Article Text

Abstract
Background The prognosis of patients with recurrent/refractory acute myelogenous leukemia (AML) remains poor and cell-based immunotherapies hold promise to improve outcomes. Natural Killer (NK) cells can elicit an antileukemic response via a repertoire of activating receptors that bind AML surface ligands. NK-cell adoptive transfer is safe but thus far has shown limited anti-AML efficacy. Here, we aimed to overcome this limitation by engineering NK cells to express chimeric antigen receptors (CARs) to boost their anti-AML activity and interleukin (IL)-15 to enhance their persistence.
Methods We characterized in detail NK-cell populations expressing a panel of AML (CD123)-specific CARs and/or IL-15 in vitro and in AML xenograft models.
Results CARs with 2B4.ζ or 4-1BB.ζ signaling domains demonstrated greater cell surface expression and endowed NK cells with improved anti-AML activity in vitro. Initial in vivo testing revealed that only 2B4.ζ Chimeric Antigen Receptor (CAR)-NK cells had improved anti-AML activity in comparison to untransduced (UTD) and 4-1BB.ζ CAR-NK cells. However, the benefit was transient due to limited CAR-NK-cell persistence. Transgenic expression of secretory interleukin (sIL)-15 in 2B4.ζ CAR and UTD NK cells improved their effector function in the setting of chronic antigen simulation in vitro. Multiparameter flow analysis after chronic antigen exposure identified the expansion of unique NK-cell subsets. 2B4.ζ/sIL-15 CAR and sIL-15 NK cells maintained an overall activated NK-cell phenotype. This was confirmed by transcriptomic analysis, which revealed a highly proliferative and activated signature in these NK-cell groups. In vivo, 2B4.ζ/sIL-15 CAR-NK cells had potent anti-AML activity in one model, while 2B4.ζ/sIL-15 CAR and sIL-15 NK cells induced lethal toxicity in a second model.
Conclusion Transgenic expression of CD123-CARs and sIL-15 enabled NK cells to function in the setting of chronic antigen exposure but was associated with systemic toxicities. Thus, our study provides the impetus to explore inducible and controllable expression systems to provide cytokine signals to AML-specific CAR-NK cells before embarking on early-phase clinical testing.
- killer cells
- natural
- cell engineering
- immunotherapy
- adoptive
- receptors
- chimeric antigen
- hematological neoplasms
Data availability statement
Data are available in a public, open access repository. Data are available upon reasonable request. Data and materials are available upon request. The bulk RNAseq data generated for this study are available at Gene Expression Omnibus repository under the accession number: GSE184552.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
- killer cells
- natural
- cell engineering
- immunotherapy
- adoptive
- receptors
- chimeric antigen
- hematological neoplasms
Introduction
Acute myelogenous leukemia (AML) is a neoplastic disorder characterized by the accumulation of malignant myeloid precursor cells in the bone marrow. AML has an aggressive clinical course in both adults and children.1 2 Intensive chemotherapeutic regimens with consolidative hematopoietic cell transplantation (HCT) remain the standard of care, but these treatments can cause significant short-term and long-term toxicities. Moreover, a subset of patients fail to respond to initial treatment or relapse after chemotherapy±HCT. For this reason, targeted therapies with non-overlapping toxicity profiles are aggressively being developed. The alpha chain of the interleukin (IL)-3 receptor (CD123) is highly expressed on both AML blasts and leukemic stem cells,3 4 and has been shown to be a safe target in clinical trials of immunotherapeutic agents.5–7 Leukemic stem cells are often resistant to chemotherapy and may be most responsible for disease initiation and relapse.3 Thus, CD123 targeting therapies could serve as valuable adjunct treatment modalities to achieve and/or sustain remission in high-risk patients with AML.
Adoptive cell transfer is a form of anticancer immunotherapy that has promise, and has been successful in the form of chimeric antigen receptor (CAR)–T cell infusions used to treat relapsed/refractory Acute Lymphoblastic Leukemia (ALL).8 CAR-T cells are also being tested against AML in several clinical trials.9 CAR-T cell therapies have associated severe toxicities including cytokine release syndrome (CRS), immune effector cell associated neurotoxicity syndrome (ICANS),10 and CAR-associated hemophagocytic lymphohistiocytosis.11 All Food and Drug Administration-approved CAR-T cell therapies originate from autologous hematopoietic starting material. The manufacturing of patient-derived T-cell products in the setting of a highly proliferative disease can be challenging.12 13 Delay in therapy associated with the time required for per-patient CAR-T manufacturing may not be possible in the setting of uncontrolled AML. In addition, intensive chemotherapy regimens, such as those administered for AML treatment, are associated with poor ex vivo T-cell expansion.14 Donor T cells can be considered as a product source; however, the infusion of allogeneic T cells is associated with the serious risk of graft-versus-host disease (GVHD).15
NK cells are immune effector cells that play a pivotal role as first-line defenders against virally infected or tumor-transformed cells. Though they act with similar cytotoxic mechanisms, NK-cell activation and function are distinct from those of T cells. NK cells express a variety of activating receptors (eg, NKG2D, NKp30, NKp46, 2B4 and CD16)16 17 that directly interact with cell surface ligands.16 18 These activating receptors associate with coreceptors (ex. DAP10, CD3ζ and FcεRIγ) to heighten directed cytotoxicity.17 The use of NK cells for immunotherapy has major advantages over the use of T cells. NK cells do not directly cause GVHD, though they may contribute inflammatory mediators that potentiate pre-existing pathology.15 19 Thus, they have the potential to be used as an off-the-shelf cellular product that can be manufactured on a large scale and can be readily available to patients. This has the advantage of decreasing production costs and preventing manufacturing associated treatment delays. Adoptive NK-cell transfer has been shown to be safe in clinical trials, without associated CRS or ICANS.20 Because of this safety profile and the expression of NK activating ligands on AML cells,18 NK-cell therapy has been tested in clinical trials. However, when used as AML treatment, NK-cell adoptive transfer induces only transient remission, and additional therapies are needed to achieve durable responses.21–24
In this study, we engineered NK cells to express CARs with intracellular (IC) domains rationally designed as those predicted to enhance NK-cell antitumor functionality. We demonstrate that NKs expressing CARs targeting CD123 show potent antigen-dependent activation and cytotoxicity. We find CAR expression to be stable and high across a panel of tested receptors incorporating NK-specific activating and costimulatory molecules, with optimal functionality associated with 2B4.ζ and 4-1BB.ζ containing CARs. CAR-NK cells did not demonstrate improved expansion or persistence when compared with unmodified NK cells in vivo. We therefore added constitutive IL-15 secretion to our CAR-NK cells and observed that this supported an activated phenotype that led to enhanced expansion and antitumor cytotoxicity. NK-cell activation translated to improved NK-cell persistence and expansion in vivo. However, constitutive cytokine secretion was also associated with severe toxicity in one animal model. Prior to clinical translation, alternate strategies to activate cytokine signaling in CAR-NK cells should be investigated with a focus to include safety.
Methods
Details about cell lines, determination of vector copy number (VCN), cytotoxicity assays, cytokine secretion measurement, and RNAseq library preparation and alignment are provided in online supplemental methods.
Supplemental material
CAR generation
CAR transgenes were designed using the CD123-specific single chain variable fragment (scFv, 26292)25 sequence and the hinge (H), transmembrane (TM), and IC domains indicated in figure 1A. Sequences were synthesized (GeneArt, Thermo Fisher Scientific) and subcloned into pSFG retroviral vectors. All sequences were validated by Sanger sequencing (Johns Hopkins Genetic Resources Core Facility).
