Article Text

Abstract
Background Intratumoral delivery of immunotherapeutics represents a compelling solution to directly address local barriers to tumor immunity. However, we have previously shown that off-target delivery is a substantial problem during intratumoral injections; this can lead to diminished drug efficacy and systemic toxicities. We have identified three variables that influence intratumoral drug delivery: injection technique, drug formulation and tumor microenvironment. The purpose of this study was to characterize the impact of modifications in each variable on intratumoral drug delivery and immunotherapy efficacy.
Methods Intratumoral injections were performed in a hybrid image-guided intervention suite with ultrasound, fluoroscopy and CT scanning capabilities in both rat and mouse syngeneic tumor models. Intratumoral drug distribution was quantified by CT volumetric imaging. The influence of varying needle design and hydrogel-based drug delivery on the immune response to a stimulator of interferon genes (STING) agonist was evaluated using flow cytometry and single cell RNA sequencing. We also evaluated the influence of tumor stiffness on drug injection distribution.
Results Variations in needle design, specifically with the use of a multiside hole needle, led to approximately threefold improvements in intratumoral drug deposition relative to conventional end-hole needles. Likewise, delivery of a STING agonist through a multiside hole needle led to significantly increased expression of type I interferon-associated genes and ‘inflammatory’ dendritic cell gene signatures relative to end-hole STING agonist delivery. A multidomain peptide-based hydrogel embedded with a STING agonist led to substantial improvements in intratumoral deposition; however, the hydrogel was noted to generate a strong immune response against itself within the target tumor. Evaluation of tumor stroma on intratumoral drug delivery revealed that there was a greater than twofold improvement in intratumoral distribution in soft tumors (B16 melanoma) compared with firm tumors (MC38 colorectal).
Conclusions Injection technique, drug formulation and tumor stiffness play key roles in the accurate delivery of intratumoral immunotherapeutics.
- drug evaluation
- preclinical
- therapies
- investigational
- translational medical research
- tumor microenvironment
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
- drug evaluation
- preclinical
- therapies
- investigational
- translational medical research
- tumor microenvironment
Background
Systemic immunotherapies represent a paradigm shifting advance in cancer care. However, the majority of patients do not demonstrate durable responses to systemic therapies such as immune checkpoint inhibitors.1 Investigations into patterns of immunotherapy resistance have highlighted multiple mechanisms including T cell dysfunction, tumor cell immune evasion tactics and immunosuppressive cells in the tumor immune microenvironment.2 While some of these barriers can be overcome through combination systemic therapies such as dual checkpoint inhibition, the toxicities associated with such multidrug regimens can be substantial.3
Intratumoral delivery of immunotherapeutics represents a compelling solution to directly address local impediments to tumor immunity. Direct delivery into target tumor lesions affords several advantages over systemic delivery, including increased local concentrations and potentially diminished systemic toxicities. Moreover, for patients with polyclonal metastases, the ability to inject multiple sites could potentially lead to a more robust adaptive immune response.
There exist a myriad intratumoral immunotherapies currently in clinical trials. In our clinical experience, we have found image-guided intratumoral immunotherapy procedures to overall be safe.4 However, we have also found that while placing a needle using image guidance into a target lesion is often straightforward, ensuring that the injected therapy is distributed throughout the target lesion without leakage into the surrounding tissue is not. Substantial variations in drug deposition accuracy can occur within the same cancer histology, the same organ and even the same patient. These deficiencies in drug delivery may result in ineffective immune activation within the target lesion because of insufficient on-target deposition, and they may result in organ injury and systemic immune toxicities due to off-target leakage. Indeed, it is entirely plausible the lack of clinical response in a clinical intratumoral immunotherapy trial may be a reflection of delivery failure rather than drug ineffectiveness.4
Thus, there are substantial implications to identify clinically translatable methods to optimize intratumoral immunotherapy delivery and efficacy. Based on our clinical observations, we have adopted a heuristic model of intratumoral drug delivery that highlights three influential variables: injection technique, drug formulation and tumor microenvironment (figure 1). The purpose of this study was to characterize the impact of modifications in each variable on intratumoral drug delivery and immunotherapy efficacy.
