Article Text

Abstract
Background Human cancers are extraordinarily heterogeneous in terms of tumor antigen expression, immune infiltration and composition. A common feature, however, is the host′s inability to mount potent immune responses that prevent tumor growth effectively. Often, naturally primed CD8+ T cells against solid tumors lack adequate stimulation and efficient tumor tissue penetration due to an immune hostile tumor microenvironment.
Methods To address these shortcomings, we cloned tumor-associated antigens (TAA) and the immune-stimulatory ligand 4-1BBL into the genome of modified vaccinia Ankara (MVA) for intratumoral virotherapy.
Results Local treatment with MVA-TAA-4-1BBL resulted in control of established tumors. Intratumoral injection of MVA localized mainly to the tumor with minimal leakage to the tumor-draining lymph node. In situ infection by MVA-TAA-4-1BBL triggered profound changes in the tumor microenvironment, including the induction of multiple proinflammatory molecules and immunogenic cell death. These changes led to the reactivation and expansion of antigen-experienced, tumor-specific cytotoxic CD8+ T cells that were essential for the therapeutic antitumor effect. Strikingly, we report the induction of a systemic antitumor immune response including tumor antigen spread by local MVA-TAA-4-1BBL treatment which controlled tumor growth at distant, untreated lesions and protected against local and systemic tumor rechallenge. In all cases, 4-1BBL adjuvanted MVA was superior to MVA.
Conclusion Intratumoral 4-1BBL-armed MVA immunotherapy induced a profound reactivation and expansion of potent tumor-specific CD8+ T cells as well as favorable proinflammatory changes in the tumor microenvironment, leading to elimination of tumors and protective immunological memory.
- immunotherapy
- local virotherapy
- lymphocyte activation
- vaccination
- lymphocytes
- tumor-infiltrating
Data availability statement
All data relevant to the study are included in the article or uploaded as online supplemental information.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
The lack of potent immune responses against solid tumors due to the poor capacity of immune cells to infiltrate or perform effector functions in the hostile tumor microenvironment (TME) is a major challenge for cancer immunotherapy.1 The concept of reprogramming the immunosuppressive TME into an inflammatory one by tumor-directed therapy has attracted much attention in recent years.2 The aim is to activate immune cells that have already homed to the tumor tissue and local lymph nodes or to recruit new immune cells to the TME, while minimizing irrelevant activation of the rest of the immune system.3 To achieve so, several strategies are being explored in preclinical models as well as in the clinic, either employing the local release and activation of biochemical signals derived from pathogen recognition and unprogrammed cell destruction or local administration of immunostimulatory monoclonal antibodies and cytokines.3
Local oncolytic virotherapy relies on the concept of tumor-targeted therapy through specific infection and destruction of tumor cells and modulation of the TME. The recent Food and Drug Administration (FDA) approval of the first-in-class oncolytic agent IMLYGIC, a modified herpes simplex virus 1 encoding human granulocyte-macrophage colony-stimulating factor (GM-CSF), for stage III melanoma patients,4 emphasized the great potential of oncolytic viruses (OVs). There is a wide spectrum of viral families that have been investigated for their oncolytic effects, including herpesvirus, poxvirus and adenovirus, among others.5
While historically tumor cell-specific replication and direct killing activity of OVs were considered the primary mode of action, initiation or augmentation of a host antitumor immune response is now known to be essential for oncolytic virotherapy.6 Hence, local virotherapy can be regarded as an in situ vaccine that leads to the release of damage-associated or pathogen-associated molecular patterns and immunogenic cell death accompanied by tumor antigen release which ultimately results in the initiation of innate and adaptive antitumor immune responses.7
Modified vaccinia Ankara (MVA)-BN is a highly attenuated vaccinia strain approved by the FDA (JYNNEOS) as a non-replicating vaccine against smallpox and monkeypox.8 In addition, a recombinant MVA-BN vaccine vector has recently been approved by the European Medicines Agency (EMA) as part of an Ebola vaccine and others are employed in clinical trials against various infectious agents as well as in immuno-oncology.9 10 MVA is a potent inducer of type I interferons (IFN)11 12 and elicits robust humoral and cellular immune responses against vector-encoded heterologous antigens.13 14 Importantly, MVA cannot replicate in human cells as its replication ability is largely restricted to embryonic avian cells.15 Thus, the excellent safety profile and immune-stimulatory properties of MVA make it a prime candidate for therapeutic interventions.16
MVA can accommodate large transgene inserts facilitating the incorporation of heterologous antigens and immune-stimulatory molecules to elicit antigen-specific T cell responses and enhance certain immune-activating pathways. CD40L-adjuvanted MVA drastically augmented innate and adaptive immune responses upon intravenous injection.17 18 Furthermore, OVs genetically altered with co-stimulatory molecules or inflammatory cytokines increased therapeutic efficacy after intratumoral (IT) therapy.5 Hence, IT treatment with MVA encoding a tumor-associated antigen (TAA) together with a costimulatory molecule might enhance antitumor immune responses in the TME.
The tumor necrosis factor receptor (TNF)-family member 4-1BB or CD137 is defined as a bona fide costimulatory molecule in T cells. 4-1BB is transiently induced on T cell receptor (TCR) stimulation and subsequent engagement of this costimulatory receptor leads to elevated levels of cytokine secretion as well as the upregulation of the antiapoptotic molecules Bcl-2 and Bcl-xL. This results in increased proliferation and protection against activation-induced T cell death which is also critical for forming immunological memory.19 4-1BB expression in tumor-infiltrating T cells (TIL),20 coupled with its capacity to promote survival, expansion, and enhanced effector function of activated T cells, has made it an alluring target for cancer immunotherapy. Indeed, stimulation of the costimulatory pathway 4-1BB/4-1BBL is beneficial in many therapeutic cancer settings including mono- or combination-therapies with agonistic 4-1BB antibodies or 4-1BBL-expressing viral vectors.21 However, systemic agonistic 4-1BB antibodies were hampered in clinical trials by severe liver toxicity.22 Therefore, the next generation of 4-1BB targeting strategies attempts to leverage the on-target liver toxicity by increasing tumor-specific targeting.23 24
In this study, we combined the immune-stimulatory properties of TAA-encoding MVA with the exquisite T cell-enhancing potential of 4-1BBL and evaluated therapeutic efficacy against solid tumors. We found that IT injection of MVA-TAA-4-1BBL exerted strong objective therapeutic responses in various unrelated tumor models. The therapy was due to strongly reactivated tumor-specific CD8+ T cells and the favorable induction of multiple proinflammatory chemokines and cytokines in the TME. Furthermore, IT MVA-TAA-4-1BBL injection induced systemic antitumor immune responses inhibiting growth of tumor deposits at distant sites. Importantly, IT MVA-TAA-4-1BBL triggered the generation of a diversified tumor-specific memory response that protected against local and metastatic recurrence.
Materials and methods
Mice and tumor cell lines
Female C57BL/6J (H-2b) and Balb/cJ (H-2d) mice aged 6–8 weeks were purchased from Janvier Labs. C57BL/6-Tg (TcraTcrb) 1100Mjb/J (OT-I) and B6.SJL-Ptprca Pepcb/BoyJ (CD45.1) mice were obtained from the University of Zurich and bred to obtain CD45.1+ OT-I mice. All mice were handled, fed, bred and maintained either in the animal facilities at Bavarian Nordic (BN), at the University of Zurich or at the University of Navarra according to institutional guidelines.