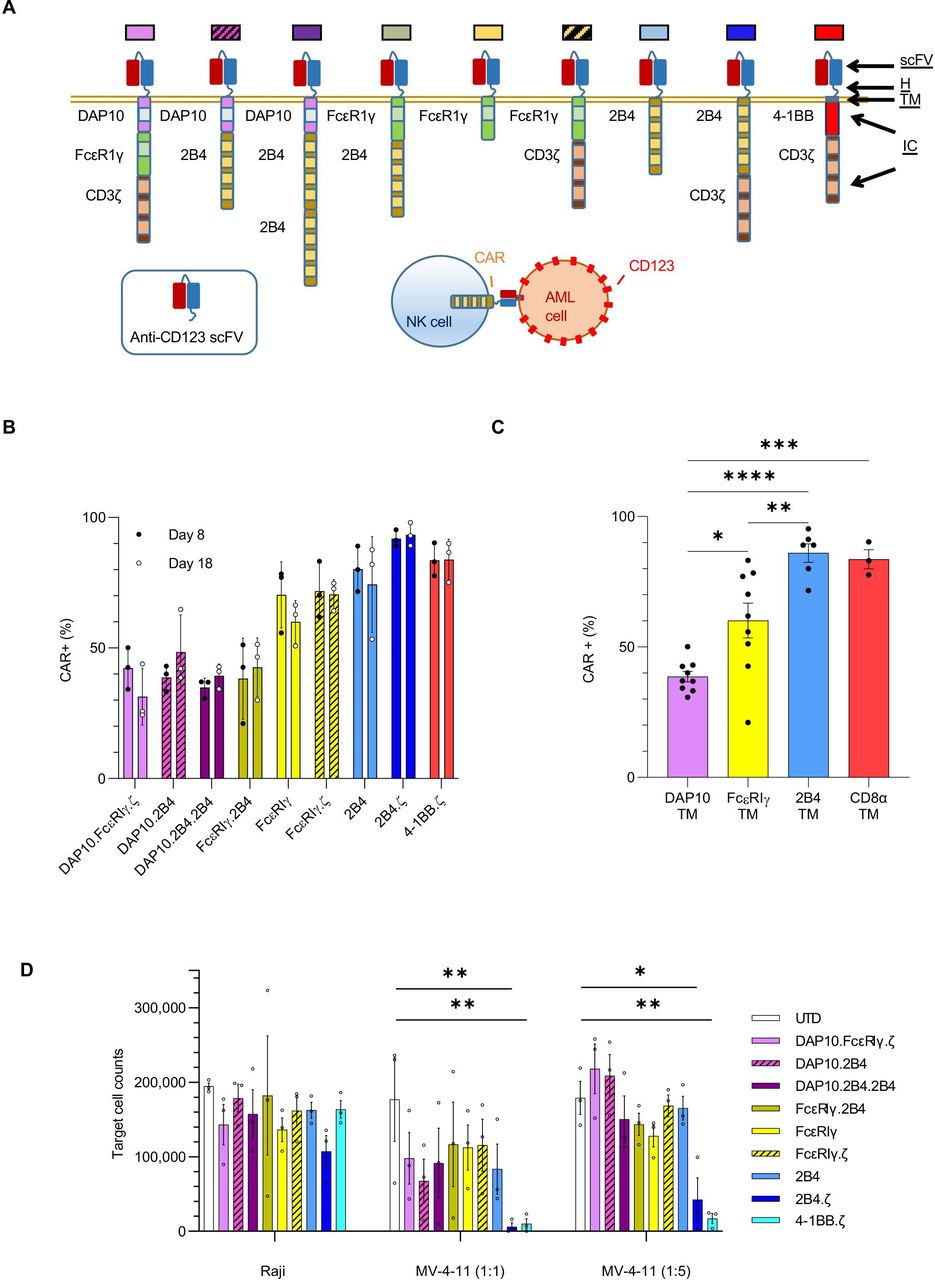
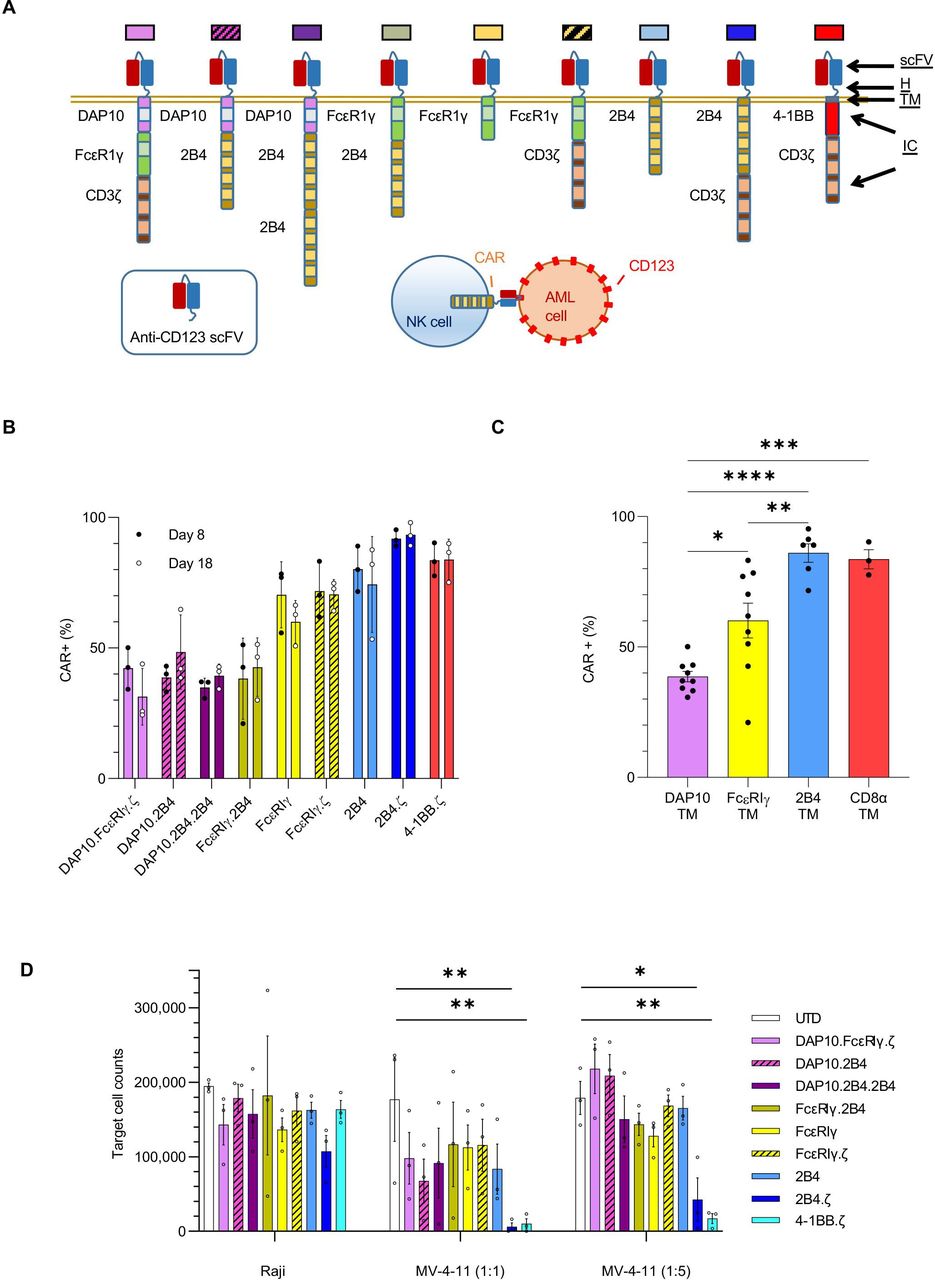
NK cells engineered with anti-CD123 CARs have antigen-specific functionality. (A) Schema of CAR design. All CARs bind CD123 via an extracellular scFv. The H, TM and IC domains of the CARs are as indicated. Colored boxes represents each particular CAR with colors carried through each figure. (B) Percentage (%) of CAR(+) NK cells detected on D8 and D18. (C) Bar plot comparing the percentage of CAR(+) NK cells with indicated TM domains on D8. (D) Absolute number of target cells measured after 72 hours of coculture with indicated NK cells. Initial target cell count was 100 000 in 1:1 and 250 000 in 1:5 E:T ratio conditions. Each bar representative of the mean±SEM; each dot is representative of individual NK cell donor. n=3 donors. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001. AML, acute myelogenous leukemia; CAR, chimeric antigen receptor; D, day; E:T, effector:target; H, hinge; IC, intracellular; NK, natural killer; ScFv, single-chain variable fragment; TM transmembrane; UTD, untransduced.