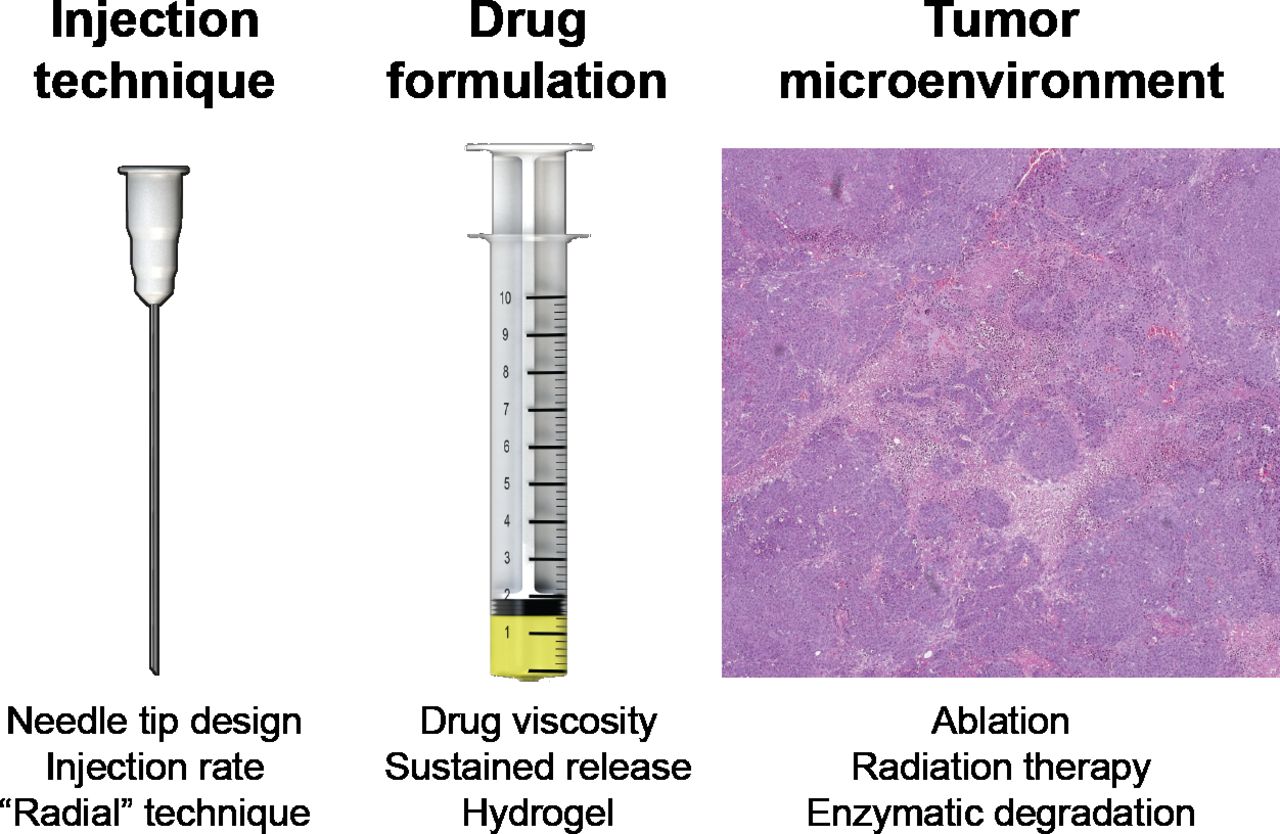
Variables that influence intratumoral immunotherapy. Based on our clinical observations, we have created a heuristic model for factors that affect intratumoral immunotherapy delivery comprising these three variables. For each variable, we have listed potential modifications that could lead to improvements in intratumoral immunotherapy delivery and efficacy.
Methods
Animal models
All animal experiments were approved by The University of Texas, MD Anderson Cancer Center’s Institutional Animal Care and Use Committee. Syngeneic rat and mouse models were used; the former model allowed for evaluation of intratumoral injections into tumors of clinically relevant sizes, and the latter models allowed for investigations into the influence of tumor stroma on injection efficacy. For the rat model, animals were acquired from a Buffalo rat colony maintained at The University of Texas, MD Anderson Cancer Center. Unless otherwise indicated as a replicate measurement, data were taken from distinct samples. A hepatocellular carcinoma (HCC) model was generated by subcutaneous implantation of 5×106 McArdle RH-7777 (ATCC) hepatoma cells in Buffalo rats. Tumor growth was monitored twice weekly with calipers for 1–2 weeks until the tumors were approximately 1 cc in volume using the formula (length×width2/2). This is a syngeneic HCC animal model in an immunocompetent rat.