The B16.OVA melanoma cell line was a kind gift of Roman Spörri (ETH Zurich). B16.F10 (ATCC CRL-6475) and CT26 wild type (CT26.WT) (ATCC CRL-2638) cell lines were purchased from American Type Culture Collection (ATCC). Tumor cells were cultured in DMEM Glutamax medium supplemented with 10% FCS, 1% NEAA, 1% Sodium Pyruvate and 1% Penicillin/Streptomycin (all reagents from Gibco) in an incubator at 37°C 5% CO2. All tumor cell lines used in experiments conducted at BN were regularly tested negative for Mycoplasma by PCR (results available on request).
Immunizations
IT injections were given into the solid tumor mass with a total volume of 50 µL containing the respective MVA recombinants. Repetitive IT injections were performed at days 0, 5 and 8 after tumor grouping, and indicated in the graphs by vertical dotted lines. When indicated, blood was collected 3 days after last IT immunization for peripheral blood immune cell phenotyping.
Statistical analysis
Statistical analyses were performed as described in the figure legends using GraphPad Prism V.7.02 for Windows (GraphPad Software, La Jolla, California, USA). For immunological data, results are presented as mean and SE of the mean. Either analysis of variance with multiple comparisons test or one-tailed unpaired Student′s t-tests were used to determine statistical significance between treatment groups. For tumor-bearing mice survival after treatment, log-rank tests were performed to determine statistical significance between treatment groups.
Results
4-1BBL potentiates IT MVA immunotherapy
IT application of poxviruses has been shown to effectively induce antitumor responses in various tumor models.25 26 However, most of these studies have been conducted with replicating viruses. Here, we tested whether local treatment of established tumors using the non-replicating poxvirus MVA encoding a TAA and the costimulatory molecule 4-1BBL would convey potent antitumor effects.
IT injections of MVA encoding the TAA ovalbumin (herein referred to as MVA-OVA) controlled tumor growth and prolonged survival of mice bearing established B16.OVA melanomas (figure 1A,B). Notably, IT administration of MVA-OVA-4-1BBL increased tumor rejection to 50% of B16.OVA tumor-bearing mice (figure 1B). Analysis of peripheral blood lymphocytes (PBLs) after the last IT injection revealed that systemic expansion of TAA-specific CD8+ T cells triggered by local MVA-OVA treatment was increased by MVA-OVA-4-1BBL IT administration (online supplemental figure 1, online supplemental file 1).
Supplemental material
Supplemental material
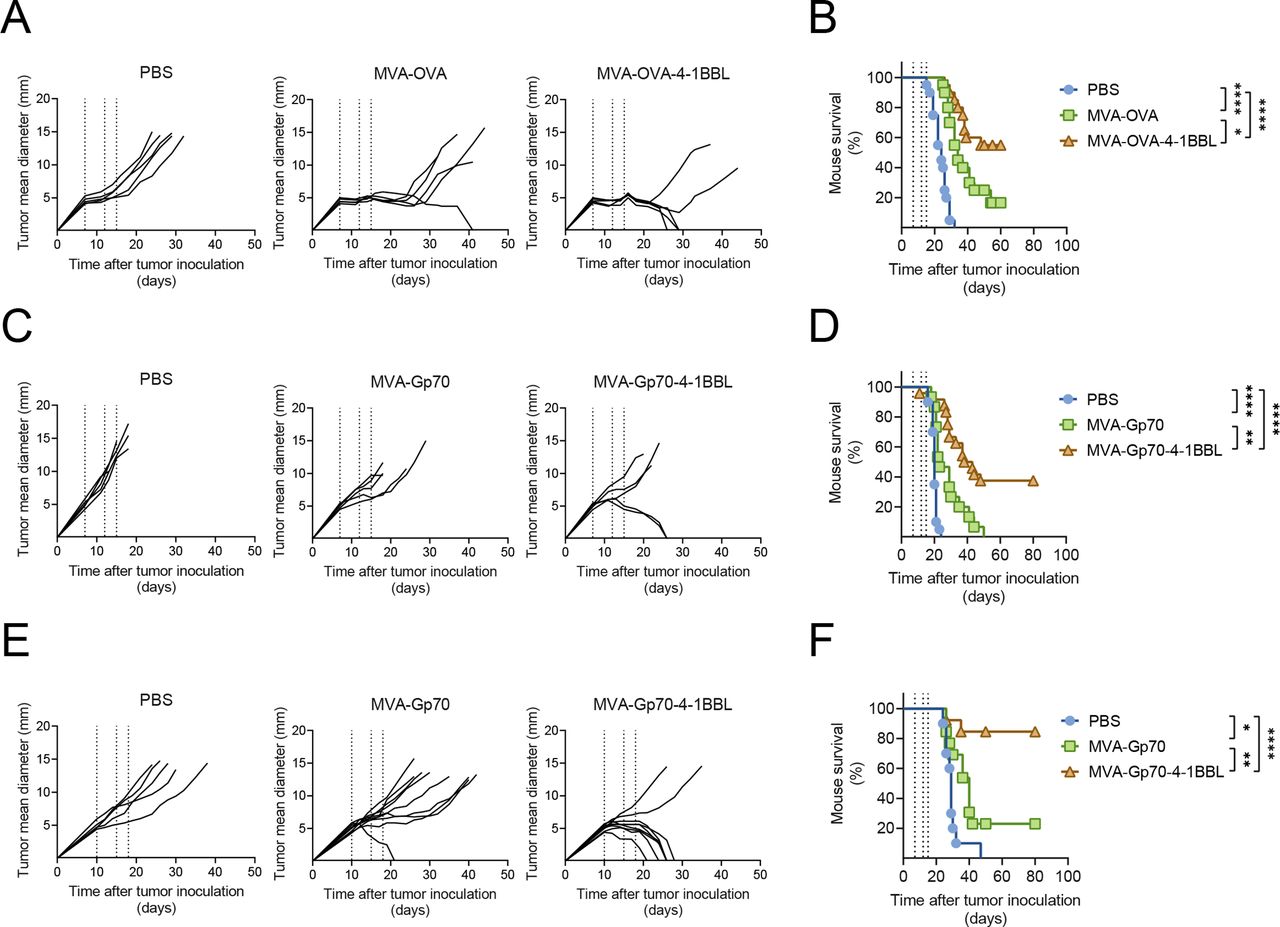
Therapeutic efficacy of intratumoral (IT) administration of MVA-TAA-4-1BBL in unrelated tumor models is independent of the choice of antigen. C57BL/6 (A–D) or Balb/c mice (E, F) received either 5×105 B16.OVA (A, B), 5×105 B16.F10 (C, D) or 5×105 CT26.WT (E, F) cells subcutaneously (SC) in the flank. 7–14 days later, when tumor volumes were above 60 mm3, mice were immunized intratumorally (IT) either with phosphate buffered saline (PBS) or with the indicated MVA constructs. IT immunization was repeated on days 4 or 5 and 8 after the first immunization (dotted lines). (A) Tumor size follow-up (n=5 mice/group) and (B) overall survival (n=20 mice/group) of B16.OVA bearing mice injected either with PBS, 2×108 TCID50 MVA-OVA or 2×108 TCID50 MVA-OVA-4-1BBL; (C) tumor size follow-up (n=5 mice/group) and (D) overall survival (n=15 mice/group) of B16.F10 bearing mice injected either with PBS, 5×107 TCID50 MVA-Gp70 or 5×107 TCID50 MVA-Gp70-4-1BBL; (E) tumor size follow-up (n=5 mice/group) and (F) overall survival (n=10 mice/group) of CT26.WT bearing mice injected either with PBS, 5×107 TCID50 MVA-Gp70 or 5×107 TCID50 MVA-Gp70-4-1BBL. (A, C, E) Data are representative of at least two independent experiments. (B, D, F) Represent overall survival of at least two merged independent experiments. Log-rank test on mouse survival was performed for figures B, D, F. *P<0.05; **p<0.01; ****p<0.0001. IT, intratumoral; MVA, modified vaccinia Ankara; OVA, ovalbumin; PBS, phosphate buffered saline; SC, subcutaneous.