CAR-NK-cell production
Healthy donor peripheral blood mononuclear cells (Anne Arundel Medical Blood Donor Center, Annapolis, Maryland, USA; Carter Bloodcare, Woodway, Texas, USA) were isolated by Ficoll density gradient centrifugation and depleted of T cells using CD3 microbeads (Militenyi Biotec, Cologne, Germany). The remaining cells were stimulated on day (D)0 with lethally irradiated K562 feeder cells26 expressing membrane bound IL-15 and 4-1BB ligand at a 1:1 ratio. Cells were maintained in SCGM media (CellGenix, Freiburg, Germany) supplemented with 10% fetal bovine serum, 2 mmol/L GlutaMAX (Thermo Fisher), and 200 IU/mL human interleukin (hIL)-2 (Biological Resources Branch Preclinical Biorepository, National Cancer Institute, Frederick, Maryland, USA). NK-cell purity was verified with flow cytometry using fluorophore conjugated antibodies against CD56 and CD3 (online supplemental table 1). NK cells were transduced on D4 of culture using transiently produced replication incompetent RD114 pseudotyped retroviral particles immobilized on RetroNectin (Clontech Laboratories, Palo Alto, California, USA).
Supplemental material
Flow cytometry
Antibodies for NK and cancer cell identification targeted CD56 and CD33 (AML cell lines) or CD19 (Raji) markers. A detailed list of all antibodies, including those used for the evaluation of immune cell phenotype is in online supplemental table 1. CAR expression analysis was performed using incubation with His-tagged recombinant CD123 protein (SinoBiological, Beijing, China) and secondary staining with αHis-PE or αHis-APC (BioLegend, San Diego, California, USA). Dead cells were excluded from analysis using LIVE/DEAD Fixable Viability Stains 780 or 575V (BD Biosciences, Franklin Lakes, New Jersey, USA). Cell enumeration was performed with CountBright counting beads (Thermo Fisher). Human Fc receptors were blocked using Human TruStain FcX (BioLegend). In samples stained with multiple BD Horizon Brilliant reagents, Brilliant Stain Buffer Plus (BD Horizon) was used. Compensation was performed with UltraComp eBeads Compensation Beads (Thermo Fisher). Cell surface antigens were quantified using microspheres of the Quantum APC Molecules of Equivalent Soluble Fluorochrome kit (Bangs Laboratories, Fisher, Indiana, USA). All samples were acquired on FACSCelesta or FACSymphony Cell Analyzers (BD Biosciences) and analyzed with FlowJo software V.10.6.1 and V.10.7.2. Cell sorting was performed on FACSMelody (BD Biosciences).
Immunophenotype analysis
Two different panels (A and B) were used for evaluation of NK-cell receptors and one panel (C) for receptor ligands. Data analysis of the multiparameter panels A and B was performed in R V.3.6.2. The median marker intensities were transformed using arcsinh (inverse hyperbolic sine) with cofactor 150.27 Non-linear dimensionality reduction on randomly selected 500 data points per sample of each panel was performed using uniform manifold approximation and projection (UMAP).28 NK-cell clusters were identified with the FlowSOM V.1.18.0 algorithm, and 40 different metaclusters were generated per panel.29 Subsequently, we manually merged hierarchically neighboring clusters similar in biology and median marker intensities. Panel A clusters do not correlate with the ones in panel B.
Serial stimulation assay
NK cells were stimulated daily with MV-4-11 cells at a 1:1 effector:target (E:T) ratio in G-rex plates (Wilson Wolf, New Brighton, Minnesota, USA) for a total of 10 days. NK-cell proliferation and cytotoxicity was measured using flow cytometric analysis. Percent (%) cytotoxicity was calculated based on the target cell numbers on the day of (Y) and the day after (X) stimulation using the formula 100×(X−Y)/(Y). Cell phenotype was evaluated at baseline, on the 1st (12 hours) and the 10th (D10) days.
RNA sequencing
On D10 of serial stimulation, coculture was depleted first of dead cells using the Dead Cell Removal Kit and next of leukemia cells with CD33 microbeads (Militenyi). NK-cell purity was verified with flow cytometry using fluorophore-conjugated CD56 and CD33 antibodies. RNA was extracted from NK cells using the RNeasy Mini Kit (Qiagen, Hilden, Germany) and RNAseq was performed (online supplemental methods). Differential expression analysis and statistical testing were performed using DESeq2 software.30Mice were injected with 1×106 MV-4-11 cells modified
Xenograft mouse model
All animal studies were carried out under protocols approved by the Johns Hopkins Institutional Animal Care and Use Committee. NSG (NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ) mice 6–8 weeks old were obtained from an internal colony that originated from the Jackson Laboratory (Bar Harbor, Maine). Mice were injected with 1×106 MV-4-11 cells modified for stable firefly luciferase (ffLuc) expression31 or 5×104 MOLM-13.ffLuc cells32 via tail vein on D0. NK-cell treatment was administered on D7 (10×106 cells) or D4, 7, and 10 (3×106 cells each). Mice were given D-luciferin (3 mg) by intraperitoneal injection, and bioluminescence (BL) was measured using IVIS Spectrum (In Vivo Imaging System). Data were analyzed using Living Image Software V.4.7.3 (PerkinElmer, Waltham, Massachusetts, USA). When indicated, peripheral blood (PB) was drawn via facial vein; red blood cells were lysed with eBioscience RBC Lysis Buffer (Thermo Fisher); and the remaining cells were analyzed with flow cytometry. Bone marrow and spleen were harvested, and tissues were analyzed with flow cytometry. Analysis of PB for cytokines (hIL-15, hTumor Necrosis Factor (TNF)-α, mouse interleukin (mIL)-6, and mIL-1β) was performed with ELISA (R&D Systems). Mice were euthanized when they exhibited >20% weight loss, hind limb paralysis, or moribund state as per protocol guidelines.
Statistical analysis
All statistical analyses was performed using GraphPad Prism Software V.9.2.0. Our comparisons included more than three groups and ordinary one-way or two-way analysis of variance (ANOVA) corrected using the method of Bonferroni. Data with variance of several logs of magnitude were log transformed (Y=log(Y)) before analysis with ANOVA. Survival of mice was estimated by the Kaplan-Meier method, and differences in survival between groups were calculated by log-rank (Mantel-Cox) test.
Results
CD123-CARs are highly expressed on the NK-cell surface
We considered NK-cell biology in our design of eight different NK-tailored CARs (figure 1A) to complement the common 4-1BB.ζ CAR.33 34 All CARs are composed of an extracellular scFv targeting CD123.25 The H, TM, and IC portion of our CARs consisted of different combinations of activating coreceptors DAP10 and FcεRIγ, the costimulatory receptor 2B4, and the ζ chain of the T-cell receptor (figure 1A). All CARs were expressed stably on the surface of primary human NK cells for at least 2 weeks in culture, with transduction efficiencies ranging from 21% to 98% (figure 1B). Representative flow cytometric plots are shown in online supplemental figure 1A). CARs encoding 2B4 or CD8α TM domains demonstrated higher transduction efficiencies (medians 89 (range 53%–98%) and 84 (range 75%–90%), respectively) than constructs containing FcεRIγ or DAP10 TM (medians 62 (range 21%–77%) and 39 (24%–64%), respectively; figure 1B,C). 2B4 and CD8α TM domains also conferred optimal CAR surface density as estimated by comparative mean fluorescence intensities (MFIs±SEM: 2B4-TM 2158±242, CD8α-TM 3254±970, FcεRIγ-TM 827±151, DAP10-TM 366±23; 2B4 and CD8α vs DAP10: p<0.0001, 2B4 vs FcεRIγ: p<0.01, CD8α vs FcεRIγ: p<0.0001, FcεRIγ vs DAP10: ns; online supplemental figure 1B). CAR-NK cells expanded from 70-fold to 213-fold within 18 days of ex vivo culture with no significant differences between generated CAR-NK-cell populations (online supplemental file 3).
Supplemental material
CD123-CAR NK cells have antigen-specific anti-AML activity in vitro
We evaluated the target specificity of our CAR-NK cells using the CD123-positive MV-4-11 and CD123-negative Raji cell lines. When challenged with MV-4-11 in coculture assays, all CAR-NK cells responded with enhanced cytokine secretion above that seen when using unmodified NK cells under identical conditions (mean percent (%) change of interferon gamma (IFN-γ) secretion; CAR-NKs (range): 65%–313% vs unmodified: 14%; online supplemental figure 2). There was no difference in cytokine production of CAR(+) and CAR(−) NK cells after coculture with CD123(−) targets (online supplemental figure 2). Next, we assessed CAR-NK-cell cytotoxicity against CD123(+) target cells in 72-hour coculture assays. We found that CARs with 2B4 or 4-1BB costimulatory and TCRζ signaling domains endowed NK cells with the greatest cytolytic activity against MV-4-11 at both 1:1 (mean % cytotoxicity±SEM: 2B4.ζ 93.8±5%, 4-1BB.ζ 89.9±6.8%) and 1:5 E:T ratios (82.9±11.6% and 93±2.6%, respectively; figure 1D). CD123(−) Raji cells were again used as controls. There was no difference between CAR-NK versus unmodified NK cell-mediated cytotoxicity against Raji cells, confirming specificity (figure 1D).
