-CD3 Bispecific molecules (GABs) with natural high affinity γ9δ2TCR can mimic the rather complex more pattern-like mode of action mediated by a γ9δ2TCR7 8 34 without the need of additional affinity maturation. GABs efficiently redirect αβT cells towards several tumor cell lines of both hematologic and solid origin, as well as primary patient material in vitro. Furthermore, we show significant reduction of tumor growth after GAB treatment in a subcutaneous myeloma xenograft model. We conclude that GABs open an avenue towards metabolic cancer targeting tumors with a bispecific format.
Methods
Generation of bispecific constructs
A customized pcDNA3-NEO vector, which allows consecutive expression of two genes of interest under their own CMV promoter, was a kind gift of Jan Meeldijk (LTI protein facility, UMC Utrecht, The Netherlands). First, the anti-CD3 scFv (OKT3)35 gene was cloned into multiple cloning site one. In addition to the antiCD3-scFv gene, the DNA fragment also contained bases encoding, a (G4S)3 flexible linker at the 5’ end and poly histidine tag on the 3’ end. At the 5’ end of the flexible linker, a BsiWI restriction site was present for the subsequent introduction of the TCR gamma chain in the vector, resulting in the TCR γ-CD3scFv fusion gene. The TCR δ-chain was cloned into the second multiple cloning site. TCR domain boundaries were used as in Allison et al.36 Most γ9- and δ2TCR sequences were reported previously,11 24 36 while other γδTCR sequences were obtained from randomly picked clones (table 1).
GAB sequences
Expression and purification of bispecifics
His-tagged GABs were expressed in 293 F cells. 293 F cells were cultured in Gibco Freestyle Expression medium, as transfection reagent Polyethylenimine (PEI) (25 kDa linear PEI, Polysciences, Germany) was used. Transfection was done using 293 F cells at a density of 1.106 cells/mL mixed with 1.25 µg DNA and 3.75 µg PEI per million cells. DNA and PEI were premixed in freestyle medium (1/30 of transfection volume), incubated for 20 min and added dropwise to the cell cultures. The cultures were maintained shaking at 37°C 8% CO2. Cell culture supernatant was harvested after 5 days and filtered through a 0.22 µm filter top (Milipore, USA). Supernatant was adjusted to 25 mM Tris (Sigma Aldrich, Germany), 150 mM NaCl (Sigma Aldrich, Germany) and 15 mM Imidazole (Merck, Germany) (pH 8). Supernatant was loaded on a 1 mL HisTrap FF column (GE Healthcare, USA) using the ÄKTA start purification system (GE healthcare, USA). Column was washed with IMAC loading buffer (25 mM Tris,150 mM NaCl 15 mM Imidazole (pH 8), and protein was eluted using a linear imidazole gradient from 21 to 300 mM in 20 CV. Fractions containing the GAB were pooled, concentrated and buffer exchanged to TBS (25 mM tris, 150 mM NaCl, pH 8) using vivaspin 4 10 kD spin columns (Sartorius, Germany). Protein was diluted 100 times in IEX loading buffer (25 mM Tris pH 8), and loaded onto a HiTrap Q HP 1 mL column (GE healthcare, USA) using the ÄKTA start purification system, for a second purification step. Column was washed with 10 column volumes IEX loading buffer, and protein was eluted using a linear NaCl gradient form 50 to 300 mM in 25 CV. Fractions containing the GAB were pooled, concentrated using vivaspin 4 10 kD spin columns (Sartorius, Germany) and examined by SDS-PAGE and staining with Instant blue protein stain (Sigma Aldrich, Germany). Protein concentration was measured by absorbance on Nanodrop and corrected for the Extinction coefficients. Protein was snap frozen and stored at −80°C and thawed before use.
Cell lines, flow cytometry, IFNγ Elispot, CD107 degranulation assay, luciferase based cytotoxicity and the animal model are reported in online supplemental methods.
Supplemental material
In vitro bone-marrow model
The 3D model was previously described in detail.20 In short: primary CD138+ were selected from the mononuclear cells (MNCs) of myeloma bone marrow from two patients by MACS separation using microbeads (Miltenyi Biotec, Germany). The CD138+ cells and the RPMI 8226 tumor cells were stained with Vybrant DiO (Thermo Fisher, USA) and seeded in Matrigel (Corning, USA) together with multipotent mesenchymal stromal cells (MSCs) and endothelial progenitor cells (EPCs), both stained with Vybrant DiD (Thermo Fisher, USA). After 4 days, T cells were stained with Vybrant DiI (Thermo Fisher, USA) and administered to the model together with CL5 or LM1 GAB (30 µg/mL) and 10 µM PAM (Calbiochem, USA). One day later, the culture medium was refreshed with medium contaning 30 µg/mL GAB. Tumor cells, T cells and stromal cells within and surrounding the matrigel were visualized 2 days later by confocal imaging. Afterwards, the Matrigel was dissolved using Dispase (Corning, USA) to retrieve the cells from the model. The cells were quantified by flow cytometry using Flow Count Fluorospheres (Beckman Coulter, USA) and normalized to mock treatment.