The mouse melanoma and colorectal cancer cell lines B16 and MC38/gp100 were cultured in RPMI 1640 supplemented with 1X HEPES, 1X glutamax, 1X insulin-transferrin-selenium, 10% heat-inactivated fetal bovine serum (FBS), 55 µM β-mercaptoethanol and 0.2% normocin (Invitrogen, Carlsbad, California, USA). These two cell lines were selected because of their disparate tumor stromal characteristics, with B16 melanoma tumors representative of ‘soft’ tumors and MC38 tumors representative of ‘firm’ tumors. Previous studies have demonstrated that interstitial pressure values within B16 melanoma tumors (~10 mm Hg)5 are much lower than those of MC38 tumors (~150 mm Hg),6 highlighting the differences in tumor stiffness between these two models; 3×105 cells were implanted subcutaneously in the dorsal and ventral subcutaneous tissue of C57BL/6NC female mice aged 6–12 weeks, and the tumors were allowed to grow for 10–14 days or until they were 6–8 mm in length as measured by calipers. Only tumors without signs of ulceration at the time of treatment were used.
Intratumoral injection technique, imaging acquisition and data analysis
Intratumoral injections were performed in a hybrid image-guided intervention suite. Injection needle placement was performed by an interventional radiologist under real-time ultrasound guidance (Acuson, Siemens). Ultrasound imaging was used to confirm that the needle tip was located within the center of the target lesion. Injection of iodinated contrast agents and/or immunotherapeutic agents was performed under live fluoroscopic imaging at 15 frames-per-second (Artis Q, Siemens). The animals were then scanned using a CT-on-rails (Miyabi, Siemens) immediately following injection; this allowed the animals to remain stationary without having to be transferred to another imaging suite or gantry. CT slice thickness in the z-axis was 0.6 mm. Volumetric analysis of the CT images was performed to quantify the percentage of tumor filled with injected drug using conventional three-dimensional (3D) imaging analysis software (iNtuition; TeraRecon, Foster City, California, USA).
The influence of injection needle design on injection drug delivery and retention was performed in the following manner. Injections were performed using either a conventional 21 gage end-hole needle (EHN; Becton Dickinson) or a multiside hole needle with no end hole (ProFusion, Cook Regentec). The ProFusion needle used in this study has a closed diamond tip to facilitate image-guided positioning within the target lesion. The distal 1 cm of the needle has 22 laser-etched holes distributed in a spiral pattern that has a characteristic echogenic pattern. The multiple parallel channels allow improved intratumoral distribution and go hand-in-hand with diminished pressure during injection. Needle placement was performed under ultrasound guidance to ensure that all of the side holes were positioned within the tumor, for a proximal side hole positioned outside of the tumor would lead to off-target leakage.
The needles were connected to syringes, and injections of an iodinated contrast agent (Omnipaque 240) were performed simultaneously into bilateral flank tumors in the rat tumor model (n=10). Injections were performed at an equal volume (1 cc) and at a constant injection rate using a dual head injection pump (Harvard Apparatus). Continuous, in-line pressure measurements were obtained during injections using a piezoelectric pressure sensor (CompassCT, Cook Regentec). Ultrasound imaging was used intermittently to ensure that the needle remained well positioned within the target lesion.
The influence of tumor stroma composition on injection drug delivery was performed in the following manner. Intratumoral injections of iodinated contrast (Omnipaque 240) were performed in the B16 melanoma and MC38 colorectal cancer mouse models described previously. Prior to injection, tumor volumes were measured with calipers using the formula length×width2/2. The intratumoral injectate volume for each tumor was drawn up to be equal to the tumor volume calculated using this technique. Injections were performed under ultrasound guidance through a 27 gage single EHN; unlike the larger rat tumors that can accommodate a clinically relevant 21G needle, a higher needle gage was necessary for injections into the smaller mouse tumors. Animals were then immediately imaged using a microCT system (Skyscan, Bruker). Volumetric analysis of the CT images was performed to quantify the percentage of tumor filled with injected drug using a semi-automated threshold-based volumetry algorithm with a conventional 3D imaging analysis software package (iNtuition; TeraRecon).