CD123-2B4.ζ CAR-NK cells have limited anti-AML efficacy in vivo
Given the superior anti-AML activity of 2B4.ζ and 4-1BB.ζ CAR-NK cells in our in vitro assays, we next evaluated their antitumor activity in a xenograft model of human AML. NSG mice were first engrafted with MV-4-11.ffLuc cells,31 then treated with CAR-NK or unmodified NK cells on D7 (figure 2A). Leukemic growth was measured with serial BL imaging. 2B4.ζ CAR-NK-cell postinjection had transient anti-AML activity, which translated into a significant survival advantage in comparison to other experimental groups (median survival in days: 2B4.ζ vs 4-1BB.ζ 63 vs 56, p<0.01; 2B4.ζ vs unmodified: 63 vs 55, p<0.05) and untreated controls (2B4.ζ vs no treatment 63 vs 58, p<0.01; figure 2B–D). Serial analysis of PB in our animals showed declining NK-cell numbers in all evaluated mice irrespective of infused NK-cell type (figure 2E). Planned bone marrow and spleen examination in a subset of mice demonstrated increasing percentages of leukemia from D15 to D22 in all treatment conditions, despite readily detectable NK cells (online supplemental figure 3). Consistent with BL data, the percentage of leukemic cells was lower in both bone marrow and spleen samples of 2B4.ζ CAR-NK cell-treated mice as compared with mice treated with UTD and 4-1BB.ζ CAR-NK cells (online supplemental figure 3).
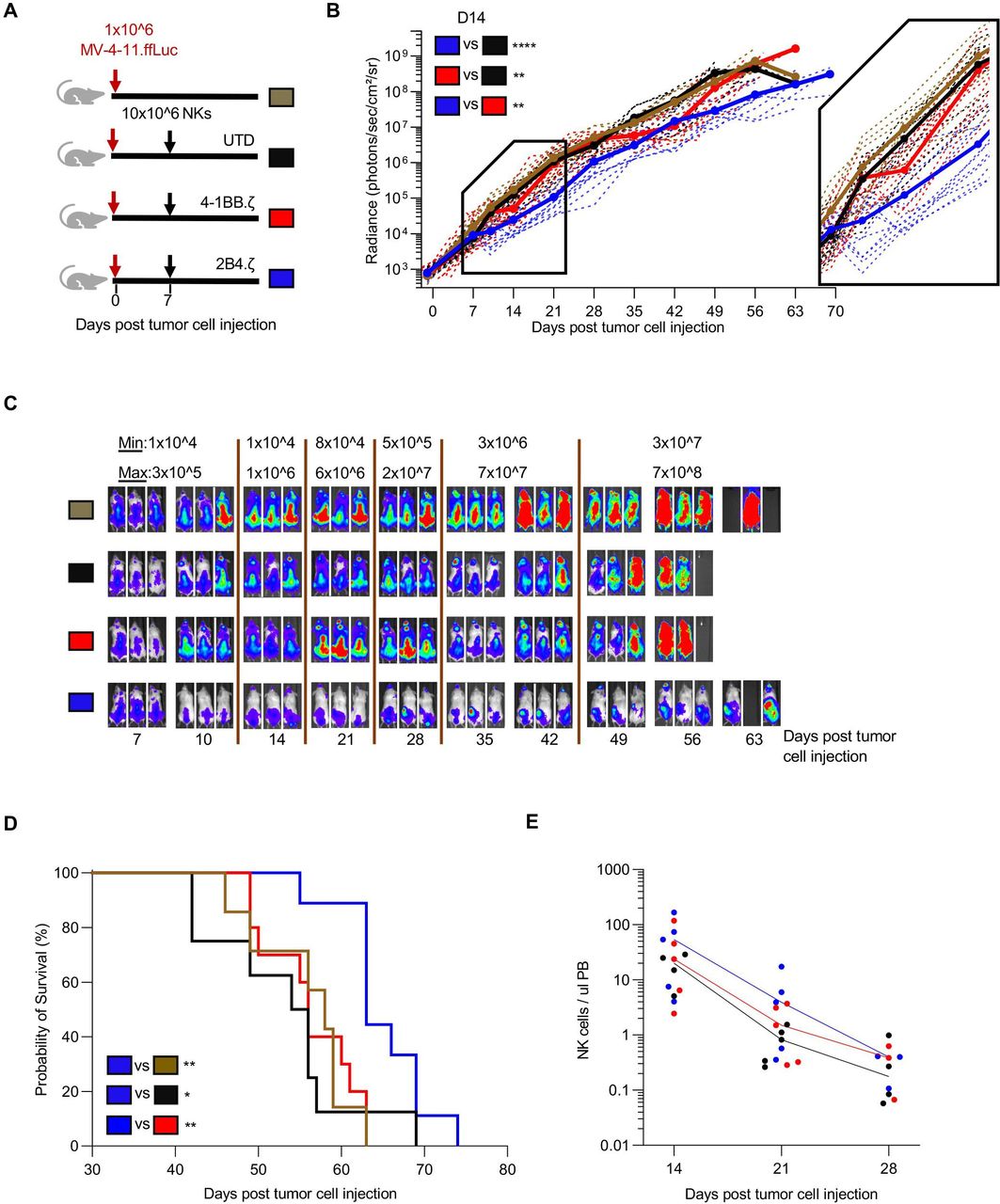
Anti-CD123.2B4.ζ CAR-NKs have transient anti-AML activity in vivo. (A) Schematic of MV-4-11 xenograft model. On D0, NSG mice were injected via tail vein with 1×106 CD123(+) MV-4-11 cells that express ffLuc (MV-4-11.ffLuc cells). In treatment groups, 10×106 NK cells were administered on D7. Cohorts: UTD/unmodified, 4-1BB.ζ CAR-NK, and 2B4.ζ CAR-NK. (B) Leukemia proliferation was monitored with bioluminescence imaging and was recorded as photons/s/cm²/sr; n=8–12 mice per group. Magnification of D7–D21 shown. (C) Representative images of three mice per condition. Minimum and maximum values of color scale are depicted at the top (min–max). (D) Kaplan-Meier survival analysis of MV-4-11 xenografts (n=8–12 mice per condition). (E) Mouse PB collected at indicated time points and analyzed via flow cytometry. Each dot represents a single mouse. Solid line: median. At later time points, NK-cell count was undetectable for all groups and is not plotted. *P<0.05, **P<0.01, ****P<0.0001. AML, acute myelogenous leukemia; CAR, chimeric antigen receptor; D, day; ffLuc, firefly luciferase; NK, natural killer; PB, peripheral blood; UTD, untransduced.