Results
Production of highly pure GABs
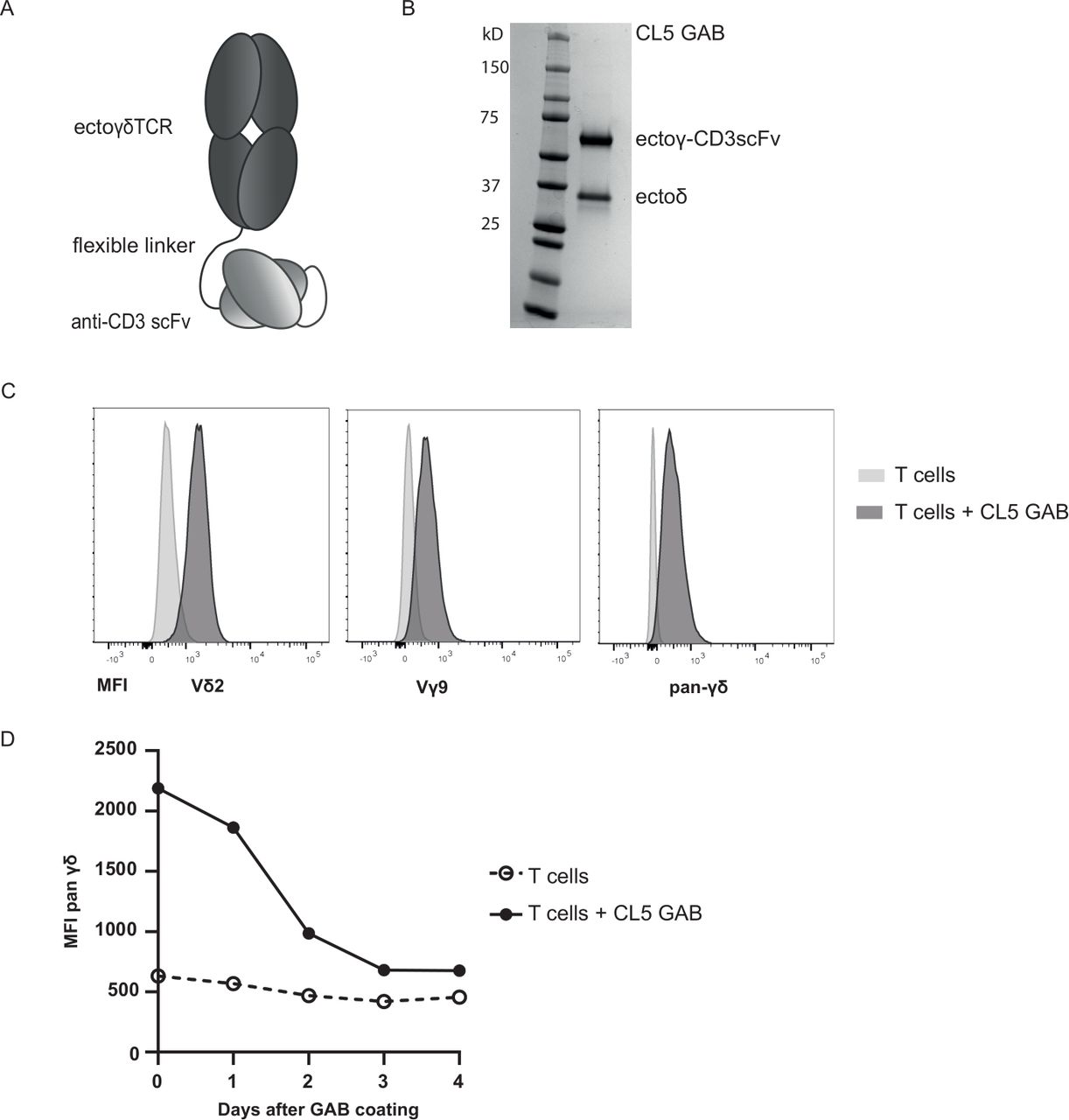
In line with the observation that antibodies and high affinity αβTCR can be linked to anti-CD3scFvs to redirect T cells to tumor cells,32 we assessed whether the γ9δ2TCR CL5 (table 1) was able to mediate antitumor reactivity in a bispecific format. Gamma delta TCR Anti-CD3 Bispecific molecules (GABs) were cloned with an anti-CD3scFv derived from the anti-CD3ε antibody OKT3 linked to the C terminus of the gamma chain of a soluble γ9δ2TCR, using a flexible (G4S)3 linker (figure 1A).

GAB design and binding to CD3+T cells. (A) Schematic representation of the GAB design, showing the extracellular γδTCR domain linked to an anti-CD3 scFv via a flexible linker. (B) Purified GAB was run on SDS-page gel and stained with coomassie brilliant blue protein stain, visualizing the ectoγ-CD3scFv and ectoδ-chain. (C,D) Coating of αβT cells with GAB (10 µg/mL (C) or 90 µg/mL (D)), followed by staining with fluorochrome labeled anti-Vγ9, Vδ2 or pan γδ antibodies. MFI was measured by flow cytometry and represented in histograms. GAB, gamma delta TCR anti-CD3 bispecific molecules
CL5 GAB was expressed in mammalian freestyle 293 F cells as secreted protein, and purified from the culture supernatant using His-tag purification, followed by a second ion exchange purification step, to ensure a highly pure protein product. As expected, the two different chains of the GAB, ectoGamma-CD3scFv and ectoDelta, were both clearly visible on gel (figure 1B). This indicates that during expression, the two separate chains of the GAB associate properly, resulting in a heterodimeric bispecific molecule.
GABs bind to αβT cells
To further address proper folding of GABs, we employed a flow cytometry based analysis. αβT cells were incubated with CL5 GAB, followed by a secondary staining using fluorochrome labeled antibodies against Vδ2, Vγ9 or panγδTCR (figure 1C). A strong and specific staining could be observed with all three antibodies, further indicating that the CD3scFv and both TCR chains are properly associated and folded. Following GAB binding on the cell surface of T lymphocytes that were coated with CL5 GAB over time, shows GAB binding up to 4 days after initial binding to CD3 (figure 1D), with a declining signal after 2 days implying, as for other bispecific molecules,37 that continuous presence of the molecule will be needed to maintain efficacy.