Immunologic profiling following intratumoral immunotherapy delivery
Syngeneic McArdle RH-7777 (ATCC) hepatoma cells were implanted in the flank of female Buffalo rats (n=10). Once tumors reached 15–20 mm in size, a STING agonist (50 μg ML RR-S2 CDA, Sigma-Aldrich) versus saline (control, n=2) was injected under ultrasound visualization using either a 21 gage EHN (Becton Dickinson; n=4) or a 21G multiside hole needle (ProFusion; n=4). Tumor tissue was harvested at 24 hours postinjection, and single cell suspensions were generated in the following manner. The tumors were dissected and cut into small pieces with a scalpel. The tissue fragments were subject to enzymatic digestion in 3 volumes of phosphate-buffered saline (PBS) containing DNAse I 1 mg/mL (Roche cat# 11284932001), Collagenase D 1 mg/mL (Roche cat# 11088882001) and Dispase II 2.4 U/mL (Roche cat# 4942078001). After incubation at 37°C for 30 min with gentle stirring, the lysate was washed with PBS+2% FBS and strained through a 40 µM nylon mesh. The red blood cells were then lysed with ammonium chloride buffer (Miltenyi cat #130-094-183). The cell suspension was washed and resuspended in PBS+2% FBS before determining the total number of cells obtained as well as their viability using Trypan blue. The leukocytes (ie, CD45+ cells) were then purified using CD45 MicroBeads (Miltenyi cat#130-109-682) following the manufacturer’s indications. The purified cells were resuspended in ice-cold PBS+2% FBS, and the total number and viability was determined with an automatic cell counter (Countess II, Thermo Fisher).
Single cell RNA sequencing (scRNAseq) of the CD45+ intratumoral leukocytes was performed in the following manner. The scRNAseq libraries were prepared by the 10X Genomics Chromium Single Cell Immune Profiling Solution based on the manufacturer’s recommendations using 10 000 purified leukocytes per sample. Samples were not pooled but rather run independently as replicates (n=2 for control, n=4 for each of the two treatment groups). The complementary DNA of single cell transcriptomes were then sequenced using the Illumina platform (NextSeq 500). The data were then analyzed using the Seurat package V.3.07 in R (R V.4.0.2, The R Foundation). We first constructed an atlas of CD45+ cells that underwent scRNAseq (n=25 399 cells). After identifying the major immune cell types, clusters were annotated based on established transcriptional profiles.8 Next, we subselected the clusters representing the myeloid populations (macrophages and monocytes) and characterized their phenotypic state using previously published gene sets for ‘inflammatory’ dendritic cells and type I interferon-stimulated genes.8 9
Hydrogel synthesis and evaluation
Multidomain peptides (MDPs) spontaneously self-assemble into beta-sheet nanofibers that result in soft hydrogels ideal for syringe injection into any location a needle can reach. MDPs have been shown to be biocompatible and biodegradable both in vitro and in vivo, and can be easily loaded with small molecules, biologics or cells for various applications. The cationic lysine-based multidomain peptide K2(SL)6K2 (or K2-MDP) was used to load and improve the biodistribution of the contrast agent iohexol as well as a STING agonist. Iohexol was acquired at a stock concentration of 240 mgI/mL (Omnipaque 240), and a series of dilutions were prepared to form various K2-MDP hydrogel formulations loaded with different concentrations of iohexol to determine its compatibility with hydrogelation. Iohexol was prepared at 120, 48 and 24 mgI/mL by diluting in 1X PBS at pH 7.2, and lyophilized K2-MDP stock peptide was prepared at 2 wt.% (20 mg/mL, or 11.2 mM) in 298 mM sucrose solution. All tested formulations were fully homogeneous and successfully formed hydrogels. A quantitative dilution intensity curve using CT imaging was also generated and allowed for the selection of an optimal concentration for in vivo experiments (60 mgI/mL within 1 wt.% hydrogel). To evaluate improvements in intratumoral drug deposition, the MDP-iohexol compound or free iohexol (100 μL each) were injected using a 29G needle at a rate of 50 μL/s under fluoroscopy, followed by immediate postinjection CT scan, into the same rat flank tumor model (n=10) as described previously.