Armoring NK cells with secreted IL-15 enhances anti-AML functionality in vitro
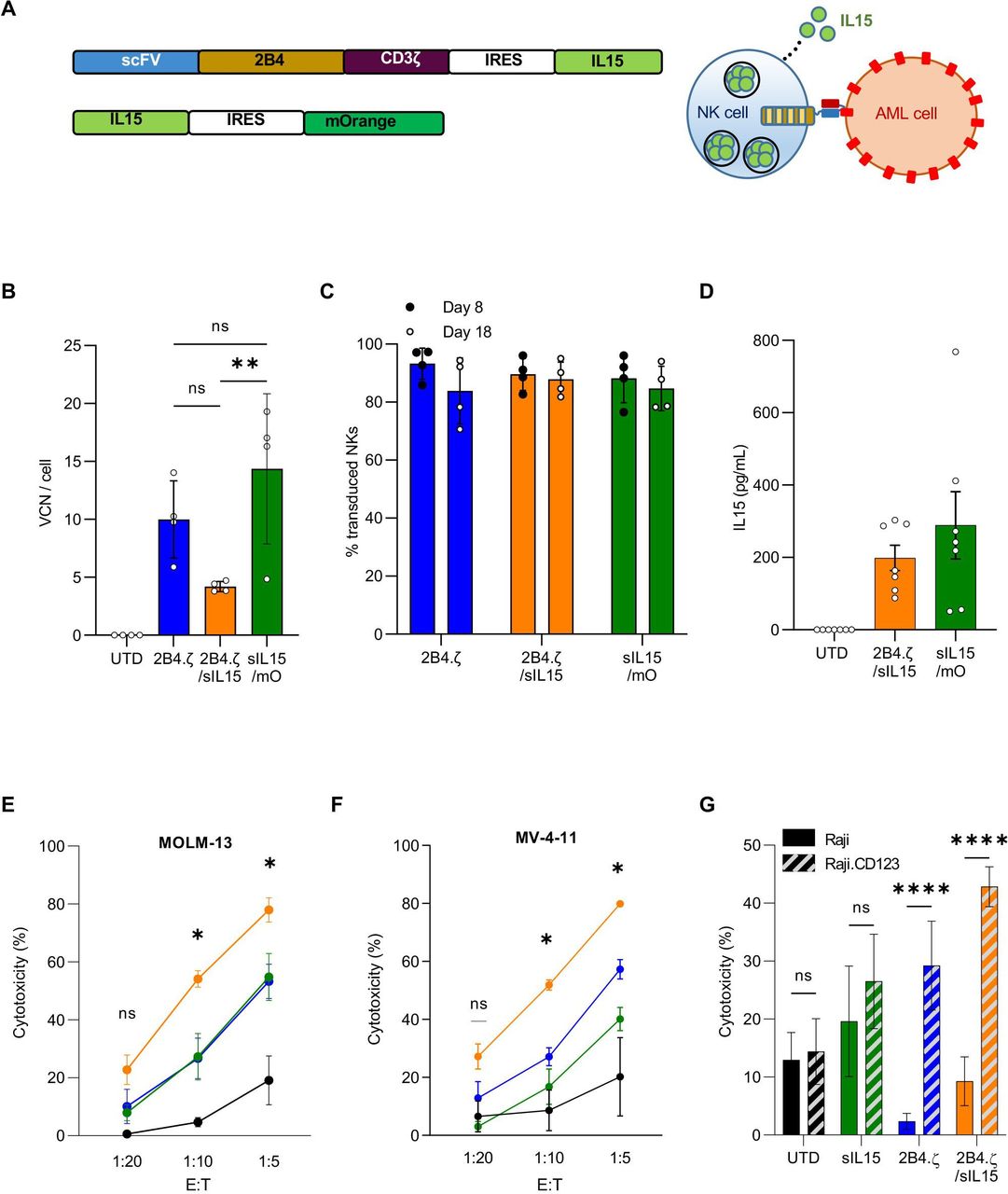
Having demonstrated that 2B4.ζ CAR-NK cells have anti-AML cytolytic capacity with short persistence, we next explored if transgenic expression of IL-15 in CAR-NK cells would support sustained anti-AML activity. To accomplish this, we cloned a sequence encoding human IL-15 downstream of an Internal Ribosome Entry Site (IRES) element into our 2B4.ζ CAR vector. We simultaneously generated a second retroviral vector encoding IL-15 and the fluorescent molecule mOrange as a control with IL-15, but not CAR expression (figure 3A). We verified transduction by measuring VCN per cell (median: 2B4.ζ 10 (range 5.8–14), 2B4.ζ/sIL-15 4.1 (range 3.8–4.7), sIL-15/mOrange 16.6 (range 4.8–19.2); figure 3B). We found no significant difference in VCN/cell or in CAR expression between NK cells transduced with 2B4.ζ or 2B4.ζ/sIL-15 encoding retroviral vectors (range CAR(+): 2B4.ζ vs 2B4.ζ/sIL-15 70%–97% vs 81%–96%, p>0.99, MFI±SEM: 2B4.ζ vs 2B4.ζ/sIL-15 2566±531 vs 2089±424; p>0.99; figure 3C and online supplemental figure 4A). Expression of IL-15 was measured with quantitative reverse transcription (qRT)-PCR (online supplemental figure 4B) and IL-15 secretion was confirmed by ELISA (figure 3D).

Simultaneous 2B4.ζ CAR expression and IL-15 secretion strengthens NK-cell cytotoxicity. (A) Schema of vectors and IL-15 secretion from CAR-NK cells. (B) Quantification of retroviral VCN in transduced NK cells. UTD NK cells served as negative controls (n=4 donors). (C) Percentage (%) of transduced NK cells in cultures on D8 (black circle) and D18 (white circle, n=4 donors). (D) NK-cell supernatant was used for quantification of IL-15 by ELISA (n=7 biological replicates using four donors). BL-based cytotoxicity assays were performed using the CD123(+) AML cell lines MV-4-11(E) and MOLM-13 (F) with ffLuc expression (n=4 donors). Asterisks indicate 2B4.ζ/sIL-15 versus 2Β4.ζ comparison. (G) Bar graph comparing percent (%) cytotoxicity of NK cells against Raji (CD123(−): solid), and Raji (CD123 (CD123(+): diagonal stripe) cancer cell lines at 1:10 E:T ratio (n=4 donors). (B-G) Mean±SEM represented. *P<0.05, **P<0.01, ****P<0.001. AML, acute myelogenous leukemia; BL, bioluminescence; CAR, chimeric antigen receptor; D, day; E:T, effector:target; ffLuc, firefly luciferase; IL, interleukin; NK, natural killer; ns, not significant; ScFv, single-chain variable fragment; sIL, secretory interleukin; UTD, untransduced; VCN, vector copy number.
We evaluated short-term cytotoxicity of 2B4.ζ/sIL-15 CAR-NKs against the CD123(+) MV-4-11, MOLM-13, and Raji.CD123 cell lines. The parental CD123(−) Raji line was used as a negative control. When compared with 2B4.ζ, the 2B4.ζ/sIL-15 CAR-NKs had higher cytotoxicity against CD123+ targets (figure 3E–G). Both 2B4.ζ and 2B4.ζ/sIL-15 demonstrated antigen-specific cytotoxicity as Raji.CD123 were more effectively killed than parental (CD123−) Raji cells (figure 3G). Target cell CD123 and IL-15Rα expressions were quantified in order to evaluate for any effect of CD123 surface density or IL-15 trans presentation on NK-cell cytotoxicity (online supplemental figure 5A). Differences in measured CD123 surface density did not correlate with observed short-term cytotoxicity, underlining the existence of additional complex mechanisms affecting NK-cell activation. Similarly, differences observed in IL-15Rα expression did not correlate with cytotoxicity (online supplemental figure 5B).
Transgenic expression of IL-15 potentiates the activation, persistence, and long-term cytolytic activity of CAR-NK cells
Using a model of chronic antigen stimulation (figure 4A), we evaluated the immune phenotype of our CAR-NK cells at baseline, after 12 hours, and on D10 of coculture with MV-4-11. Cells were counted daily, and AML was repleted to maintain a 1:1 E:T ratio. We used flow cytometry to measure surface expression of markers in two different panels (panels A and B), then performed hierarchical clustering of NK-cell subsets (figure 4B,C, and online supplemental figure 6). In panel A, we observed similar population distributions in the 2B4.ζ CAR and unmodified NK cells with cluster shifts from earlier (12 hours) to later (D10) time points. Specifically on D10, there was an increase in the percentage of 2B4.ζ and unmodified NK cells populating clusters defined by lower surface expression of activating receptors NKG2D and NKp30 (clusters 19, 25, and 31; figure 4B,C, and online supplemental figure 7). For 2B4.ζ/sIL-15 CAR-NK cells, only minimal changes in population density of these subsets were observed on D10 (figure 4B,C). The unique 2Β4.ζ/sIL-15 immunophenotype is highlighted by comparison of the MultiDimensional Scaling (MDS, global) and UMAP (cluster-specific) plots in online supplemental figure 8.

IL-15 maintains NK cell-activated phenotype in a model of chronic antigen stimulation. (A) Schematic representation of our serial stimulation assay. D0 was the day of the initial seeding of the coculture; D1 was the first; and D10 was the last day of cell quantification. Immunophenotypical analysis of effector and target cells performed at baseline (before coculture), 12 hours after first stimulation, and on D10 (n=1 donor). Transcriptomic analysis performed on D10 (n=3 donors). (B) Heatmap of flow cytometry data showing expression of 15 different NK-cell surface markers. Heatmap coloring represents arcsinh transformed median marker intensity. (C) Bar plots of relative abundance of the 32 population subsets found in each sample. (D) NK-cell counts over a period of 10 days. Initial seeding count was two million NK cells (mean±SEM, n=3 donors). Asterisks indicate 2B4.ζ/sIL-15 versus 2Β4.ζ and sIL-15 versus UTD comparison. (E) Heat map of the percent (%) NK-cell cytotoxicity. Each column represents the specific day and each row a unique biological replicate (n=3 donors). D, day; IL, interleukin; MFI, mean fluorescence intensity; NK, natural killer; sIL, secretory interleukin; UTD, untransduced.