GABs induce pAg-dependent tumor recognition by αβT cells which is influenced by variations in the Vδ2 TCR chain
γ9δ2T cells are known to recognize SCC9 cells, a squamous cell carcinoma cell line. This recognition can be enhanced by treating tumor cells with pamidronate (PAM), which causes an increase in the intracellular phosphoantigen (pAg) levels by inhibiting the mevalonate pathway.24 To test whether GABs can also induce recognition of this cell line, αβT cells and SCC9 target cells were coincubated with and without PAM, and CL5 or LM1 GAB. LM1 GAB was generated to serve as negative control, LM1 GAB harbors a γ9δ2TCR where the CDR3 region of the δ-chain is replaced by a single alanine, making the γ9δ2TCR non-functional.24 As anticipated, CL5—but not LM1 GAB, induced recognition of SCC9 target cells by αβT cell in the presence of PAM (figure 2A), suggesting that the mode of recognition by GABs is comparable to recognition mediated by γ9δ2TCRs expressed at a cell membrane.11

GABs induce pAg dependent tumor recognition by αβT cells. (A–E) IFNγ production was measured by elispot, if separate spots could not be distinguished, spot count was set to a maximum value of 800. (A) αβT cells were co-incubated with SCC9 target cells in the presence or absence of PAM (100 µM) and LM1 or CL5 GAB (10 ug/mL), values are corrected for T cells only (n=4). (B) T cells were incubated with SCC9 target cells, PAM (100 µM) and an increasing concentration of GABs derived from different Vγ9Vδ2TCRs. A representative experiment is shown. (C) γ-chain HV4 mutations shown to hamper TCR binding were tested in the GAB format, αβT cells and target cells were co-incubated with the wildtype or mutant GABs (10 µg/mL) and PAM (100 µM) n=2. (D) AJ8 GAB (10 µg/mL) was co-cultured with T lymphocytes, HL60, HEK293FT WT or BTN3A1 knockout cells with and without PAM (100 µM) n=1 in duplo. (E) CL5 and AJ8 GAB (10 µg/mL) were tested in a coculture of T cells and a larger panel of target cell lines with and without the addition of PAM (100 µM), and compared with mock GAB LM1 n=2. Error bars represent SD, significance was calculated using a multiple T test (A) or one-way ANOVA (C–E). * P<0.05,**p<0.001, ***p<0.0001. ANOVA, analysis of variance; GAB, gamma delta TCR anti-CD3 bispecific molecules; PAM, pamidronate.
We11 24 and others38–40 reported on the impact of changes in the CDR3 region of δ2TCR chains on TCR function. To assess the impact of variations in the CDR3 region of the δ2TCR chain on GAB activity, we generated a larger panel of GABs, derived from previously published γ9δ2TCRs11 24 36 and randomly picked γ9δ2T cell clones, varying in CDR3δ-chain (table 1). To assess activity, the different GABs were coincubated with αβT cells and SCC9 target cells in the presence of PAM. Most GABs efficiently induced an IFNγ response, though activity substantially differed between different constructs (figure 2B), although all showed similar binding to αβ T cells (online supplemental figure S1). GABs in which the CDR3δ were reduced to one alanine (LM1) did not induce IFNγ production at any concentration (figure 2B). Titrating GAB concentrations allowed for further analysis of the differences in efficacy between the different CDR3δ sequences. We observed large differences in GAB activity with an EC50 of 0.8 µg/mL for the best performing GAB to an EC50 of 25 µg/mL for the lowest activity. EC50 of several non-active or very low active receptors could not formally be assessed (figure 2B and table 2).
EC50 of GAB for IFNγ release
γ-TCR loop and BTN3A are critical for GAB mediated αβT cell activation
The HV4 region between the CDR2 and the CDR3 of the γ-chain is critical for γ9δ2TCR activity by binding to BTN2A1 expressed on target cells.9–11 To assess whether GABs also depend on this mode of action, we focused on GAB CL5, one of the most active TCR sequences from the tested panel, and introduced two mutations in the γHV4 region of CL5 GAB (E70D72→K70L72 (CL5EDKLGAB) and H85→R85 (CL5HR GAB)), reported to cause loss of activity in membrane expressed γ9δ2TCRs.41 CL5, CL5EDKL and CL5HR GABs were added to a coculture of αβT cells with the well-described breast cancer cell line (MDA-MB231) or multiple myeloma cell line (RPMI 8226)11 24 in the presence of PAM. CL5EDKL and CL5HR GAB lost activity, assessed by IFNγ production, when compared with the wild type CL5 GAB (figure 2C), highlighting the importance of the γHV4 region for target cell engagement by GABs.
BTN3A has also been recognized as a crucial factor in phosphoantigen dependent γ9δ2TCR reactivity. Loss of BTN3A membrane expression on target cell leads to a complete loss of membrane-bound γ9δ2TCR reactivity to pAgs.11 42 By testing GAB mediated recognition of HEK293FT WT and BTN3A knockout, we confirmed that GAB induced recognition after PAM treatment also depends on BTN3A expression (figure 2D). These findings support the assumption that there is a similar binding mode between membrane-expressed γ9δ2TCRs and GABs, both depending on encounter of BTN2A1 through the γ-chain and a second signal, which is pAg and BTN3A depended.
GABs retarget αβT cells to a wide variety of tumor cells
Next, we addressed whether GABs can redirect αβT cells to a broader variety of tumor cells, and whether GABs with different EC50 against SCC9 target cells also have different activities against a broader range of hematological and solid tumor cells. GABs with lower (AJ8) and higher (CL5) EC50 or the negative control LM1 GAB were coincubated with αβT cells and previously defined panel of tumors targets cells.43 A significant increase in IFNγ production was observed for CL5 and AJ8 GABs against most tumor targets except for HL60 and MDA-MB157, while LM1 GAB did not induce cytokine secretion (figure 2E). For most cell lines, CL5 GAB had a slightly higher activity compared with AJ8 GAB, although not always significant. Isolated CD4+ and CD8+ αβT cells induced IFNγ release after coincubation with CL5 GAB (online supplemental figure S2A). However, as expected, we observed that the relative contribution of CD4+ and CD8+ αβT cells differed between donors and target cells, with CD4+ αβT cells producing more cytokines in general.
