Article Text

Statistics from Altmetric.com
The prognosis of patients with relapsed/refractory (R/R) acute lymphoblastic leukemia (ALL) remains poor, particularly for those relapsing after allogeneic hematopoietic cell transplantation (alloHCT).1 Novel agents such as inotuzumab ozogamicin or blinatumomab achieve increased response rates, but these are generally transient unless followed by alloHCT. Chimeric antigen receptors (CAR) targeting CD19 have shown promising results in R/R ALL, and one of these products (tisagenlecleucel) has been approved for the treatment of patients with R/R ALL up to 25 years of age.2
In 2013, we designed our own CAR19 construct, comprising a single chain variable fraction sequence from the A3B1 hybridoma and the 4-1BB/CD3z signaling domains (online supplemental figure 1). After scaling up both lentiviral and cell production,3 4 the Spanish Medicines Agency (AEMPS) approved our product (ARI-0001 cells) and the CART19-BE-01 trial.5 In October 2019, once the recruitment was completed, the AEMPS approved a compassionate use program (CUP) for patients fulfilling the same inclusion/exclusion criteria. The trial’s preliminary results were published elsewhere.5 Here, we present the long-term results of all consecutive patients with R/R ALL recruited into the CART19-BE-01 trial and the subsequent CUP. Detailed information on ARI-0001 cell manufacturing, inclusion/exclusion criteria, primary and secondary endpoints and assessment criteria can be found elsewhere.5
Supplemental material
Before ARI-0001 cell infusion, patients received fludarabine at 30 mg/m2/day plus cyclophosphamide at 300 mg/m2/day on days −6, –5, and −4 followed by ARI-0001 cells. The first 15 patients received a single intravenous infusion of 0.5–1×106 ARI-0001 cells/kg (adults) or 5×106 ARI-0001 cells/kg (children) on day 0. The following 38 patients (23 from the CART19-BE-01 trial and 15 from the CUP) received 1×106 ARI-0001 cells/kg regardless of age: the first fraction (10%) on day 0, followed by the second (30%) and third (60%) fraction. The second fraction was administered 24–48 hours after the first, and the third 24–48 hours after the second, only if the patient had no signs or symptoms of CRS (online supplemental figure 2). The reason for this amendment to the protocol were three cases of fatal toxicity (two patients, aged 11 and 19, who died of refractory CRS, and one patient, age 35, who died of pseudomembranous colitis as a complication of grade 4 CRS).5 The entire patients’ disposition is depicted in online supplemental figure 3.
Adverse events and response rates are presented with 95% exact Clopper-Pearson CIs. The possible association between CRS (all grades and grade ≥3), tumor burden at screening (<5% vs ≥5% blasts in the bone marrow (BM)) and type of administration (single dose vs fractionated) was assessed using Fisher’s exact test. We also analyzed the impact on progression-free survival (PFS) and overall survival (OS) of the following variables: age (<25 vs ≥25 years), type of administration, tumor burden and loss of B-cell aplasia (BCA), the latter as a time-dependent covariate. PFS/OS curves were plotted using the Kaplan-Meier method for time-fixed covariates and the Simon-Makuch method for BCA loss. Landmark analyses were performed to identify the most appropriate timepoint for BCA loss. Univariate Cox regression was used to evaluate the impact of these covariates on PFS/OS, and those with Benjamini-Hochberg adjusted p values lower than 0.1 were introduced into a multivariate Cox regression. Schönfeld residuals were used to check the proportional hazards assumption. The trial was registered at clinicaltrials.gov (NCT03144583).
Fifty-three patients with R/R ALL received therapy with ARI-0001 cells: 38 in the context of the CART19-BE-01 trial and 15 as part of the CUP. The median age was 30 years (range, 3–68), while 24 (45%) patients were female. Baseline characteristics of the entire population are displayed in online supplemental table 1). The data cut-off date was March, 2021, when all infused patients had a minimum follow-up of 100 days or had experienced disease relapse or death before that date.