Next, we evaluated other independent, established tumor models. IT administration of MVA encoding the endogenous retroviral antigen Gp7027 (herein referred to as MVA-Gp70) resulted in antitumor effects in B16.F10 melanomas (figure 1C,D). Interestingly, IT MVA-Gp70-4-1BBL markedly prolonged tumor growth control and significantly improved mouse survival (figure 1C,D). Similar results were observed in CT26.WT tumor-bearing mice after IT immunization with either MVA-Gp70 or MVA-Gp70-4-1BBL (figure 1E,F). Local administration of MVA-Gp70-4-1BBL resulted in over 80% rejection of CT26.WT tumors. Restimulation of PBLs with Gp70-derived peptides revealed a robust induction of IFNγ by p15E604-611 (H-2Kb-restricted Gp70 peptide)- and AH16-14 (H-2Kd-restricted Gp70 peptide)-specific CD8+ T cells on MVA-Gp70-4-1BBL IT regime in B16.F10 and CT26.WT tumor-bearing mice, respectively (online supplemental figures S1B and S1C), respectively). Of note, over 70% of C57BL/6 mice that were cured on MVA-OVA-4-1BBL or MVA-Gp70-4-1BBL IT treatment developed vitiligo (online supplemental figure 1 D and E).
CD8+ T cell induction by 4-1BBL adjuvanted MVA dictates antitumor immune responses
We have observed a relationship between tumor growth control and the expansion of TAA-specific CD8+ T cells in the blood upon IT MVA-TAA-4-1BBL administration. Hence, we first interrogated whether 4-1BB ligation affects the quality of antigen-specific CD8+ T cell activation in vitro. Coculture of OT-I CD8+ T cells and MVA-OVA-4-1BBL infected B16.F10 cells led to significantly increased frequencies of Granzyme B- and IFNγ−expressing OT-1 CD8+ T cells compared with MVA-OVA -infected counterparts (online supplemental figures S2A and S2B). Furthermore, substantial production of IFNγ, TNFα and GM-CSF was detected in the supernatant of cocultures of OT-I CD8+ T cells and MVA-OVA-4-1BBL -infected B16.F10 cells (online supplemental figure S2C).
Supplemental material
Next, we analyzed T cell activation and dynamics directly in the TME and the tumor-draining lymph node (TdLN) after IT MVA injection. An increase in the number of CD8+ T cells in the TdLN was observed already 3 days after IT immunization with MVA vectors and further expanded by day 7 (figure 2B). The number of CD8+ T cells infiltrating the TME peaked by day 7 after IT MVA injection (figure 2A). Importantly, in both organs, MVA-OVA-4-1BBL significantly elevated the number of infiltrating CD8+ T cells as compared with MVA-OVA (figure 2A,B). A similar expansion kinetics was detected for CD4+ T cells, however these cells were not further enhanced by 4-1BB ligation (online supplemental figures S3A and S3B).
Supplemental material

CD8+ T cell expansion and dependency on IT MVA-OVA-4-1BBL. C57BL/6 mice received 5×105 B16.OVA cells subcutaneously in the flank. Ten days later when tumor volumes were around 80 mm3, mice were grouped and IT injected with either PBS, 2×108 TCID50 MVA-OVA or MVA-OVA-4-1BBL. 1, 3 and 7 days after immunization, mice were sacrificed for further analysis (n=5–11 mice/group). (A) Number of CD8+ T cells per Mg tumor; (B) number of CD8+ T cells per tumor-draining lymph node (TdLN); (C) number of OVA257-264 –specific CD8+ T cells per Mg tumor; (D) number of OVA257-264 –specific CD8+ T cells per TdLN; (E) Number of B820-27 –specific CD8+ T cells per Mg tumor; (F) number of B820-27 –specific CD8+ T cells per TdLN; (G, H) When B16.F10 tumor volumes were above 60 mm3, mice received PBS or were immunized IT with 5×107 TCID50 of MVA-Gp70-4-1BBL. IT immunization was repeated on day 5 and 8 after the first immunization (dotted lines). Mice received 200 µg of either IgG2b or anti-CD8 antibody intraperitoneally (IP) at day −2, 2, 6 and 10 after immunization; (G) tumor size follow-up (n=8 mice/group) and (H) overall survival (n=8 mice/group). Data in A–F expressed as mean±SEM A–F two-way ANOVA comparing cell numbers in analyzed organs on treatment. *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001. Log-rank test on mouse survival was performed for (H). **P<0.01; ***p<0.001. ANOVA, analysis of variance; IP, intraperitoneally; IT, intratumoral; MVA, modified vaccinia Ankara; n.s., non-significant; OVA, ovalbumin; PBS, phosphate buffered saline; SC, subcutaneous; SEM, SE of the mean.
Analysis of antitumor responses revealed a peak of expansion of OVA-specific CD8+ T cells in the TdLN 3 days after MVA IT injection. By day 7, OVA-specific CD8+ T cells dropped in the TdLN but were significantly expanded in the TME (figure 2C,D). By contrast, MVA-specific CD8+ T cells increased in the tumor and the TdLN on day 7 (figure 2E,F). Notably, numbers of OVA- as well as MVA-specific CD8+ T cells were significantly elevated by 4-1BBL adjuvanted MVA (figure 2C–2F). Lag3 and PD-1 are highly expressed on functionally impaired TILs, thereby contributing to tumor-mediated immune suppression.28 Indeed, OVA-specific CD8+ TILs in untreated tumors expressed high levels of both Lag3 and PD-1. Expression of both surface markers by OVA-specific CD8+ TILs was decreased after treatment (online supplemental figure S3C and SD). In addition, IT MVA injection led to a significant reduction of CD4+ regulatory T cells (Treg) in the TME (online supplemental figure S3E). This resulted in an elevated OVA-specific CD8+ T cell (Teff) to Treg ratio (online supplemental figure S3F).
Driven by our findings, we investigated the contribution of CD8+ T cells to MVA-Gp70-4-1BBL-mediated antitumor effects. Repetitive IT injections using MVA-Gp70-4-1BBL led to tumor growth control and prolonged survival of B16.F10 tumor-bearing mice as compared with PBS (figure 2G,H). Antibody depletion of CD8+ T cells resulted in complete loss of therapeutic efficacy of IT MVA-Gp70-4-1BBL injection. In addition, depletion of both CD4+ T cells and natural killer (NK) cells increased MVA-Gp70-4-1BBL-mediated antitumor effects (online supplemental figure S4). Together, these results determine a central role for CD8+ T cells in the therapeutic responses to MVA adjuvanted with 4-1BBL.