As in blood up to 5% of the CD3+T lymphocytes are comprised of Vδ2+T cells, we next investigated GAB activity in combination with Vδ2+ and αβT cells side by side. Vδ2+ and αβT cells were isolated from a healthy donor and IFNγ release was measured after a coculture with two recognized (RPMI8226, SCC9) and one unrecognized cell line (ML-1) with and without CL5 GAB and in the absence or presence of PAM (online supplemental figure S2B). LM1 GAB was added as extra control to the Vδ2+T cells. As expected, the Vδ2+T cells alone recognized the positive target cell lines after PAM treatment; surprisingly however, this recognition was lower compared with αβT cells coincubated with CL5 GAB. Activity of Vδ2+T cells was not blocked by the addition of the mock LM1 GAB, and addition of GAB CL5 did not lead to a further increase in activation of the Vδ2+T cells. These data imply that GABs will most likely not activate Vδ2+T cells, which could be a consequence of the differences in CD3 composition of Vδ2+T cells versus αβT cells.44 This is also in line with the previous observation that Vδ2+T cell expansion protocols usually do not use CD3 engagers, but rather rely on agents that directly engage the TCR, such as phytohaemagglutinin (PHA).4 43
To this point, IFNγ production was used as a readout for GAB activity. However, the clinical activity of bispecific molecules comes through their ability to mediate killing of target cells. Therefore, as the next step, we assessed CD8+ αβT cell-mediated toxicity by using a degranulation assay detecting surface expression of the lysosomal-associate membrane glycoptrotein-1 (LAMP-1/CD107a) by flow cytometry (FC). αβT cells were cocultured with three different target cell lines and CL5, AJ8 or negative control LM1 GAB, with and without PAM for 7 hours (figure 3A). As an extra control, αβT cells and GABs were incubated together without target cells. Similar to the IFNγ release data, GABs induced degranulation of CD8+ αβT cells on binding to a target cell line in a PAM dependent manner, while no upregulation of CD107a was observed when coincubated with the negative control cell line HL60. To formally assess the ability of GABs to kill tumor targets, we employed a luciferase-based cytotoxicity assay. RPMI 8226 and SCC9 tumor cells stably transduced with a luciferase gene were cocultured with GABs and αβT cells at different effector to target (E:T) ratios. After a coculture of 16 hours, the bioluminescence was measured by adding beetle luciferin to the coculture. The amount of viable cells was determined by comparing the bioluminescence signal to untreated target cells (figure 3B). Both CL5 and AJ8 GAB efficiently induced up to 60%–80% lysis of the tumor cells at the lower effector to target cell ratios, while LM1 GAB had as little activity as αβT cells alone.
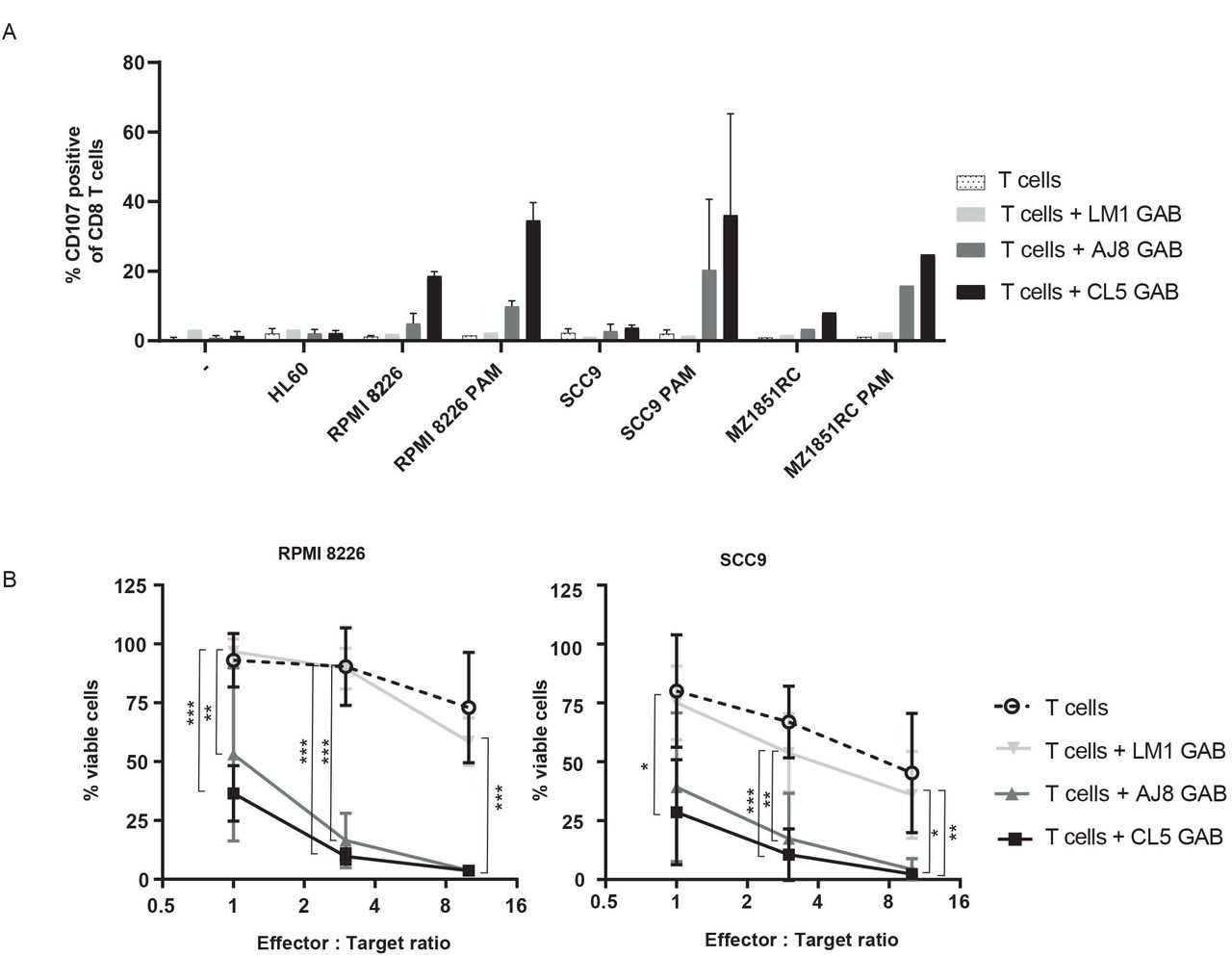
GABs induce T cell mediated lysis of cancer cell lines. (A) CD8+T cell degranulation was measured by staining with CD107a antibody during a 7-hour coincubation of T cell effector and three different target cell lines in the presence and absence of GAB (10 µg/mL) and PAM (100 µM). Golgistop was added during the incubation. N=2 (for LM1 GAB and MZ1851RC N=1) significance was not calculated because of amount of data points. (B) T effector and luciferase transduced RPMI 8226 and SCC9 target cells were coincubated for 16 hours in the presence and absence of GAB (10 µg/mL) and PAM (100 µM) at different E:T ratios. Percentage viable cells were determined by comparing luminescence signal to untreated target cells, representing 100% viability. N=3, error bars represent SD, significance was calculated using a one-way ANOVA. * P<0.05, **p<0.001, ***p<0.0001. ANOVA, analysis of variance; GAB, gamma delta TCR anti-CD3 bispecific molecules.