Patients received ARI-0001 cells a median of 55.5 days (range, 27–216) after inclusion, and the median vein-to-vein time (from apheresis to infusion) was 43 days (range, 21–190). The original target dose was infused to all except 3 (5.7%) patients who received 0.1–0.4×106 ARI-0001 cells/kg due to CRS. CRS was reported in 56.6% (95% CI 42.3%–70.2%) of patients, being grade ≥3 in 11.3% (95% CI 4.3% to 23%) and requiring treatment with tocilizumab and steroids in 20.7% and 11.3% of patients, respectively. Patients with ≥5% lymphoblasts in the BM had a higher incidence of CRS (any grade: 82% vs 39%, p=0.0022; grade ≥3: 27% vs 0%, p=0.0036) compared with those with <5% lymphoblasts. Moreover, the incidence and severity of CRS was also associated with single dose versus fractionated administration of ARI-0001 cells (any grade: 87% vs 45%, p=0.0064; grade ≥3: 27% vs 5%, p=0.047). Neurotoxicity was observed in 13.2% (95% CI 5.5% to 25.3%) of patients, with one self-limited grade ≥3 occurrence (1.9%). No new second malignancies have been reported apart from a previously notified case of myelodysplasia (1/53; 1.9%).5
The safety profile of ARI-0001 cells was comparable to that of similar products, with grade ≥3 CRS/neurotoxicity rates lower than 5%.2 6 Moreover, both fractionated administration and tumor burden were significantly associated with the incidence of CRS, in keeping with similar studies.7–10 Of note, two patients enrolled in the CUP experienced grade ≥3 CRS with the first fraction (0.1×106 cells/kg), but successfully recovered after treatment with tocilizumab. Both patients had a high tumor burden (>90% blasts in the BM) at study inclusion, and yet they could receive therapy while avoiding irreversible toxicity.
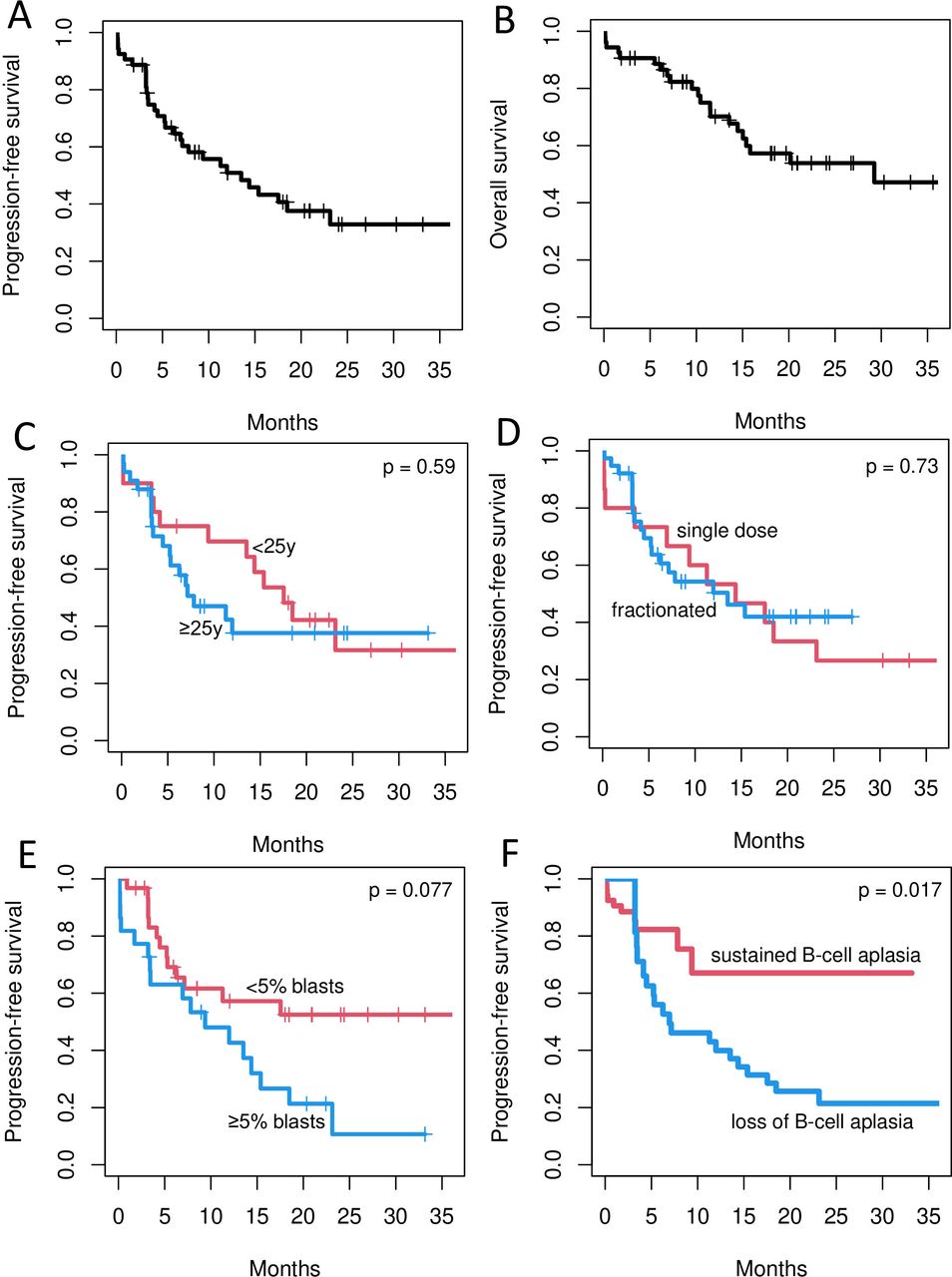
The measurable residual disease (MRD)-negative CR rate was 88.6% (95% CI 77.0% to 95.7%) at day +28% and 79.2% (95% CI 65.9% to 89.2%) at day +100. All three patients who received less than 1×106 ARI-0001 cells/kg due to toxicity achieved an MRD-negative CR. All evaluable patients (n=50) developed absolute BCA that lasted for a median of 4.2 months (95% CI 3.32 to 7.53 months). PFS was 50.9% (95% CI 38.4% to 67.4%) and 32.9% (95% CI 20.6% to 52.6%) at one and 2 years, respectively, while the 1-year and 2-year OS were 70.2% (95% CI 58.1% to 84.8%) and 53.9% (95% CI 40.5% to 71.8%). Progressive disease has occurred in 27 (50.9%; 95% CI 36.8 to 64.9%) patients at a median of 5.3 (range, 0.2–23.1) months. Tumor cells expressed CD19 in 24 (89%) of these relapses, while three (11%) were CD19-negative. ARI-0001 cells served as a bridge to alloHCT in three (6%) patients. Second ARI-0001 infusions were documented in nine patients (three more than previously reported5: four due to CD19 +relapse and five in patients with early BCA loss). These resulted in transient responses and brief periods of BCA, but one of these responses allowed the patient to receive a second alloHCT.
Subgroup analyses are depicted in figure 1 and online supplemental table 2. By univariate analysis, only two variables had a potential impact on PFS: tumor burden (<5% vs ≥5% lymphoblasts in the BM at screening), with a 2-year PFS of 52.5% (95% CI 36.4% to 75.7%) vs 10.7% (95% CI 2.1% to 54.4%) and an HR of 2.14 (95% CI 1.04 to 4.42) for patients with 5% or more blasts (adjusted p=0.077). On the other hand, loss of BCA had an HR of 4.41 (95% CI 1.59 to 12.2), adjusted p=0.0172. Both variables (tumor burden and loss of BCA) were also confirmed in the multivariate model, with an HR of 2.05 (95% CI 1.004 to 4.17) for patients with 5% or more blasts at screening (p=0.0484) and an HR of 4.32 (95% CI 1.57 to 11.86) for patients with loss of BCA (p=0.0045). Regarding OS, none of the covariates evaluated had sufficient impact to justify a multivariate analysis. Seeing that BCA loss had such an impact on PFS, we performed a series of landmark analyses to identify the most appropriate cut-off for clinical practice. We chose 3 and 6 months as potential landmark times because the median time to BCA loss was 4.2 months in our series. According to these analyses, the 3-month time point was the closest to statistical significance (HR 1.83; 95% CI 0.82 to 4.11; p=0.15, (online supplemental table 3).