Clustering of NK cells based on expression of markers included in our second receptor panel (panel B) again supported the maintained NK-cell activation to D10 in 2B4.ζ/sIL-15 CAR-NK cells (predominant clusters 16, 20, and 26 and minor clusters 11, 13, 14, 15, 21, and 23; online supplemental figures 6–9). Differences were again observed in 2B4.ζ and unmodified NK cells on D10 compared with earlier time points (increasing percentage of cells populating clusters 2, 4, 17, and 18, and decreasing percentages of 14, 16, 20, nd 26; online supplemental figure 6). Clusters 14, 16, 20, and 26 expressed higher levels of LFA-1, CD69, TRAIL, TIM-3, NKG2A, and KLRG1 compared with clusters 2, 4, 17, and 18 (online supplemental figure 9). Negligible differences were observed in PD-1, LAG-3, FASL, 2B4, and NKG2C expression. Taken together, 2B4.ζ/sIL-15 CAR-NKs had a higher percentage of NK cells populating clusters defined by higher surface expression of LFA-1 (adhesion/activation receptor), CD69 (activation marker), TRAIL (death receptor), and TIM-3 (commonly upregulated after NK-cell activation), as well as NKG2A and KLRG1 (inhibitory receptors; figure 4B,C, and online supplemental figures 6–10). Overall, the data from both antibody panels suggest that with continuous antigen stimulation, IL-15 preserves an activated CAR-NK-cell phenotype associated with high cytotoxicity. We also analyzed the expression of NK-cell activating and inhibitory ligands. AML cells expressed high levels of MICA/MICB (NKG2D ligands), CD112 and CD155 (DNAM-1, PVRIG, CD96, and TIGIT ligands) and PDL1 (PD-1 ligand), but not ULBP1 (NKG2D ligand) or galectin-9 (TIM-3 ligand, online supplemental figure 11).
Next, we evaluated NK-cell cytotoxicity and persistence in our model of chronic antigenic stimulation (figure 4A). While unmodified and 2B4.ζ CAR-NK cells sharply diminished in number over the course of this assay, NK cells genetically engineered to express IL-15 (2B4.ζ/sIL-15 and sIL-15) survived throughout the experiment (n=3, figure 4D). As predicted by the short-term cytotoxicity assay, 2B4.ζ CAR-NK cells had high initial anti-AML activity. However, their killing potential decreased as 2B4.ζ CAR-NK-cell counts declined. Cytotoxicity was completely abrogated by D5 (figure 4E). IL-15 activation alone (without CAR expression) also had an early antitumor effect that was not sustained. Only the combination of CAR expression and IL-15 secretion led to continued NK cell-mediated anti-AML cytotoxicity (figure 4E and online supplemental figure 12).
NK cells secreting IL-15 exhibit a highly proliferative and activated transcriptomic signature after chronic antigen stimulation
We evaluated the transcriptional programs of our NK cells on the 10th day of continuous antigen stimulation. NK cells were isolated, and RNA libraries were prepared and used for RNAseq analysis. Samples clustered by IL-15 secretion, with overlap between 2B4.ζ/sIL-15 and sIL-15 conditions. These clearly separated from NK cells that did not secrete IL-15 (UTD and 2B4.ζ, figure 5A). Differential gene expression analysis (DESeq2) of 2B4.ζ/sIL-15 versus 2B4.ζ and sIL-15 versus UTD NKs identified in the IL-15-secreting NK-cell differences in expression of genes in the pathways of DNA replication, cell cycle progression, and NK cell-mediated cytotoxicity (figure 5B,C). Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis revealed cell cycle progression as the top-ranked pathway for both comparisons (figure 5C). No biologically relevant pathways were found to be significantly enriched when comparing IL-15-secreting CAR-NK cells to non-CAR sIL-15 NKs (online supplemental figures 13A,B). Hierarchical clustering of differentially expressed genes also revealed upregulation of genes involved predominantly in cell cycle progression, chemokine, and cytokine signaling in IL-15-secreting NK conditions (figure 5D and online supplemental table 2). We used this dataset to evaluate differential expression of molecules of biological relevance. 2B4.ζ/sIL-15 and sIL-15 NK cells had higher expression of genes encoding for NK activating receptors (NCR2 (NKp44), NCR3 (NKp30), KLRC2 (NKG2C), KLRC4-KLRK1 (NKG2D), CD226 (DNAM-1), and FCGR3A (CD16)), adaptor molecules (FCER1G (FcεRIγ)), death receptor ligands (TNFSF10 (TRAIL)), granzyme (granzyme A (GZMA), proinflammatory cytokines and chemokines (IFNG (IFN-γ), CCL1, CCL3, CCL4, XCL1, XCL2, and CCL3L3), activation markers (CD69), proliferation markers (MKI67 (Ki-67)), antiapoptosis regulators (BCL2), and adhesion molecules (ITGB2 (integrin-β2), CD2, and CD53; figure 5E and online supplemental figure 13C). We also observed upregulation of select inhibitory receptors (NKG2A, CEACAM1, and LILRB1) including inhibitory killer Ig-like receptors (KIRs; KIR2DL1, KIR2DL2, KIR2DL3, KIR2DL4, and KIR3DL1) as well as ‘checkpoint’ molecules (HAVCR2 (TIM-3) and TIGIT, figure 5E and online supplemental figure 13C). KIRs are subject to extensive regulatory splicing and there is an inability to distinguish KIR with identical extracellular domains but functionally disparate IC tails by flow cytometry, making transcriptional analysis necessary and complementary for study of KIR expression. Analysis of the chemokine receptor expression showed upregulation of CCRL2, CCR1, CCR5, and CCR6 with similar or lower expression of CX3CR1, CXCR3, and CXCR4 in IL-15-secreting NKs (online supplemental figure 13C).