To extend our findings to GABs harboring sequences published by others,9 38 45 46 a second set of five GABs were generated (table 1) and tested for ability to induce target cell lysis after coincubation with αβ T lymphocytes and the multiple myeloma target cell line RPMI 822611 in the presence of PAM. As benchmark, we used the previously identified GABs with lower (AJ8) and higher (CL5) EC50 and as negative control LM1 GAB. Again, we observed differences in activity, GABs harboring sequences from CL5 were superior to all other tested GABs. Only the GABs derived from DGSF68 and MOP TCR were not significantly different from the lysis induced by AJ8 GAB, while the other three tested GABs were inferior to the AJ8 GAB (online supplemental figure S3).
GABs are active against primary leukemia but not against primary healthy tissues
To test whether GABs can mediate recognition of primary tumors such as primary acute myeloid leukemia (AML), αβT cells were cocultured with AJ8 GAB and primary AML blasts of four patients, with and without PAM. GABs induced a significant increase in IFNγ production on PAM treatment against two out of the four patient samples (figure 4A).

GABs induce recognition of primary AML samples but not of healthy hematopoietic cells or fibroblasts. (A) αβT cells were incubated with AML blasts with or without 100 µM PAM, and 10 µg/mL AJ8 GAB. IFNγ production was measured by elispot after 24 hours. Fold change in IFNγ production on addition of pamidronate was calculated N=1. (B) αβT cells were cocultured with healthy hematopoietic cells or fibroblasts and LM1 or CL5 GAB (10 ug/mL). Target cells were activated as indicated, or stressed by irradiation or a combination treatment with cyclophosphamide (Cyclo) and fludarabine (Fluda). IFNγ release was measured by elispot. The figure represents pooled data from four different target cell donors (CD19+ and CD14+) or two donors (CD34+ and fibroblasts). αβT effector cells were derived from four different donors (CD19+ and CD14+) or two donors (CD34+ and fibroblasts). Error bars represent SD, significance was calculated using a multiple T test. * P<0.05, **p<0.001, ***p<0.0001. GAB, gamma delta TCR anti-CD3 bispecific molecules.
Given the broad activity of GABs, we next assessed their ability to sense healthy tissues in a resting or stressed situation. To this end, we isolated B cells, monocytes and CD34+ cells from a healthy donor, and tested reactivity of CL5 and LM1 GAB against these cells and against healthy donor-derived fibroblasts in an IFNγ release assay. Recognition of the cells was tested in resting and also activated or stressed conditions, such as after irradiation or chemotherapy treatment. Neither CL5 nor LM1 GAB induced recognition of healthy cells, in resting, activated or stressed conditions, while the positive control, RPMI 8226 tumor cells, induced cytokine release when incubated with CL5 GAB (figure 4B).