{kind=link}
Progression-free (PFS) and overall survival (OS) of patients with relapsed/refractory acute lymphoblastic leukemia treated with ARI-0001 cells. A, B show the PFS (A) and OS (B) of the entire population. C–F depict the PFS of patients according to age (C) (red curve: <25 years; blue curve: ≥25 years); type of administration (D) (red curve: single dose; blue curve: fractionated); percentage of blasts in the bone marrow (E) (red curve: <5%; blue curve: ≥5%); and loss of B-cell aplasia (F) (red curve: no; blue curve: yes). P values refer to the Benjamini-Hochberg-adjusted univariate analysis.
With more patients and longer follow-up, we identified two covariates of potential predictive value: tumor burden in the BM and loss of BCA. This contrasts with our previous report, where this potential effect was not fully evident.5 The adverse impact of tumor burden has been documented for other therapies, including alloHCT and CART19-cells,8 10 and is therefore not surprising. The importance of BCA loss is, on the other hand, more controversial. Our strategy was associated with a relatively short in vivo ARI-0001 cell survival (median 4.2 months) and most relapses had a CD19-positive phenotype, all in keeping with other studies.8 10 Consequently, further immune manipulation may be justified in patients with a short-lived BCA, perhaps less than 3 months, although this potential timepoint needs further validation.
In conclusion, both tumor burden and BCA loss appeared to have a significant impact on PFS and could guide clinicians in the management of patients after cell infusion. The validity of the fractionated cell administration was also confirmed and will be explored in a phase 2 trial. The results of the CART19-BE-01 trial led to the approval by the AEMPS of ARI-0001 cells for the treatment of patients with R/R ALL older than 25 years of age (Hospital Exemption). To the best of our knowledge, this is the first purely academic CART19-cell product approved in any European country for any indication. This also makes Spain the only country in Europe where patients with R/R ALL have at least one approved CART19-cell product regardless of age (tisagenlecleucel for younger patients; ARI-0001 cells for older patients).
Data availability statement
Not applicable (this trial started recruitment in July 2017).
Ethics statements
Patient consent for publication
Ethics approval
The CART19-BE-01 clinical trial and subsequent compassionate use program were approved by the Spanish Medicines Agency (AEMPS) and both Institutions’ Review Boards and Ethics Committees. All participants provided written informed consent.
Acknowledgments
We are very grateful to Prof. Josep Maria Ribera (Hospital Germans Trias i Pujol, Badalona), who critically read our manuscript. We thank the patients who participated in the study and their families, friends, caregivers and referring physicians. We are very grateful to Drs. Miguel Ángel Perales (Memorial Sloan Kettering Cancer Center, New York), Shannon Maude (Children’s Hospital of Philadelphia), Juan José Lasarte (Centro de Investigación Médica Aplicada, Navarra) and Michael Schmitt (Universitäts Klinikum Heidelberg), members of the CART19-BE-01 trial DSMB. We are also indebted to the study staff and health care providers at Hospitals Clínic and Sant Joan de Déu, specifically to Anna Boronat, Raquel Martín-Ibáñez, Unai Perpiñá, Josep M. Canals, Vanina Rodríguez and Guillermo Suñé, who participated in the initial development of the construct; Judit Pich, Gabriela Recalde, Leticia Pereira and Joan Albert Arnáiz, from the Clinical Trials Unit; Andrea Scalise, Joaquín Sáez-Peñataro, Ferran Torres and Sandra Castaño, from the Department of Pharmacology; and Josep Maria Campistol, Antoni Castells, Aurea Mira, Manel del Castillo, Miquel Pons and Marc Roda as the main representatives from both hospitals.