Next, to determine the influence of hydrogel-based immunotherapy delivery, a MDP-STING agonist gel formulation was prepared as previously described.10 The MDP-STING agonist gel formulation versus free STING agonist (50 μg ML RR-S2 CDA, Sigma-Aldrich in both arms) versus saline control was injected into the same rat flank tumor model, and flow cytometry of CD45+ immune cells from the harvested tumors was performed 48 hours following injection. Flow cytometry analysis was performed in the following manner. Expression of cell surface antigens was evaluated using the following antibodies: CD45-PE/Dazzle594 (BioLegend cat# 202223), CD3-VioBlue (Miltenyi cat# 130-102-677), CD8-FITC (BD Pharmingen cat# 554856) and CD4-PE/Cy7 (BD Pharmingen cat# 561578). In brief, non-specific binding was first blocked by treating the samples with an anti-CD32 antibody (BD Pharmingen, cat# 550270) at 1:200 at 4°C for 5 min. The antibody dilutions were prepared in PBS+2% FBS containing Fixable Viability Dye eFluor 780 (eBioscience cat # 65-0865-14) at 1:1000. The cells were incubated with the antibodies at the dilutions recommended by the makers during 30 min at 4°C in the dark. The cells were washed twice, resuspended in PBS+2% FBS and analyzed by flow cytometry with a Gallios 561 flow cytometer (Beckman Coulter, California, USA). Quantification of tumor necrosis was performed on H&E-stained sections using QuPath.11
Results
Delivery as a function of injection needle design
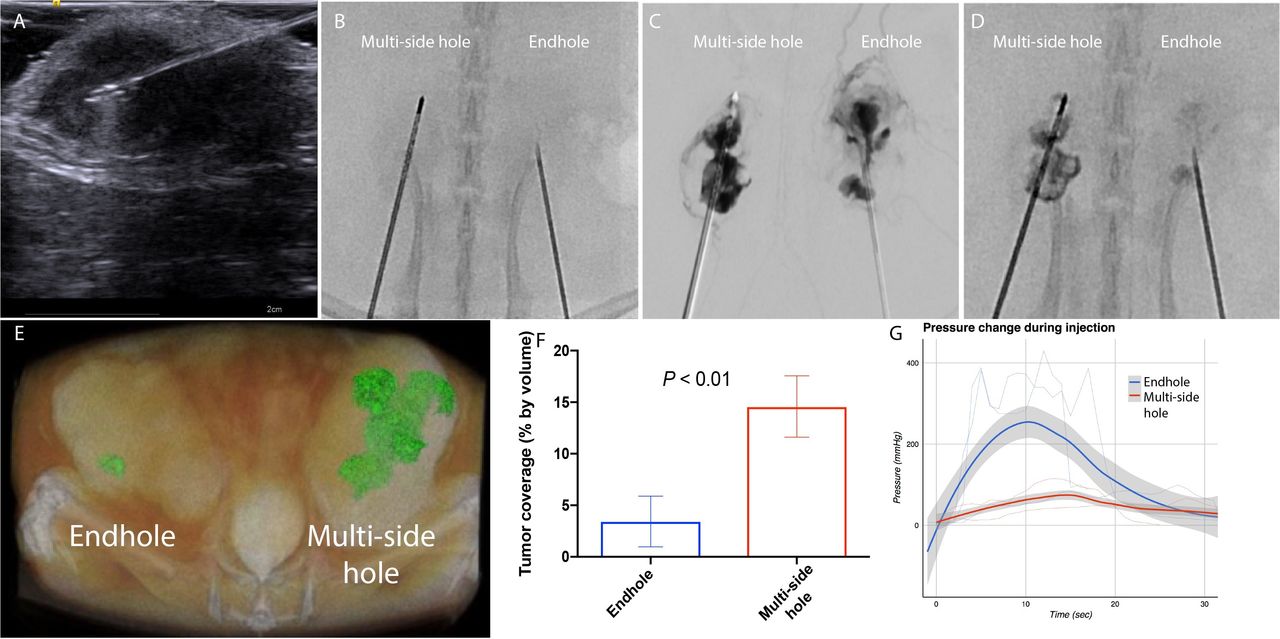
There exist a wide variety of clinically available injection needle designs. While single EHNs are the most common, we hypothesized that a needle without end holes but rather multiple side holes (MSHNs) would result in improved intratumoral drug deposition. Simultaneous injection of 1 cc iodinated contrast agent through a 21 gage EHN and a 21 gage MSHN under live fluoroscopy in a bilateral flank rat tumor model was performed (figure 2, online supplemental video 1). Intravasation (unintentional systemic delivery by leakage of the injected drug into the tumor vasculature) was notably greater with the EHN injection than with the multiside hole needle. Immediate postinjection CT-confirmed improved retention within the tumor with the multiside hole needle. These findings correlated well with clinical observations from iodinated intratumoral immunotherapy agents that identified intravasation and peritumoral leakage to be common with EHNs (online supplemental figure 1).4 Concurrent measurements of interstitial pressure during injection revealed an expected relative decrease in pressure during injection with the MSHN compared with the EHN.