Supplemental material
IT injected MVA localizes to the tumor and induces changes in the tumor microenvironment
Having established that T cells rapidly expanded in the TdLN after IT MVA injection raised the question whether replication-deficient MVA would reside exclusively at the site of injection or transit to other organs. Six hours after IT injection of MVA encoding luciferase (herein referred to as MVA-Luc), high bioluminescence was detected within B16.F10 tumors and decreased upon time (figure 3A,B). Ex vivo analysis of tumor, TdLN and non-draining lymph node (NdLN) 6 hours after IT injection revealed that bioluminescence was not only detected in the tumor, but also in TdLN (figure 3C). We confirmed these results using a MVA vector expressing a soluble form of the human growth factor FMS-like tyrosine kinase 3 ligand (huFlt3L) (online supplemental figure S5A). Furthermore, we determined the presence of MVA-derived genomic DNA (gDNA) in tumor, TdLN, NdLN and peripheral organs 6 hour after IT injection. Similarly, MVA gDNA was detected at high amounts in B16.F10 tumors with minimal appearance in the TdLN (figure 3D). Of note, minute amounts of MVA gDNA were detected in lung, liver, spleen and blood (figure 3E). The use of certain 4-1BB agonists has been associated with severe liver damage in preclinical models29 30 and clinical trials.22 IT administration of MVA-Gp70-4-1BBL neither resulted in elevated alanine aminotransferase (ALT) levels (figure 3F) nor in increased liver weight, hepatic CD8+ T cell infiltration, proliferation or cytotoxicity (online supplemental figure S5B-F). All these features were observed when using IV injection of the anti-4-1BB clone 3H3 as positive control.
Supplemental material

MVA localization and induction of inflammation on IT MVA injection. (A–C) C57BL/6 mice received 5×105 B16.F10 cells SC. (A–C) Five days after tumor inoculation, mice were grouped (n=3 mice/group) and administered IT either with PBS or with 5×107 TCID50 MVA-Luc. (A, B) 6, 24 and 48 hours after IT injection, mice received 150 µg luciferin IP. 10 min later, mice under inhaled anesthesia were assessed for 2 min for bioluminescence using an in vivo imaging system. (A) Representative dorsal images of assayed mice 6 hour after it injection; (B) Photons per second per diameter (ph/s/cm²/sr) in gated regions of interest. Data graph represents two independent experiments (n=3 mice per group) (C) 6 hour after IT injection, mice received 150 µg Luciferin intraperitoneally and were immediately sacrificed to harvest tumor, TdLN and NdLN. Then bioluminescence of the individual organs was assessed (ph/s/cm²/sr). Presented data are representative of two independent experiments. (D, E) C57BL/6 mice received 5×105 B16.OVA cells SC. When tumors reached 60 mm3, mice were grouped (n=4–5 mice/group) and administered IT either with PBS or with 2×108 TCID50 MVA. Six hours after IT injection tumor, TdLN, NdLN (D) and blood, spleen, lung and liver (E) were snap-frozen and viral DNA was extracted from tissue lysates. Gene Copies (gcs) of the MVA gene MVA082L in the different organs is shown. (F) Assessment of liver damage. Schematic representation. Briefly, when B16.F10 tumor volumes were above 60 mm3, mice were injected it with 2×108 TCID50 of MVA-Gp70-4-1BBL on days 0, 5 and 8. As positive control, naïve C57BL/6 mice received 500 µg of anti-4-1BB antibody (clone 3H3) IV twice per week (n=5–8 mice). Mice were bled at the indicated time points and ALT serum levels were determined. (G) C57BL/6 mice received 5×105 B16.OVA cells. When tumors reached 60 mm3, mice were grouped (n=10 mice/group) and administered IT either with PBS or with 2×108 TCID50 MVA, MVA-OVA or MVA-OVA-4-1BBL. Six hours after IT injection, tumors were extracted and tumor lysates processed. Concentration (pg/mL) of indicated cytokines/chemokines in tumor lysates is shown. Data in B–G expressed as mean±SEM. (B–F) Two-way ANOVA was performed. *P<0.05; ***p<0.005; ****p<0.0001. (G) One-way ANOVA was performed. *P<0.05; **p<0.01; ****p<0.0001. ALT, alanine aminotransferase; ANOVA, analysis of variance; GC, gene copies; IP, intraperitoneally; IT, intratumoral; IV, intravenous; Luc, luciferase; MVA, modified vaccinia Ankara; NdLN, non-draining lymph node; OVA, ovalbumin; PBS, phosphate buffered saline; SC, subcutaneous; SEM, SE of the mean; TdLN, tumor-draining lymph node.
We demonstrated that after IT application MVA infection is primarily constrained to the tumor and thus virus-induced antitumor immune responses most likely originated in the tumor. Therefore, we hypothesized that IT injection of MVA-TAA-4-1BBL might induce changes in the TME. IT injection of B16.OVA tumors either with MVA or MVA-OVA led to an upregulation of the proinflammatory molecules IFNα, TNFα, CXCL10 and CCL2 compared with PBS. This effect was significantly increased by MVA-OVA-4-1BBL (figure 3G; online supplemental figure S6A). Interestingly, cytokines such as IFNγ and GM-CSF were almost exclusively induced by 4-1BBL adjuvanted MVA (figure 3G).
Supplemental material
Replicating viruses induce death of infected tumor cells and immune cells.5 Infection with MVA, MVA-TAA or MVA-TAA-4-1BBL enhanced B16.OVA and CT26.WT tumor cell death in vitro (online supplemental figure S6B). Macrophages which have been shown to be preferentially infected by MVA31 were effectively killed, whereby this effect was significantly increased by 4-1BBL adjuvant (online supplemental figure S6B). Cell death results in the release of intracellular proteins such as High Molecular Group Box 1 (HMGB1) that are sensed by innate immune cells and contribute to the initiation of immune responses.32 A significant increase of HMGB1 was detected after MVA infection of tumor cells or macrophages irrespective of 4-1BBL (online supplemental figure S6C).
Our results suggest that local injection of MVA resulted in IT expression of MVA encoded genes, leading to the induction of inflammation, cell death of infected cells and release of immunogenic mediators in the TME.