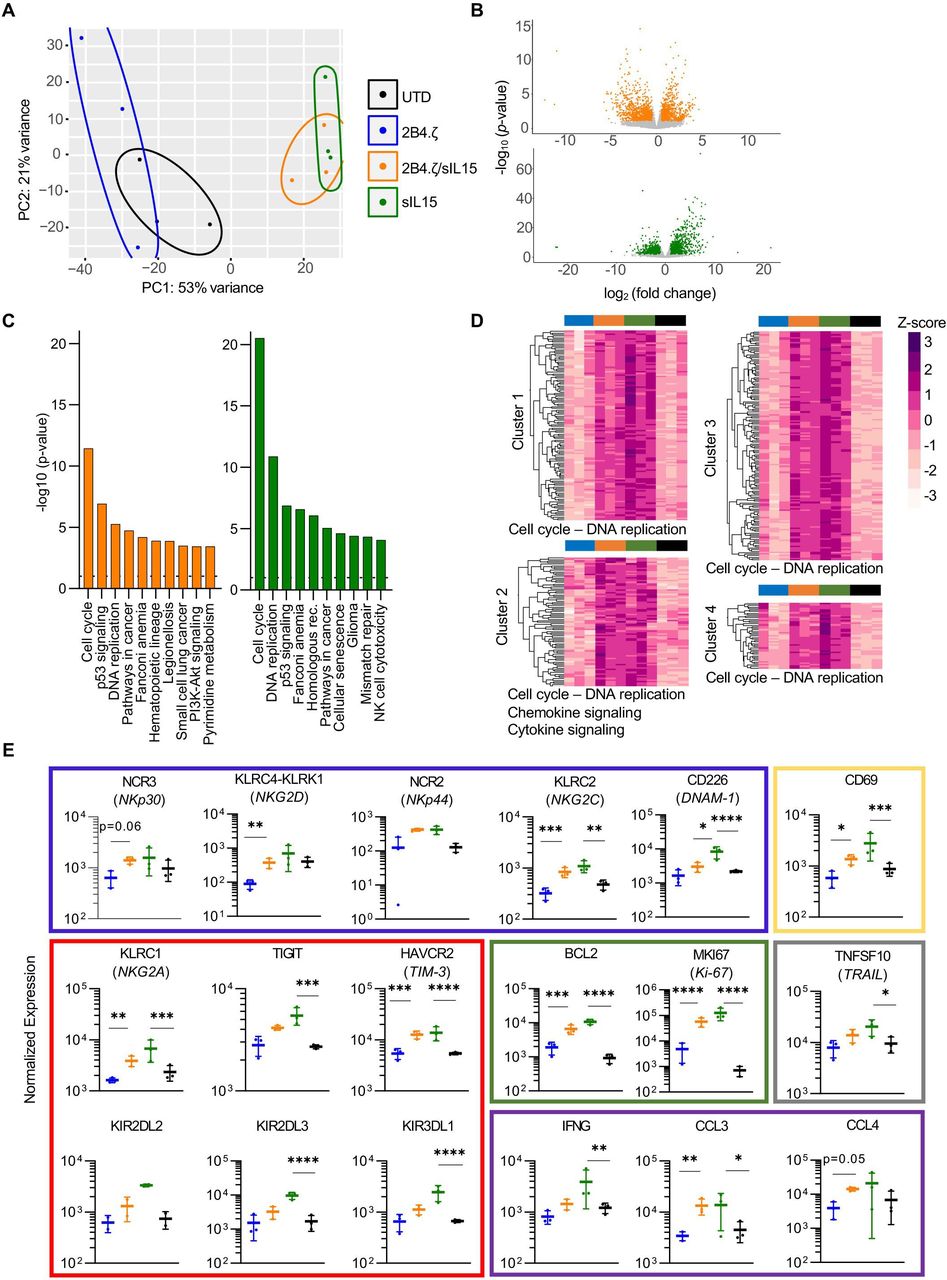
IL-15 stimulation of NK cells upregulates genes important to cell cycle progression, NK-cell activation, and cytotoxicity. (A) PCA of NK-cell transcriptome on D10 of the chronic antigen stimulation. Each dot represents a unique NK-cell donor (n=3 donors). (B) Volcano plots representing differentially regulated genes in 2B4.ζ/sIL-15 compared with 2B4.ζ (orange) and sIL-15 compared with unmodified cells (green). Gray dots are those not meeting criteria: p value ≥0.05; fold change > 2 or <1/2. (C) Bar plot depicting the top 10 significantly different KEGG 2021 human gene set enrichment pathways. Dotted line at a p value of 0.05. (D) Heatmap and hierarchical clustering performed on differentially expressed genes. Biologically relevant clusters and enriched pathways (shown on the bottom of the cluster) represented. Z-score scale bar at right. (E) Dot plots displaying the normalized expression of genes encoding activating receptors (blue rectangle), inhibitory receptors/checkpoint molecules (red), proliferation/antiapoptosis markers (green), inflammatory mediators (purple), death receptor ligands (gray) and activation (yellow) markers (n=3 independent donors). *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001. IL, interleukin; NK, natural killer; PCA, principal component analysis; sIL, secretory interleukin; UTD, untransduced.
Constitutive IL-15 expression improves NK cell in vivo persistence but causes lethal toxicity
We next evaluated IL-15-secreting NK cells in two AML xenograft models. In our first experiment, we used our MV-4-11 xenograft model (figure 2A) and our previous NK-cell dosing regimen (online supplemental figure 14A). Surprisingly, MV-4-11 engrafted mice treated with 10×106 IL-15-secreting NK cells had early mortality (median survival (days): 2B4.ζ/sIL-15: 25, sIL-15: 21; n=5 mice each group; online supplemental figure 14B). Circulating human IL-15 and NK-cell counts both increased in the 3 weeks from NK-cell injection to death (online supplemental figure 14C,D). Therefore, in our next experiment we decreased our treatment dose of infused NK cells in order to evaluate whether we could mitigate the observed treatment associated mortality. We compared a single decreased dose of 2B4.ζ/sIL-15 CAR-NK cells with multiple doses of 2B4.ζ CAR-NK cells (figure 6A). Infusion of a single dose of 3×106 2B4.ζ/sIL-15 CAR-NK and three doses of 3×106 2B4.ζ CAR-NK cells both had transient AML control (figure 6B,C). Three doses of 2B4.ζ CAR-NKs prolonged survival, as compared with mice in all other treatment groups (median survival in days; 2B4.ζ vs UTD and untreated; 71 vs 48 and 49, p<0.001; figure 6D). In contrast, even at the lower dose, toxicity and premature death of mice treated with IL-15-secreting NK cells again occurred (median survival in days; 2B4.ζ/sIL-15 vs untreated; 26 vs 49, p<0.001; sIL-15 vs untreated; 21 vs 49, p<0.001; figure 6D). Analysis of PB showed in vivo expansion of 2B4.ζ/sIL-15 and sIL-15 NK cells, with declining NK-cell counts again observed in non-IL-15 secreting NK-cell cohorts (figure 6E). Bone marrow and spleen analysis at necropsy of mice treated with 2B4.ζ/sIL-15 CAR-NKs revealed high numbers of infiltrating NK cells (online supplemental figure 15A,B). We identified increasing systemic hIL-15 in mice treated with 2B4.ζ/sIL-15 and sIL-15 NKs (figure 6F). In addition, high levels of hTNF-α, low levels of mIL-1β and negligible levels of mIL-6 were detected in the blood of mice treated with 2B4.ζ/sIL-15 CAR-NK cells (online supplemental figure 15C–E).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
IL-15 secreting CAR-NK cells can cause lethal toxicity in an AML xenograft model. (A) Schematic of NK-cell dosing in MV-4-11.ffLuc model. (B) MV-4-11 proliferation was monitored with BLI. Representative images of mice. The minimum and maximum values of the color scale are indicated (min–max). (C) BL representative of leukemia proliferation was recorded as photons/s/cm²/sr . Dotted lines: individual mice; solid lines: mean (n=5–7 mice per group; two experiments performed for UTD and 2Β4.ζ cohorts). (D) Kaplan-Meier survival analysis. (E) Mouse PB was collected at indicated time points and analyzed via flow cytometry. NK-cell numbers per microlitre of mouse PB were tracked starting on D13 of the experiment. Each dot represents cell numbers from a single mouse; line is at median. Asterisks indicate 2B4.ζ/sIL-15 versus 2Β4.ζ comparison. (F) Human IL-15 from PB of MV-4-11 engrafted mice drawn at the indicated time points was quantified (pg/mL) with ELISA. Asterisks indicate 2B4.ζ/sIL-15 versus 2Β4.ζ comparison. (G) Schematic of NK-cell dosing in MOLM-13.ffLuc model. (H) MOLM-13 proliferation was monitored using BLI (n=5 mice per group). Representative images, (I) radiance, and (J) Kaplan-Meier survival analysis. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001. AML, acute myelogenous leukemia; BLI, bioluminescence imaging; CAR, chimeric antigen receptor; D, day; ffLuc, firefly luciferase; IL, interleukin; NK, natural killer; PB, peripheral blood; sIL, secretory interleukin; UTD, untransduced.
We further evaluated our CAR-NK cells in a more aggressive MOLM-13 xenograft model (figure 6G). A single dose of 3×106 2B4.ζ/sIL-15 CAR-NK cells and three doses of 3×106 2B4.ζ CAR-NK cells again had equivalent transient antitumor control (figure 6H1). In contrast to the lethal toxicity seen in the MV-4-11 model, both treatment strategies prolonged survival in MOLM-13 engrafted mice (median survival in days: 2B4.ζ vs UTD 26 vs 20, p<0.01; 2B4.ζ/sIL-15 vs UTD 27 vs: 20, p<0.01; figure 6J). Analysis of PB at necropsy showed circulating systemic hIL-15 and hTNF-α, but negligible levels of mIL-1β and mIL-6 (online supplemental figure 16A–D).