Favorable efficacy toxicity profile of GABs in the bone marrow niche
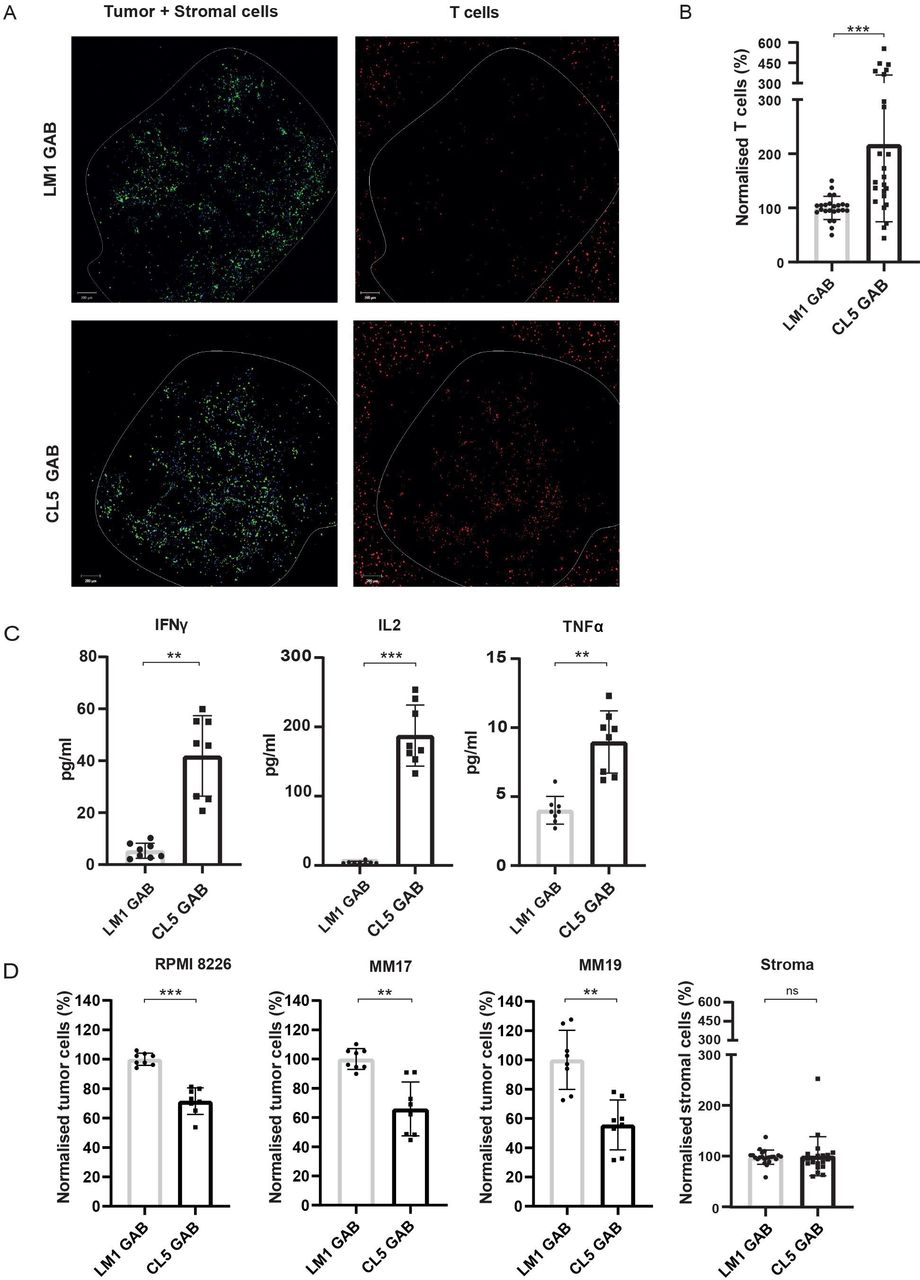
In vivo, the tumor microenvironment is often important for survival and proliferation of tumor cells. Therefore, we tested whether GABs can also eradicate primary tumor material without harming healthy tissues in a more natural environment, using a previously described 3D bone marrow niche model.20 In this model, mesenchymal stem cells (MSC) and endothelial progenitor cells (EPC) are used as stromal support for the growth of a multiple myeloma (MM) cell line (RPMI 8226) or primary CD138+ MM cells derived from patients. CD138+ MM cells from two patients, and the MM cell line RPMI 8226 were stained and seeded in matrigel together with MSCs and EPCs. After 4 days, labeled αβT cells, together with CL5 or LM1 GAB and PAM were added to the model. One day later, fresh medium with GABs was added to the model to ensure constant GAB coating on the αβT cells. After 2 days, visualizing αβT cells infiltrated into the tumor bearing matrigel by confocal microscopy indicated an increased αβT cell infiltration in the presence of Cl5, but not LM1 GAB (figure 5A). This observation was supported by a subsequent FC based quantification of the αβT cells present in the matrigel (figure 5B). To further study specific αβT cell activation by GABs, we measured several cytokines in the supernatant of the 3D model containing primary MM tumor cells. Next to IFNγ, we also observed a significant increase in the levels of other Th1 cytokines, IL2 and TNFα for the CL5 GAB condition (figure 5C).

GABs mediate recognition and lysis of primary multiple myeloma in a 3D model. The RPMI 8226 tumor cell line or primary MM patient material was cultured in a 3D bone marrow niche consisting of matrigel and stromal cells. After 4 days, αβT cells were added together with PAM (10 µM PAM) and GAB (30 µg/mL). (A) Confocal images showing cell localization within and around the 3D model (boundaries indicated by the white line) with the tumor and stromal cells, respectively, in green and blue and T cells in red. (B) Two days after addition of the T cells, the matrigel was dissolved to retrieve the cells from the model. αβT lymphocytes were quantified by flow cytometry and normalized to mock treatment. (C) Cytokines were measured in the supernatant by luminex. (D) Tumor and stromal cells were collected from the dissolved matrigel and quantified by FC. Cell numbers were normalized to mock treatment. Significance was calculated by a paired T test. *P<0.05, **p<0.001, ***p<0.0001. N=4 with technical duplo’s. GAB, gamma delta TCR anti-CD3 bispecific molecules
The most important measurement remains the elimination of tumor cells. Therefore, the amount of tumor cells remaining in the model after CL5 GAB treatment was determined by FC analysis and cell numbers were normalized to treatment with mock LM1 GAB. Treatment with CL5 GAB induced a signification reduction of CD138+ MM cells compared with the mock treatment with LM1 GAB, for both patient samples and the MM cell line RPMI 8226 (figure 5D). Healthy stromal cells were also quantified, showing no differences between CL5 or LM1 GAB treatment (figure 5D), suggesting that surrounding healthy tissues are not affected by the treatment with active GAB.