Local MVA immunotherapy controls tumor growth of distant untreated lesions
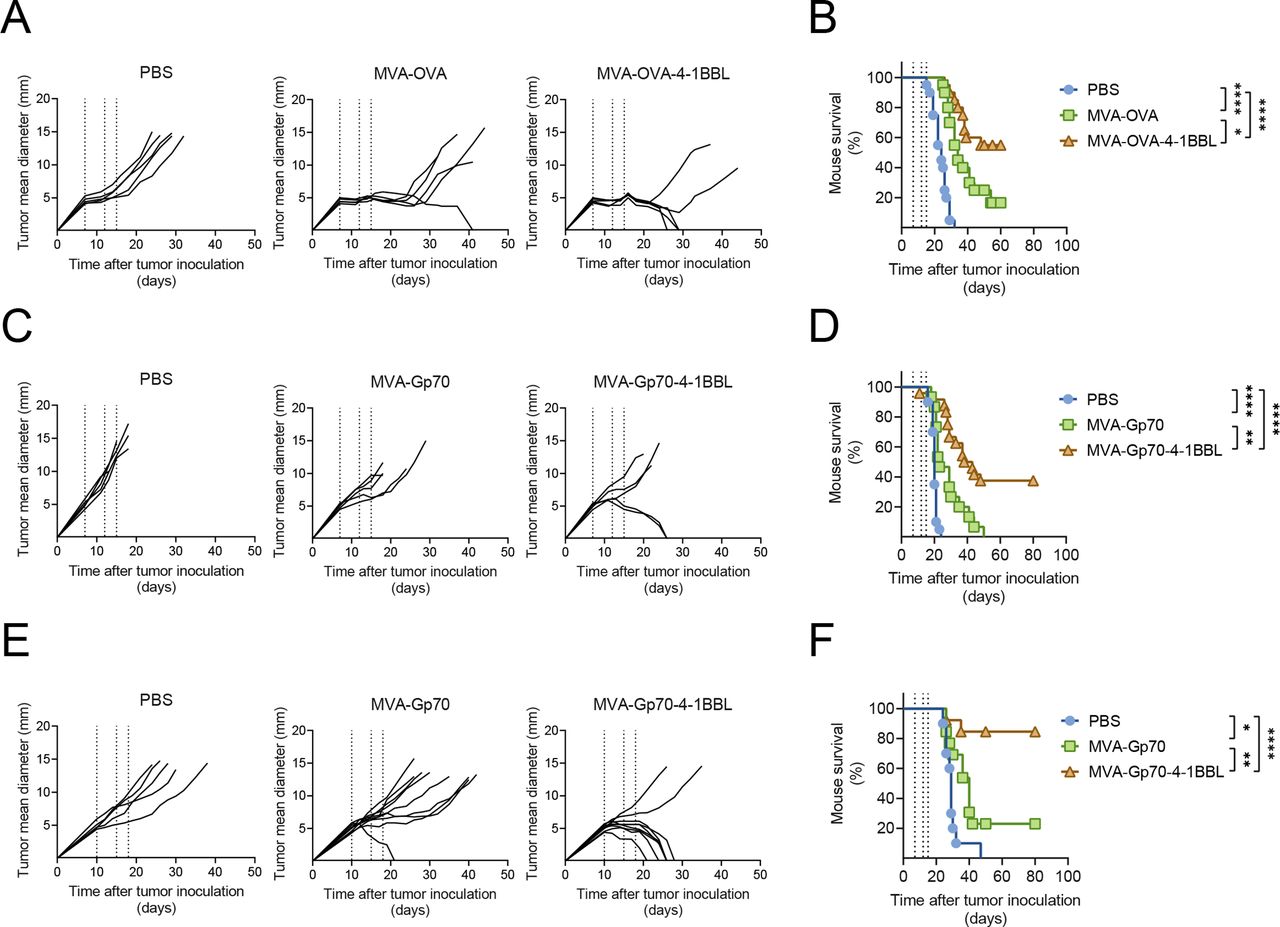
As local treatment with MVA-TAA-4-1BBL not only induced robust tumor-specific T cell responses in the TME but also in the blood, we next assessed the systemic antitumor potential of IT MVA immunotherapy on distant tumor deposits. CT26.WT tumor cells were implanted subcutaneously to the right and the left flank of Balb/c mice (figure 4A). IT injection of MVA-Gp70 delayed tumor growth as compared with PBS (figure 4B,C). IT MVA-Gp70-4-1BBL injection resulted in clearance of the treated tumor in 6/10 CT26. WT tumor-bearing mice (figure 4D). Importantly, local administration of both MVA-Gp70 and MVA-Gp70-4-1BBL led to tumor growth delay and, in some cases, complete tumor clearance of the untreated tumor lesions (figure 4C,D). These data demonstrate the effective induction of antitumor immune responses against distant, untreated tumor lesions by IT MVA immunotherapy.

intratumoral MVA-immunotherapy induces rejection of untreated lesions. (A–D) Bilateral tumor model. (A) Experimental layout. Balb/c mice received 5×105 and 1×105 CT26.WT tumor cells SC into the right and left flank, respectively. Five days later, right flank tumors were immunized IT either with PBS or with the indicated MVA constructs. IT immunization was repeated on days 5 and 8 after the first immunization (arrows). (B) Tumor size follow-up (n=10 mice/group) of the treated and untreated tumor after PBS IT injection. (C) Tumor size follow-up (n=10 mice/group) of the treated and untreated tumor after 5×107 TCID50 MVA-Gp70 it injection. (D) Tumor size follow-up (n=10 mice/group) of the treated and untreated tumor after 5×107 TCID50 MVA-Gp70-4-1BBL it injection. IT, intratumoral; MVA, modified vaccinia Ankara; PBS, phosphate buffered saline; SC, subcutaneous.
IT MVA-TAA-4-1BBL treatment protects from local tumor rechallenge and induces epitope spreading
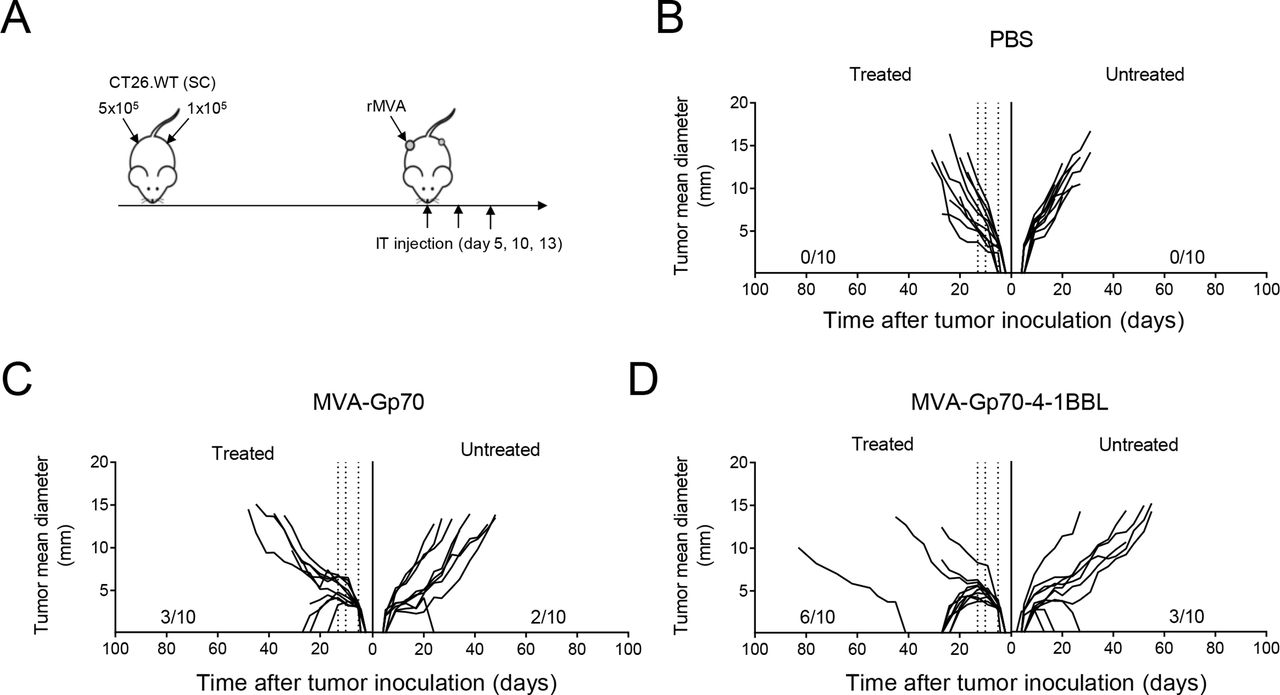
One of the main goals of cancer vaccines is to achieve long-term protective immunological memory to prevent tumor recurrence. Therefore, we first assessed whether IT MVA-TAA-4-1BBL-induced antitumor responses generate immunological memory that protects against local tumor rechallenge (figure 5A). Naïve mice used as controls rapidly grew tumors in 100% of mice. Mice that were previously cured after IT MVA-OVA treatment had a high prevalence for tumor regrowth of 60% on rechallenge. In contrast, about 80% of mice (9/11) that previously received IT MVA-OVA-4-1BBL were resistant to secondary tumor growth (figure 5B). Similar results were obtained in mice that were cured after MVA-Gp70-4-1BBL treatment and local rechallenge with B16.F10 cells. About 55% of pretreated mice remained tumor-free after B16.F10 tumor cell implantation (online supplemental figure S7A). Hence, IT MVA-TAA-4-1BBL treatment induced strong protective immunological memory against local tumor rechallenge.