Discussion
Herein we describe the generation and functional evaluation of chimeric receptors targeting the AML-associated antigen CD123 and expressed in primary human NK cells. The molecular domains distal to the extracellular scFv consisted of various combinations of NK-specific activation moieties. We identified a 2B4.ζ CAR as having similar in vitro functionality to the well-studied 4-1BB.ζ CAR,35 with high surface expression, antigen-specific activation, and cytotoxicity. The in vivo antitumor effect was more pronounced when AML-engrafted mice were treated with 2B4.ζ CAR-NK cells as compared with those treated with 4-1BB.ζ CAR-NK cells, which suggests a potential additive effect of 2B4 and CD3ζ signaling. However, this in vivo effect was short-lived and was accompanied by circulating NK-cell decline. We thus engineered our 2B4.ζ CAR-NK cells with an IL-15 transgene to promote IL-15-mediated activation, proliferation, and survival. Secretion of IL-15 stimulated CAR-NK-cell expansion both in vitro and in vivo, and enhanced short-term and long-term anti-AML cytotoxicity. This bolstered activation profile was confirmed by immunophenotypical and transcriptomic analysis in the setting of chronic stimulation. However, in one in vivo AML model, constitutive IL-15 secretion caused dramatic NK-cell expansion and high levels of circulating human proinflammatory cytokines, and was associated with early death. Treatment with three doses of non-IL-15-secreting CAR-NK cells prolonged survival without systemic toxicity, but antitumor efficacy was transient.
CARs are used to enhance and redirect immune effector cells against cancer cells. CAR-T cells have been extensively investigated in preclinical and clinical models of AML.9 However, there are limited preclinical animal studies36 37 and only three active clinical trials (NCT04623944, NCT05008575, and NCT02742727) testing CAR-NK cells as AML therapy. There is a single completed anti-AML CAR-NK-cell trial with evaluable data (NCT02944162). This study tested an engineered anti-CD33 CAR-NK92 cell line in three patients with relapsed and refractory AML. The CD33-CAR NK92 cell infusion was safe, but the treatment had minimal antitumor efficacy.38 We aimed to enhance the molecular functionality of our CAR by using NK cell-specific receptor domains. We identified the 2B4TM-2B4-CD3ζ CAR as optimal for NK-cell expression and activation. The end result of 2B4 downstream signaling includes synergy with other NK activating receptors.17 39 Protein interactions including 2B4 occur inside membrane (‘lipid’) rafts. We chose to use the 2B4 TM domain to localize the CAR within lipid rafts40 and found that CARs containing a 2B4 TM domain had high surface density and stable CAR expression. Tandem CD3ζ was also important, highlighted by the observed functional differences between 2B4.ζ and 2B4 CAR-NK cells.
To our knowledge, we describe the first study investigating both the in vitro and in vivo anti-AML functionality of peripheral blood-derived CAR-NK cells (PB-NKs) targeting CD123. We chose the PB of healthy donors as the source of our CAR-NK cells. PB-NKs can be isolated through apheresis and expanded in a large scale using feeder cells.26 PB-NKs are functionally mature, with high activating receptor expression and cytotoxic potency. PB-NKs also display higher levels of KIRs compared with other NK-cell products derived from alternative sources. KIR expression is indicative of more complete NK-cell licensing.41 Donor-derived, allogeneic PB-NK cells have therapeutic potential due to their relative safety, immediate availability once manufactured and stored, and reduced manufacturing costs as compared with per-patient manufacture of autologous cell therapy products.12 13 Historically, one challenge facing PB-NK-cell engineering was that of poor viral and non-viral genetic modification.42 With our method, we are able to achieve high levels of PB-NK transduction. All of our CARs were stably expressed on the surface of primary NK cells, though inter-CAR variability in surface density was observed.
NK cells have a natural life span of approximately 2 weeks in humans. The success of adoptively transferred cellular therapies for cancer is determined, in part, by effector cell persistence. The use of systemic cytokine supplementation is one common strategy employed to support prolongation of NK-cell survival. The short half-life of infused IL-15 necessitates frequent or continuous administration, and systemic toxicity is common.43–46 IL-15 super agonists, like N-803 (formerly known as ALT-803), have a longer half-life and can mimic physiological IL-15 trans presentation, but administration can cause inflammatory toxicities.47 The administration of N-803 has been effective at promoting NK-cell proliferation and antitumor efficacy against hematological malignancies, with expected associated fever, chills, and injection site rashes observed.48–51 Another strategy that has been successful in specifically supporting in vivo CAR-NK-cell survival is engineering constitutive activating cytokine expression.52 This approach has been shown to be safe in a clinical trial using CD19-CAR NK cells against CD19+ lymphoid malignancies.53 The demonstrated safety profile motivated us to also test transgenic IL-15 expression with a goal of enhanced in vivo CAR-NK cell persistence. We found NK cells subject to activation with constantly available IL-15 exhibited enhanced and sustained in vitro and in vivo functionality. Though we concluded this to be resultant from specific activation above baseline, it is possible that our NK cells had been rendered ‘cytokine addicted’54 due to ex vivo culture conditions that include supplemental IL-2. In this case, the cohorts of cells without engineered IL-15 secretion may have become dysfunctional once removed from a state of trophic cytokine availability.
In our study, treatment with IL-15 secreting CAR-NK cells caused early death in mice engrafted with MV-4-11 AML. A likely cause of the observed systemic toxicity is severe inflammation due to the dramatic NK-cell proliferation associated with high levels of circulating IL-15 and other proinflammatory cytokines. A CRS-like syndrome triggered by murine monocytes or other immune cells is unlikely due to our inability to detect common murine proinflammatory cytokines. IL-15 stimulation of accelerated leukemic growth was not observed. Clinical signs associated with hyperinflammation (such as weight loss and hunching) are also seen in GVHD and have been observed in our premorbid mice. NK cells have the potential to exacerbate subclinical T cell-mediated acute GVHD.19 We believe that classically defined GVHD is a less likely cause of our observed toxicity due to the absence of human or murine T cells in our NSG model. However, the possibility of lethal toxicity due to NK-cell alloreactivity against mouse cells cannot be excluded.
We observed that IL-15-secreting CAR-NK cells both prolonged and shortened survival in two xenograft models of AML. Notably, the median survival of mice treated with the same dose of 2B4.ζ/sIL-15 NK cells was similar in each model (MOLM-13: 27 days, MV-4-11: 26 days). This could potentially be explained by the aggressiveness of the MOLM-13 model, as these mice died of widespread disease at roughly the same time as we recorded the onset of treatment-associated mortality. Our observation of unexpected toxicity begs for expanded study of novel ways to stimulate CAR-NK-cell cytokine receptor pathways while circumventing systemic cytokine delivery. Localization of IL-15 with membrane tethering55 or targeted delivery through the use of oncolytic viruses56 are alternative therapeutic strategies. Controllable cytokine expression using engineered inducible systems and safety switches also holds promise. Specific activation of intrinsic gamma-cytokine receptor signaling without the use of a pharmacological agent is another strategy that has the potential to sustain NK-cell function with an improved safety profile.
In conclusion, our data underscore the critical nature of IL-15 signaling as a stimulant of CAR-NK-cell long-term survival and anti-AML functionality. Our in vivo data highlight the limitations in murine modeling and the necessity for caution if the use of IL-15 secreting NK cells is planned in human trials of CAR-NK-cell adoptive transfer. Continued therapeutic development is imperative, to include testing of alternate and potentially safer methods of cytokine pathway activation.
Supplemental material
Data availability statement
Data are available in a public, open access repository. Data are available upon reasonable request. Data and materials are available upon request. The bulk RNAseq data generated for this study are available at Gene Expression Omnibus repository under the accession number: GSE184552.
Ethics statements
Patient consent for publication
Ethics approval
No human samples were collected for this study. Human cells and cell lines were deidentified and used under a secondary use protocol approved by the institutional review board. All animal studies were carried out under protocols approved by the Johns Hopkins Institutional Animal Care and Use Committee.
Acknowledgments
We thank Dr Stephen Gottschalk and Dr Peter Chockley for helpful discussion, review, and advice. We also thank the Johns Hopkins University Genetic Resources Core Facility for DNA sequencing, the Experimental and Computational Genomics Core for RNAseq data generation, and the Bloomberg-Kimmel Institute Cytometry Center for multiparameter flow cytometry data acquisition.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors IC and CB conceived of the work, developed the methodology, interpreted the data, and wrote the manuscript. IC, AM, JWR, AT, RR, AF, CR, SY, EES, and CB performed the experiments. IC, WJH, MAK, and CB analyzed the data. IC, RV, and CB performed statistical analysis. All authors revised the manuscript. CB is responsible for overal manuscript content as guarantor.
Funding This work was supported by the Emerson Collective Cancer Research Fund (CLB and IC).
Competing interests CLB and IC have pending patent applications describing the use of engineered natural killer cells to enhance tumor targeting. WJH is a coinventor of patents with potential for receiving royalties from Rodeo Therapeutics/Amgen, is a consultant for Exelixis, and receives research funding from Sanofi.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.