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Twitter @ainitaoliver
Contributors VO-M, SR, MJ and JD designed the clinical trial and wrote the paper; JE, GC and AU-I provided significant contribution to the study planning and analysis; VO-M, JE, LM, EG, MD-B, MM, LGR-L, AO-C, AM-R, MR, PC, SF and JD looked after adult patients; SR, AA-S, AC, AF and IJ looked after pediatric patients; DB-R, EAG-N, ME-R, JRO, MP and MJ manufactured ARI-0001 cells and monitored B-cell aplasia; MC and MJ designed the CAR construct and produced the lentiviral vector; NV, MA and MT performed measurable residual disease tests; JC, EG-R and ML performed leukocytaphereses; JD performed the statistical analysis; ET, GC and SV coordinate the Advanced Therapies Unit at Hospital Clínic. All authors reviewed the study data, read and approved the manuscript.
Funding This study was funded by CatSalut, Projecte ARI and grants cofinanced by the Instituto de Salud Carlos III–Subdirección General de Evaluación y Fomento de la Investigación Sanitaria–and Fondo Europeo de Desarrollo Regional (FEDER) PICI14/122, PI13/676, PIE13/33, PI18/775. VO-M was a recipient of a research grant from FEHH and JD was a recipient of a research grant from the Generalitat de Catalunya (PERIS IPFE SLT006/17/301).
Competing interests VO-M: Consultant or advisory role (Kite Gilead, Celgene, Novartis), travel grants (Kite Gilead, Celgene, Novartis, Roche, Takeda, Janssen), honoraria (Kite Gilead). SR: Consultant or advisory role (Novartis, Jazz, Shire/Servier, Amgen, Celgene/Bristol-Myers, Kite Gilead), travel grants (Novartis, Jazz, Shire/Servier, Amgen, Celgene/Bristol-Myers, Kite Gilead), honoraria (Novartis, Jazz, Shire/Servier, Amgen, Celgene/Bristol-Myers, Kite Gilead). AA-S: Consultant or advisory role (Novartis), travel grants (Novartis), honoraria (Novartis). EG: Consultant or advisory role (Kite Gilead, Janssen, Genmab), research funding (Kite Gilead, Janssen, Roche). MD-B: Consultant or advisory role (Celgene, Novartis, Jazz, Astellas). AC: Consultant or advisory role (Novartis, Celgene), travel grants (Novartis, Celgene), honoraria (Novartis, Celgene). AF: Consultant or advisory role (Novartis), travel grants (Novartis), honoraria (Novartis). LGR-L: Travel grants (Kite Gilead, Amgen, Janssen). ML: Honoraria (Grifols, Fresenius Kabi), research funding (Terumo BCT, Maco-Pharma). AM-R: Consultant or advisory role (Bristol Myers Squibb, Abbvie), travel grants (Kite Gilead, Roche, Takeda, Janssen, Abbvie), honoraria (Abbvie). EG-R: Honoraria (Novartis). PC: Consultant or advisory role (Kite Gilead, Celgene, Janssen), travel grants (Kite Gilead, MSD, Janssen). MT: Consultant or advisory role (Novartis). CFDL: Consultant or advisory role (Janssen, Celgene/Bristol-Myers, GSK), honoraria (Janssen, Celgene/Bristol-Myers, Amgen, GSK), research funding (Janssen, Celgene/Bristol-Myers, Amgen, Takeda). GC: Consultant or advisory role (Celgene, Novartis). JE: Consultant or advisory role (Abbvie, Novartis, Celgene, Astellas, Jazz, Daiichi Dankyo, Roche, Amgen, Pfizer), travel grants (Celgene, Roche, Astellas, Daiichi Dankyo), research funding (Novartis, Celgene). AU-I: Consultant or advisory role (Kite Gilead, Celgene/Bristol-Myers, Miltenyi), travel grants (Kite Gilead, Celgene/Bristol-Myers). MJ: Consultant or advisory role (Kite Gilead, Grifols), honoraria (Kite Gilead, Grifols).
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.