IT MVA-TAA-4-1BBL treatment protects from local tumor rechallenge and induces epitope spreading. (A) Experimental layout. Naïve C57BL/6 mice or long-term survivors (12–36 weeks after tumor clearance) of figure 1A,B were rechallenged SC into the tumor-naïve flank of cured mice with 5×105 B16.OVA cells. Peripheral blood was analyzed by flow cytometry before (day −6) and after (day 7) after rechallenge. Blood, spleen, NdLN and TdLN mononuclear cells were analyzed on day 41 after tumor cell inoculation. (B) Percentage of tumor-free mice over time is displayed (n=5–11 mice/group). Number of tumor-free mice per group is shown. (C) Frequency of peripheral blood CD44+ OVA257-264 Dex+ CD8+ T cells pre-B16 and post-B16.OVA rechallenge of naïve mice and long-term survivors after IT MVA-OVA or MVA-OVA-4-1BBL treatment. (D) Frequency of CD44+ OVA257-264 Dex+ CD8+ T cells in blood, spleen, NdLN and TdLN. (E) Frequency of splenic CD44+ IFNγ+ TNFα+ IL2+ CD8+ T cells after restimulation with OVA257-264 peptide. (F) Frequency of CD62L- CD127+ CD69- OVA257-264 Dex+ T cells (TEM) in blood, spleen, NdLN and TdLN (left). Frequency of CD62L+ CD127+ OVA257-264 Dex+ cells (TCM) in blood, spleen, NdLN and TdLN (middle). Frequency of CD62L- CD127+ CD69+ OVA257-264 Dex+ (TRM) in blood, spleen, NdLN and TdLN 41 days after B16.OVA cell challenge (right). (G) Experimental layout. Naïve C57BL/6 mice (n=5) or long-term survivors (12–36 weeks after tumor clearance, n=18) that rejected B16. OVA tumors on IT MVA-OVA-4-1BBL were rechallenged SC into the left flank with 5×105 B16.F10 cells. (H) Tumor size follow-up. (I) Overall survival. data in A–H expressed as Mean±SEM. (C–F) Two-way ANOVA was performed. *P<0.05; **p<0.01; ***p<0.005. (E) one-way ANOVA was performed. *P<0.05; **p<0.01; ***p<0.001. Log-rank test on mouse survival was performed for figure 1. ***P<0.001. ANOVA, analysis of variance; IFNγ, interferon-γ; IL2, interleukin 2; IT, intratumoral; MVA, modified vaccinia Ankara; NdLN, non-draining lymph node; OVA, ovalbumin; PBL, peripheral blood lymphocyte; SC, subcutaneous; SEM, SE of the mean; TdLN, tumor-draining lymph node; TNFα, tumor necrosis factor-α; TRM, resident memory T cells.
OVA-specific CD8+ T cells could be readily detected prior to rechallenge in mice that had rejected the tumor after IT treatment with MVA-OVA-4-1BBL, but not with MVA-OVA (figure 5C). Seven days after tumor cell injection, the OVA-specific T cell population was significantly expanded, indicative of effective tumor recognition (figure 5C). Splenocyte OVA257-264 peptide restimulation showed that IT MVA-OVA-4-1BBL therapy induced a large population of multicytokine-producing antigen-specific CD8+ T cells (figure 5E). Analysis of spleen, blood, TdLN and NdLN on day 41 after tumor rechallenge revealed an accumulation of OVA-specific CD8+ T cells in all organs analyzed (figure 5D). Memory subset examination33 revealed that OVA-specific TCM cells were equally distributed over all organs, while TEM cells were mainly found in blood, spleen and TdLN but not in the NdLN (figure 5F). Next, we analyzed tissue-resident memory T cells (TRM) that have been shown to play a key role in antitumor immunity. Strikingly, we could also detect a significant population of resident memory T cells (TRM) exclusively located in the TdLN (figure 5F). Likewise, p15E-specific CD8+ T cells in the blood were detected pre and post rechallenge of mice that cleared primary B16.F10 tumors on IT MVA-Gp70-4-1BBL treatment (online supplemental figure S7B). Furthermore, we could identify p15E-specific TCM, TEM and TRM cells at day 42 post B16.F10 rechallenge (Suppl. Figure S7C-E), whereby the latter were exclusively found in the TdLN (Suppl. Figure S7E). Our results demonstrate that IT MVA-TAA-4-1BBL immunotherapy led to the induction of a diversified population of tumor-specific memory CD8+ T cells encompassing TCM, TEM as well as TRM cells.
We next addressed whether MVA-TAA-4-1BBL IT treatment induced the diversification of epitope specificity from the defined TAA in the vaccination regime, a phenomenon known as epitope spreading. To test this, we rechallenged mice that rejected B16.OVA tumors on IT MVA-OVA-4-1BBL with B16.F10 cells, which do not express OVA (figure 5G). Interestingly, significant delay in tumor growth (figure 5H) and mouse survival (figure 5I) was observed. These results indicate that IT MVA-TAA-4-1BBL-mediated immune responses do not only restrict to the initially TAA encoded in the vaccine but also extend to other, tumor-specific antigens.
IT MVA-TAA-4-1BBL cured mice are resistant to systemic tumor rechallenge
IT MVA-TAA-4-1BBL injection induced systemic immune responses that mediate control of local recurrent tumors and untreated lesions. We reasoned that the tumor-specific T cell memory generated by IT MVA injection might also protect against metastatic recurrences (figure 6A). Macroscopic quantification of tumor nodules in the lung after intravenous CT26.WT tumor cell injection showed the development of multiple lesions in naïve mice. No macroscopic metastatic lesions were found in the lungs of mice that were previously cured with IT MVA-Gp70 or MVA-Gp70-4-1BBL (figure 6B). T cell analysis revealed an enlarged population of multifunctional AH1-specific CD8+ T cells in MVA-Gp70-4-1BBL cured mice (figure 6C).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
intratumoral (IT) MVA-TAA-4-1BBL treated mice are resistant to subsequent systemic tumor rechallenge. (A–C) Systemic tumor rechallenge. (A) Experimental layout. Naïve Balb/c mice or long-term survivors of figure 1E,F were rechallenged IV with 2×105 CT26.WT cells. Spleen and lungs were analyzed on day 19 after tumor cell injection. (B) Representative Photographs of lungs after fixation in Bouin′s solution on day 19 after tumor cell transfer into naïve or cured mice. Total number of macroscopic pulmonary metastasis was evaluated (n=3–13 mice/group). (C) Frequency of splenic CD44+ IFNγ+ TNFα+ IL2+ CD8+ T cells after restimulation with AH16-14 peptide 19 days after rechallenge. (A–C) n=3–13 mice/group. (B, C) Data are expressed as mean±SEM. One-way ANOVA was performed *P<0.05; **p<0.01; ***p<0.001. ANOVA, analysis of variance; IFNγ, interferon-γ; IL2, interleukin 2; IV, intravenous; MVA, modified vaccinia Ankara; SEM, SE of the mean; TNFα, tumor necrosis factor-α.