GABs control tumor growth in vivo
To test whether treatment with GABs can also affect tumor growth in vivo, we established a xenograft model by injecting RPMI 8226 multiple myeloma cells subcutaneously (s.c) into NSG mice. For this in vivo experiment, we generated RPMI 8226 B2M knock-out cells that we injected s.c, as in previous experiments intravenously injected WT RPMI 8226 cells were rejected when coengrafted with human PBMC, most likely due to allo-reactivity (online supplemental figure S4). One week after tumor cell injection, mice received an intravenous injection of human PBMCs (figure 6A). Next, the mice were randomized over two groups, based on tumor size, and received treatment for seven consecutive days with CL5 GAB or the mock LM1 GAB. Moreover, an additional group in which mice received tumor and PBMCs but no GABs was included as extra control to monitor coengraftment of PBMCs and tumor in NSG mice. Tumor volume was measured three times per week for 30 days. Treatment with CL5 GAB significantly decreased tumor growth compared with the control group treated with LM1 GAB (figure 6B). Furthermore, mice treated with LM1 GAB showed similar tumor outgrowth compared with the PBMC only group. Persistence of GABs bound to αβ T cells in the blood was determined by flow cytometry 1, 2 and 8 days after GAB injection by calculating absolute number of αβTCR- and αβTCR/γδTCR double positive (GAB coated) T cells. figure 6C shows that 24 hours after the first GAB injection (day 10) and 48 hours after the last GAB injection (day 17), around 30% of the total αβTCR positive cells are αβTCR/γδTCR double positive, meaning that there are still GABs bound to the T cells. Furthermore, we found that 8 days after the last GAB injection (day 23), this double positive population was no longer present.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
In vivo control of tumor growth by GABs. (A) Schematic representation of experimental design. NSG mice were irradiated at day −1, and injected subcutaneous (s.c) with 10*106 RPMI 8226 tumor cells 1 day later. After 7 days, the mice were randomized over three groups, based on tumor size (N=10). From day 9 to 15, mice in two groups were treated with one intravenous injection per day of CL5 or LM1 GAB (2,7 mg/kg). Tumor size was measured three times per week for 3 weeks after start of the GAB treatment (B) and is plotted as percent change in tumor volume compared with the initial tumor volume at the start of the GABs treatment. (C) Amount of αβTCR single positive and αβTCR/γδTCR double positive cells in the mice was determined by flow cytometry on day 10, 17 and 23 after tumor injection, which corresponds to 24 hours after the first GAB injection and 48 hours and 8 days after the last GAB injection. Data are shown as mean of percentage of total αβTCR positive cells. PBMC only N=4, LM1/CL5 GAB N=10. Error bars represent SEM, significance was calculated by mixed-effects model with repeated measures. * P<0.05, **p<0.001, ***p<0.0001. GAB, gamma delta TCR anti-CD3 bispecific molecules.
Discussion
In this study, we developed a novel bispecific T cell engager format, gamma delta TCR Anti-CD3 Bispecific molecules (GABs), based on the fusion of a soluble γ9δ2TCR to an anti-CD3 scFv. With GABs, we introduce the targeting of cancer as a metabolic disease to the field of bispecific T cell engagers. GAB activity against tumor but not healthy tissues was observed when using naturally occurring high affinity γ9δ2TCR and relied, as for membrane bound γ9δ2TCR, on the complex orchestration of BTN2A1 and BTN3A1 and was modified by intracellular phosphoantigen levels.8 11 12
Most T cell engagers use tumor targeting domains with binding affinities in the nanomolar range, a 10–100-fold affinity maturation has been reported to further enhance activity.37 47 For T cell engagers with an αβTCR as tumor binding domain, affinity maturation from the micromolar to picomolar range is needed to overcome the rather low overall avidity mediated by a low density of tumor associated molecules within the context of MHC molecules, in order to create functional T cell engagers.48 Therefore, it was initially surprising that a γ9δ2TCR is active in the bispecific format without artificial affinity maturation, while natural αβTCR showed only a little activity.32 Most recent studies estimated the binding affinity of the γ9 chain to BTN2A1 to be around 40 µM9 which is in the range of αβTCRs.49 However, the number of BTN2A1 molecules that are present on the cell surface for binding to the γ9 TCR chain is most likely substantially higher compared with tumor associated antigens in HLA complexes, potentially generating a higher avidity for γ9δ2TCR based T cell engagers compared with αβTCRs. This however does not explain why, in our data set, only a selected group of defined γ9δ2TCR clones was active in the GAB format.
The reported affinity of the γ9 chain to BTN2A1 (9) is presumably an underestimation of the binding affinity of the γ9δ2TCR to its complete interacting complex, as the TCR binding is not solely mediated by the γ9-chain. This assumption is supported by our previous observation that apart from the γ-chain, variations in the CDR3 region of the δ2 chain also contribute substantially to the overall functional avidity of γ9δ2 TCRs once expressed in a T cell.11 24 δ2TCR sequences that were previously reported to mediate high overall efficacy when expressed at the cell membrane,11 also mediated high activity when used in the GAB format, for example, CL5 and A3. Vice versa, sequences which mediated lower efficacy in the TEG format were even poorer performers in the GAB format, for example, A1. Thus, as both the γ9-chain and δ2-chain contribute to the affinity of a γ9δ2 TCR to its complex, a careful selection of δ2TCR sequences is needed guarantee a functional GAB.