Supplementary video
Supplemental material
Supplemental material

End-hole needle (EHN) versus multiside hole needle (MSHN). In a bilateral flank rat model of hepatocellular carcinoma, simultaneous injection through a MSHN versus EHN was performed. Needles were placed under ultrasound guidance (A). Under live fluoroscopy, iodinated contrast was injected using a dual head contrast injector (B). Digital subtraction angiography (C) during the injection shows increased intravasation with EHN compared with MSHN. A 5 min postinjection image reveals improved intratumoral retention with MSHN compared with EHN (D). This finding was confirmed by CT scan (E), with an approximately threefold improvement using MSHN over EHN (F). In-line interstitial pressure measurements demonstrated a smaller increase in peak pressure during injection with MSHN compared with EHN (G).
Influence of immunotherapy drug delivery on drug function
We next tested our hypothesis that improvements in intratumoral immunotherapy drug delivery will yield improvements in drug efficacy. To do so, we performed image-guided injections of a STING agonist using EHNs versus multiside hole needles in a rat model of HCC. CD45+ intratumoral immune cells were isolated 24 hours postinjection, and scRNAseq was performed (figure 3). Unsupervised cluster analysis identified multiple discrete immune populations. Cluster characterization was performed using individual gene expression feature plots and heat maps. Given the direct mechanism of action of STING agonists on dendritic cells,12 we focused our analysis on this subpopulation. There were significantly increased expression of type I interferon-associated genes and ‘inflammatory’ dendritic cell gene signatures in the MSHN treatment arm relative to ENH and control arms.
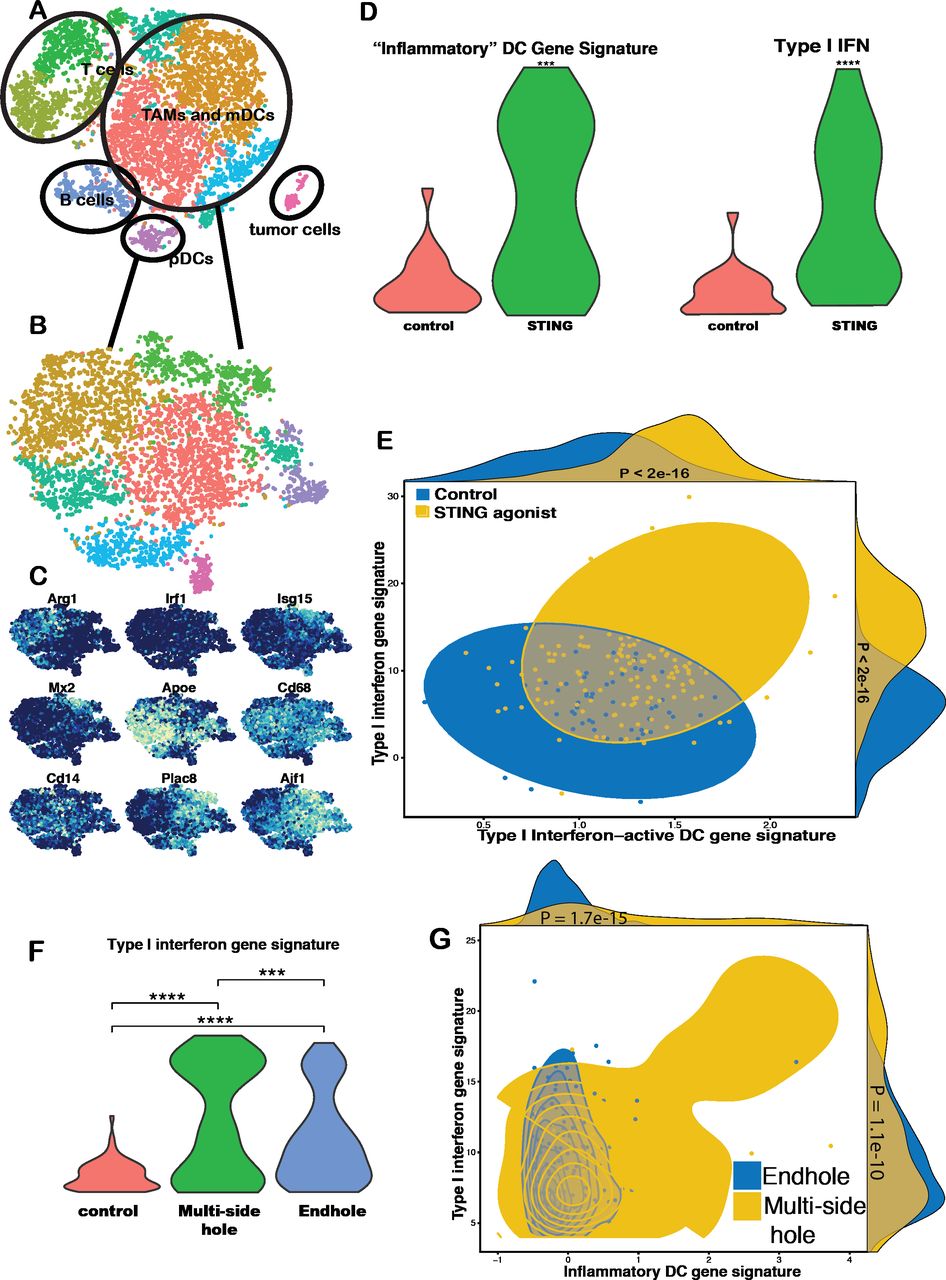