Taken together, local immunotherapy using MVA genetically modified to express a tumor antigen together with the costimulatory molecule 4-1BBL conveyed strong antitumor activity by combining innate and adaptive immune activation. This not only resulted in the induction of systemic antitumor effects but also in the generation of a potent tumor-specific memory response that protected against local and systemic tumor rechallenge.
Discussion
In the present study, we took a novel approach for tumor-directed virotherapy and used a non-replicating MVA genetically modified to express TAAs and the costimulatory molecule 4-1BBL. This combines the excellent immune-stimulatory properties of MVA and its high safety profile with the immune-activating potential of 4-1BBL. IT MVA-TAA-4-1BBL injection activates a sequence of immediate and long-term immune events, ultimately resulting in tumor eradication. The induction of multiple proinflammatory mediators by IT MVA treatment is indicative of a fundamental alteration of the previously immune-suppressive TME that facilitates the re-activation and expansion of tumor- specific T cells. MVA-encoded 4-1BBL triggered drastic qualitative and quantitative changes in cytotoxic antitumor immune responses that were essential for both therapeutic efficacy and formation of local and systemic long-term immunologic memory against the primary tumor.
We show for the first time that IT injection of active, non-replicating MVA conveys potent therapeutic antitumor effects. Interestingly, it has been reported that IT delivery of heat-inactivated MVA but not MVA induced strong antitumor effects mainly depending on the activation of cytotoxic T cells.26 In contrast to this study, we utilized active MVA encoding for TAA with or without additionally expressing 4-1BBL. Importantly, MVA-TAA alone was already effective in delaying tumor growth and the adjuvantation with 4-1BBL significantly improved therapeutic efficacy, leading to rejection of established tumors within multiple models. Moreover, the antitumor effect was independent of the choice of tumor antigen. Apart from the model antigen OVA, we investigated the endogenous retroviral protein Gp70 for its immunogenic potential as TAA. Endogenous retroviral elements are epigenetically silenced in healthy tissues but re-activated and expressed in various cancers.34 Likewise, Gp70 is highly expressed in several murine tumor cell lines.35 In humans, there is growing evidence that these transposable elements such as endogenous retroviral sequences might have potent immunogenic properties and therefore represent excellent TAA targets for cancer immunotherapy.36 37 Given the self-nature of Gp70,38 the strong therapeutic effects obtained by IT MVA-Gp70-4-1BBL treatment in Gp70-expressing tumor models imply that local MVA therapy cannot only induce the rejection of tumors expressing neoantigens but also break peripheral tolerance to endogenous self-antigens.
IT virotherapy repurposes virus-induced inflammation and cell death to alter the immunosuppressive TME.5 This cascade of events would enhance antitumor-specific immunity. Likewise, our data show that MVA infection promotes tumor cell death and hence HMGB1 release, similar to oncolytic vaccinia virus.39 Moreover, IT injection of MVA elicited a strong inflammatory response within the TME which was accompanied by the induction of multiple MVA-related cytokines and chemokines.40 IT application of 4-1BBL-adjuvanted MVA strongly increased the concentration of IFNγ and GM-CSF in B16.OVA tumors. This infers that the induction of those molecules is downstream of 4-1BB signaling. Indeed, in vitro activation of OVA-specific CD8+ T cells by MVA-infected tumor cells led to the production of large amounts of IFNγ and GM-CSF exclusively in the presence of MVA-encoded 4-1BBL. Interestingly, a fraction of tumor-infiltrating CD8+ T cells in murine and human cancers expresses 4-1BB, indicative of previous antigen encounter and tumor specificity.20 41 Therefore, it is tempting to speculate that the production of both proinflammatory mediators within the TME is unleashed by direct interaction of pre-existing antigen-specific CD8+ T cells on encounter with MVA-OVA-4-1BBL infected cells. Furthermore, T cell activation and subsequent T cell-mediated cytotoxicity in tumor tissue could further propagate immunogenic cell death in the TME.42
Our data demonstrate a central role for the induction of cytotoxic CD8+ T cells on IT MVA-based immunotherapy. In contrast to this, systemic antibody depletion of CD4+ T cells improved therapeutic efficacy alone or in combination with IT MVA-Gp70-4-1BBL. This is in line with published data showing B16.F10 tumor growth retardation on anti-CD4 treatment and enhancement of agonistic 4-1BB antibody-mediated therapy.43 The depletion of immunosuppressive CD4+ Treg was implicated in the improved antitumor response. Similar effects were observed when NK cell depletion and MVA-based immunotherapy was combined. Even though NK cells are potent mediators of antitumor immunity, they do play a regulatory role in antiviral T cell immunity. CD8+ T cells can be directly killed by NK cells in vivo,44 thereby suppressing the ability of antiviral CD8+ T cells to control infection.45 We hypothesize that excessive NK cell activation by IT MVA-TAA-4-1BBL injection eliminates pre-existing tumor-specific CD8+ T cells infiltrating the tumor. Then, in the absence of NK cells, tumor-infiltrating CD8+ T cells would get better primed by antigen-presenting cells or directly by MVA-4-1BBL and exert stronger antitumor responses.
Analysis of T cell expansion on IT MVA-TAA-4-1BBL treatment revealed differences in the kinetics of tumor-specific (OVA) and virus-specific (MVA) CD8+ T cells. While OVA-specific CD8+ T cells peaked as early as day 3 after injection, we observed the increase of MVA-specific CD8+ T cells by day 7. As expected, MVA-directed responses were generated de novo from the naïve circulating CD8+ T cell pool in the periphery. In contrast, OVA-specific CD8+ T cells were expanded by IT MVA injection most likely from an antigen-experienced population of tumor-specific CD8+ T cells located in or in close vicinity of the tumor. The rapid reactivation and expansion of tumor-resident T cells after IT immunotherapy was reported previously.46 In support of this assumption, we observed the downregulation of Lag-3 and PD-1 on tumor-specific CD8+ T cells in the tumor after IT administration of MVA-TAA-4-1BBL which implicates the re-acquisition of potent effector functions on treatment.
Together, our results support the idea that IT MVA-TAA-4-1BBL therapy cannot only generate and recruit de novo T cell responses from the periphery but also reinvigorate and boost antigen-experienced T cells resident in the tumor or local LN. Thus, various tumor categories might benefit from our IT administration, irrespective if they are already associated with pre-existing T cell responses (hot tumors) or lack significant T cell infiltrates (cold tumors) thus depending on de novo activation of peripheral T cells.47
The expansion of tumor-specific CD8+ T cells was not only observed in the tumor but also in the TdLN and even preceding their appearance within the tumor. We, therefore, investigated MVA-encoded antigen distribution and potential T cell priming in the TdLN and other organs upon IT MVA administration. We addressed this by performing a comprehensive analysis of the localization of MVA within different organs after IT injection. Protein expression by MVA and MVA gDNA was mostly confined to the tumor site. However, MVA-encoded soluble hFlt3L, MVA-encoded luciferase or MVA gDNA were also detected in the TdLN, although at significantly lower amounts compared with the tumor. Hence, the TdLN could also serve as a priming site for tumor-specific T cells. Our results are in concordance with previous work showing that MVA localizes in the paracortical region of the draining LN after footpad injection of MVA.48 In agreement with this, no protein or gDNA was found in the NdLN. Interestingly, we could detect low amounts of viral DNA in the spleen, blood, lung and liver suggesting some leakage of the 4-1BBL-expressing virus or processed viral DNA from the tumor into the blood or lymphatic systems. We did not observe signs of liver damage on local MVA-TAA-4-1BBL injection compared with systemic agonistic 4-1BB-antibody. Sandin et al reported for agonistic CD40 antibody to enhance hepatotoxicity when given IV, but not IT.49 Hence, our results highlight both the safety and specificity of IT MVA-TAA-4-1BBL injection.