Transforming cold into hot tumors is a key success factor for immune therapies.50 Novel αβTCR based biologics have been reported to warm ‘cold’ tumors.51 By using a 3D bone marrow niche model for primary MM cells,20 we provide evidence that γ9δ2TCR, when provided in the GAB format, can initiate infiltration of immune cells into the tumor microenvironment. This was further confirmed by the in vivo model, showing that GABs can reduce tumor growth of a subcutaneously growing RPMI 8226 tumor.
Furthermore, as the used 3D model was comprised of healthy MSC and EPC to guarantee survival and proliferation of MM cells in vitro,20 this model also allowed us to assess the impact of GABs on healthy tissues and extended our in vitro safety data for GAB. These current data confirm the previously reported lack of toxicity of targeting BTN2A1 and BTN3A1 when using a high affinity γ9δ2TCR in the TEG format17 20 23 24 or when administering BTN3A1 targeting antibodies.15
In this report, we tested the reactivity of GABs to patient material from several patients with AML and found that GABs were reactive to two out of the four samples. This observation is in line with our previous report assessing larger tumor panels, including 16 patients with AML, which suggest that approximately 50% of all tumor cells are recognized by primary γδT cells or TEGs.52 Mode of action studies investigating requirements for γ9δ2TCR mediated tumor cell recognition, conducted in order to elucidate this differential tumor recognition, pointed to multiple factors such as pAg dependent rearrangement of the BTN2-BTN3 complex involving RhoB and the intracellular B30.1 domain of BTN3A1 (9–11). However, these studies also imply that a yet to be defined second ligand, binding to the CDR3δ is most likely involved. Thus, although a lot of knowledge has been obtained over the past years, tumor recognition mediated by a γ9δ2TCR cannot be fully explained and predicted yet.11 Therefore, further investigation into the complex γ9δ2TCR mediated target cell recognition, and the identification of novel biomarkers that can help identifying patient populations that are susceptible to γδ based therapies will be key for a successful clinical translation.12
The GAB format outperformed natural γ9δ2T cells, as reported previously for TEGs,11 43 most likely reflecting the careful selection of a high affinity γ9δ2TCR in the GAB or TEG design. Despite this superior activity, a limiting factor for γ9δ2TCR mediated target cell recognition remains the requirement for pAg accumulation, also GAB mediated recognition of many cancer cells required additional treatment with amino-bisphosphonates to increase pAg levels. To elucidate why tumor cells differ in the dependence on PAM to enhance γ9δ2TCR recognition further investigation will be needed, but it is most likely a consequence of different availabilities of all the characterized key components for γ9δ2TCR binding, including, but not limited to, the intracellular accumulation of pAgs. The dependence on increased intracellular pAg levels for recognition of many tumors does however imply that γ9δ2TCR based therapeutic strategies most likely need to be combined with amino-bisphosphonate treatment, a state of the art drug safely combined with many different treatments including γ9δ2T infusions.12
In conclusion, we have shown that a γ9δ2TCR bispecific format can mimic the rather complex metabolic cancer targeting usually mediated by membrane bound γ9δ2TCR,7 8 34 though requires a very careful selection of the used sequences and then allows for the introduction of the unique tumor targeting potential of γ9δ2T cells to the field of bispecific T cell engagers. Our findings imply also that, in contrast to previously reported data for αβTCR derived bispecifics, selecting an endogenously occurring high affinity γ9δ2TCR for use in a bispecific format could omit the need for affinity maturation. Since the use of affinity matured TCRs poses the risk of altering the TCR specificity or introducing cross-reactivity,53 54 using a therapy based on the endogenous TCR affinity could be a preferred strategy. This approach might overcome cumbersome engineering efforts and provide with GABs and TEGs, two complementary or even additive strategies as reported for CAR-T and bsAbs55 to harvest the potential of the new universe of targets extracted from γδT cells.
Data availability statement
Data sharing not applicable as no datasets generated and/or analyzed for this study.
Ethics statements
Patient consent for publication
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
EvD and PHL contributed equally.
Correction notice This article has been corrected since it was first published. The Abstract section has now been added.
Contributors EvD, DB, and JK wrote the paper. EvD, PHL, AM, AV, EK, SH, JGB, SvD, MN, LG, IJ, TS, ZS, and DB performed experiments. JK is guarantor. All authors approved the final manuscript.
Funding We thank the staff of the Flow Core Facility and the Multiplex Core Facility at the UMC Utrecht. We kindly thank Professor Erin Adams (The University of Chicago) for providing the CD277 KO HEK293T cell line and Halvard Boenig (Institute for Transfusion Medicine and Immunohematology, Goethe University, Frankfurt a. M., Germany) for providing feeder cells. Funding for this study was provided by ZonMW 43400003 and VIDI-ZonMW 917.11.337, KWF 2013-6426, 2014-6790 2015-7601, 2018-11393, 2018-11979, 2020-13403 to JK, KWF 2018-11393 2020-13403 to ZS, KWF 11979 and Marie Curie 749010 to DB.
Competing interests JK is shareholder of Gadeta (www.gadeta.nl). JK, ZS, DB, AV, and EvD are authors on the following patent applications: 2013: WO 2013 /147606 A1 Combinatorial gamma 9 delta 2 T cell receptor chain exchange, (C. Grunder, JK); 2017: 20170319674 Use of antibodies for enrichment of engineered t cells with exogenous immune receptors and antibodies for use in depletion of engineered t cells (JK) ; 2017: WO/2017/2017/212074 Novel method for identifying delta T cell (or gamma T cell) receptor chains or parts thereof that mediate antitumor or anti-infectious response (JK and colleagues; 2017: WO/2017/212072 Human leukocyte antigen restricted gamma delta T cell receptors (JK and colleagues), 2017/ 62508807 (ZS & JK) Composition and methods for cell targeting therapies; 2018/ 6044506 (EvD, DB, JK): GABs as next generation of immune therapy
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.