Single cell RNA sequencing analysis of injection technique. In a rat model of hepatocellular carcinoma, a STING agonist (50 μg ML RR-S2 CDA) was injected using an endhole or multiside hole needle. Tissue samples were harvested at 24 hours, and single cell RNA sequencing was performed on the intratumoral CD45+ population. Unsupervised cluster analysis (tSNE) identified multiple discrete immune populations (A). Myeloid TAMs and plasmacytoid dendritic cells (mDCs and pDCs) were then analyzed (B), and cluster characterization was performed using individual gene expression feature plots (C). Gene set enrichment analysis revealed a significant increase in type I interferon gene expression and ‘inflammatory’ DC gene signatures in tumors that underwent STING injection compared with control (D, E). Violin plots of Z-scores for the STING injection samples showed were significantly greater compared with control (D). Moreover, compared with endhole needles, there was a cluster of DCs with a significant increase both of these gene signatures when STING injections were performed with a multiside hole needle based on violin plot of Z-sores (F) and scatter plots of Z-scores (G). ***P<0.001; ****p<0.0001. TAMs, tumor-associated macrophages.
Drug delivery as a function of drug composition
Next, we evaluated whether modifications in the injectate composition could have an influence on intratumoral delivery and the subsequent immune response (figure 4). Soluble drugs in low viscosity fluids localize poorly to solid organ metastases. We hypothesized that drugs embedded in a MDP-based hydrogel would exhibit significantly improved delivery and retention within tumors following percutaneous delivery. Fluoroscopic imaging during in vivo injection of free iohexol demonstrated leakage of drug along the tumor capsule as well as venous intravasation, as expected; in comparison, there was substantially improved localization of the MDP-iohexol compound within the tumor without intravasation. This was confirmed by subsequent CT imaging which demonstrated an approximately fivefold improvement in the percentage of tumor volume containing the injected drug (38% vs 8%, p<0.001).