An important aspect of tumor-directed immunotherapy is the generation of a systemic antitumor immune response that eradicates distant metastases and induces long-term tumor immunity. We showed that local MVA-TAA-4-1BBL treatment across different tumor models not only elicited immune responses within the TME, but also led to systemic antigen-specific CD8+ T cell responses in the blood, including multifunctional memory populations. In addition, our data obtained from the bilateral tumor model unambiguously demonstrated that IT MVA injection resulted in significant antitumor effects in untreated lesions. A clear contribution of 4-1BBL adjuvantation on the untreated tumor, however, could not be observed in this experimental setup.
Moreover, across many individual experiments and different TAAs employed, mice that had rejected melanomas on IT MVA-TAA-4-1BBL treatment developed vitiligo. Vitiligo is a pigmentation disorder with focal loss of melanocytes in the skin caused by autoreactive CD8+ T cells.50 These cells have been induced by IT MVA-TAA-4-1BBL virotherapy, most likely through antigen spread, a phenomenon that describes the diversification of epitope specificity from the initial focused, dominant epitope-specific immune response, for example, Gp70 or OVA. In support of this hypothesis, IT MVA-OVA-4-1BBL cured mice that were rechallenged with B16.F10 tumor cells lacking the primary rejection antigen OVA showed significantly prolonged survival compared with naïve mice.
Together, the antitumor response triggered by local MVA-TAA-4-1BBL administration was associated with system-wide immunity against the primary tumor. Furthermore, our results support the notion that MVA IT treatment induces tumor antigen spread which is a desirable feature of cancer immunotherapy as it broadens the antitumor response and prevents the likelihood of tumor escape by TAA loss.
The ability of the immune system to maintain memory of previous antigen encounters is the basis for long-term immunity. Here, we defined the components of immunological memory induced on IT MVA administration. Circulating TAA-specific CD8+ T cells were detected in mice several months after tumor clearance regardless of the tumor model or mouse strain used. CD8+ T cell frequencies were significantly increased when 4-1BBL-adjuvanted MVA was used. We found that mice that were previously cured with IT MVA-TAA-4-1BBL were more resistant to subcutaneous tumor rechallenge with B16.OVA or B16.F10 than MVA-TAA treated counterparts. Analysis of tissues from those mice showed that T cell memory subsets were not only found in the circulation but also in multiple anatomical sites, suggesting immune surveillance. Increased frequencies of antigen-specific CD8+ TCM and TEM subsets were detected in spleen and blood after local tumor rechallenge of cured mice that previously received MVA encoding 4-1BBL. It is well established that 4-1BBL/4-1BB signals are particularly potent in enhancing the expansion and maintenance of CD8+ effector and memory T cells.51 Likewise, MVA-encoded 4-1BBL costimulation enhanced the activation and effector function of tumor-specific cytotoxic T cells which resulted in the formation of a potent and diverse memory compartment.
In addition to circulating CD8+ TCM and TEM subsets, resident CD8+ TRM cells have been shown to cooperate in antitumor immunity.52–54 Interestingly, cured mice after IT 4-1BBL-adjuvanted MVA showed increased frequencies of tumor-specific TRM cells exclusively in the TdLN after local rechallenge either with B16.OVA or B16.F10. Although CD8+ TRM cells were first identified in the tissues, they can also migrate from the tissues and accumulate in the draining LN of mice on antigen reencounter.54–56 In line with our results, 4-1BB has been shown to promote the establishment of an influenza-specific CD8+ TRM pool in the lung after intranasal immunization.57 We postulate a relationship between the expansion of tumor-specific CD8+ TRM cells in the TdLN and the better response to local secondary tumor rechallenge by cured mice on IT 4-1BBL adjuvanted MVA.
In cancers, memory CD8+ T cells are often dysfunctional due to suboptimal differentiation or maintenance conditions and chronic antigen exposure.58 This phenomenon is associated with the inability to secrete IL-2 and TNFα.59 60 Importantly, IT 4-1BBL adjuvanted MVA generated a highly competent CD8+ T cell memory pool, that on reencounter of tumor antigen expanded and produced significant amounts of IFNγ, TNFα and IL-2 compared with IT MVA in all rechallenge models tested.
In summary, we describe a novel therapeutic platform based on the local injection of a non-replicating MVA expressing a TAA in conjunction with 4-1BBL. IT virus injection induced profound proinflammatory changes in the TME leading to reactivation and expansion of tumor-specific CD8+ T cells. In addition, we demonstrated the generation of a diverse CD8+ T cell memory population protecting from local and systemic tumor rechallenge. Together with the excellent safety profile of MVA, our preclinical data provide a strong rationale for exploring this approach in the clinic.
Supplemental material
Data availability statement
All data relevant to the study are included in the article or uploaded as online supplemental information.
Ethics statements
Ethics approval
Animal experiments were approved by the animal ethics committee of the government of Upper Bavaria (Regierung von Oberbayern, Sachgebiet 54, Tierschutz) and were carried out in accordance with the approved guidelines for animal experiments at Bavarian Nordic. Bilateral tumor treatment, as well as luciferase biodistribution experiments, were conducted at CIMA, University of Navarra (Pamplona, Spain) in compliance with the Association for Assessment and Accreditation of Laboratory Animal Care International (AAALAC). The experimental design was approved by the Ethics Committee for Animal Testing of the University of Navarra.
Acknowledgments
We would like to thank Jana Haug, Kerstin Laemmermann, Ronny Kassub, Ismail Housni, Markus Feigl, Rodrigo Carrasco-León, Nuria Ardaiz, Leire Arrizabalaga and Stephan Rambichler for excellent technical performance of experiments. In addition, we would like to thank Yvonne Krause and Kerstin Zehentbauer for great technical support in handling the animal facility and Vaccine Generation for producing virus stocks. We would also like to thank Dr Karin Weber from the Medizinische Kleintierklinik for performing ALT assays.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors Experiment design: MH, FG, HL, HH and JM-E. Virus generation: SW. Experiment performance: MH, RG, FG, GF, BB and JM-E. Data analysis and interpretation: MH, FG, GF, MS, PB, HL, HH and JM-E. Manuscript writing and scientific discussions: MH, MS, PB, PC, IM, HL, HH and JM-E. Authors cosupervised this work: HH and JM-E.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests MH, RG, FG, GF, BB, SW, PC, MS, HL, HH and JM-E are or have been employees of Bavarian Nordic. IM reports advisory roles with Roche-Genentech, Bristol-Myers Squibb, CYTOMX, Incyte, MedImmune, Tusk, F-Star, Genmab, Molecular Partners, Alligator, Bioncotech, MSD, Merck Serono, Boehringer Ingelheim, Astra Zeneca, Numab, Catalym, Bayer, and PharmaMar, and research funding from Roche, BMS, Alligator, and Bioncotech. PB reports advisory roles with Ferring, Tusk and Moderna, research funding from Sanofi, and Bavarian Nordic and speaker honoraria from Ferring, BMS, MSD, Novartis, Boehringer Ingelheim and AstraZeneca.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.
Linked Articles
- Oncolytic and local immunotherapy