Multidomain peptide (MDP) hydrogel-based intratumoral delivery. In a flank rat model of hepatocellular carcinoma, intratumoral MDP hydrogel containing iohexol demonstrates excellent retention within the target tumor on real-time fluoroscopic imaging (A) and CT imaging (B, blue asterisk) compared with free iohexol (B, red asterisk), with a greater than fourfold improvement in intratumoral delivery (C). Histological evaluation at 48 hours following intratumoral injection of saline (D), free aqueous phase STING agonist (E), MDP hydrogel only (F) and MDP-STING agonist (G) demonstrate geographic areas of necrosis corresponding to the sites of MDP deposition. Tumor necrosis was seen with MDP hydrogel alone, in addition to free STING and MDP-STING injections (H). Flow cytometry of intratumoral CD45+ cells at 48 hours (I) and 96 hours (J) after treatment demonstrate an increase in CD8+ T cells in all three treatment arms at both time points. CD3 subsets were gated on CD45+ cells, and the CD4/CD8 subsets were gated on CD3+ cells. There was a similar increase in both CD4+ and CD8+ T cells after intratumoral delivery of free STING and MDP-STING (K). *P<0.05;**p<0.01. ns, not significant.
Comparison of the tumor-infiltrating leukocytes from subcutaneous rat HCC and mouse colorectal tumors treated with a MDP-STING agonist versus those treated with the free STING agonist alone and MDP hydrogel alone showed several significant differences. We found that 48 hours after intratumoral drug delivery, histological analysis was notable for large geographic areas of necrosis corresponding to intratumoral deposition of the MDP hydrogel for both hydrogel alone and MDP-STING agonist. Flow cytometry revealed that in both rat (figure 4) and mouse models (online supplemental figure 2), the tumors that received the free STING agonist had on average more than twice the percentage of CD3+ cells as compared with the hydrogel formulation (13.03% vs 5.41%, p=0.050, two-tailed Student’s t-test; data from mouse model). However, in the mouse MC38 tumor model, it was the MDP-STING agonist treated tumors that demonstrated the highest percentage of CD3+ cells that were CD8+. At 96 hours after treatment, all but the saline-treated tumors showed increases in the percentages of CD3+ cells when compared with 48 hours. Remarkably, the tumors that received hydrogel only had a very significant increase in the percentage of CD8+ cells, indicating that the MDP itself elicits an immune response that can conceal that of the therapeutic agent it carries.
Supplemental material
Influence of tumor microenvironment on drug delivery
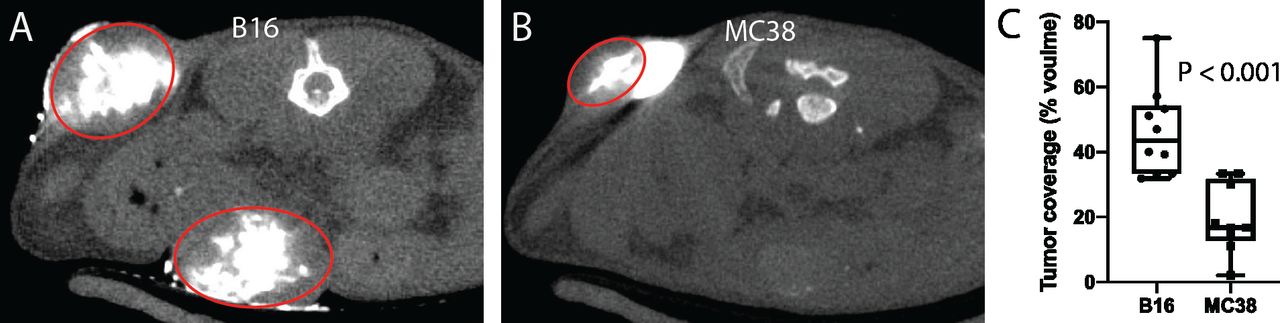
We hypothesized that tumor stroma and microenvironmental characteristics have a significant effect on intratumoral drug delivery. To test this hypothesis, we evaluated the efficacy of intratumoral injections in two mouse models of cancer with differing internal composition, namely, B16 melanoma and MC38 colorectal cancer. Subcutaneous tumors were injected with a volume of iodinated contrast equal to the volume of the tumor and then immediately imaged with microCT (figure 5). Volumetric analysis of the injected tumors revealed a greater than twofold improvement in intratumoral distribution in B16 melanoma tumors compared with MC38 (46.2%±13.5% vs 19.5%±10.7%, p<0.001).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Influence of tumor microenvironment on intratumoral drug delivery. Intratumoral injections of an iodinated contrast agent were performed in B16 melanoma and MC38 colorectal cancer subcutaneous tumors in mice (tumors circumscribed by red ovals). Immediate postinjection microCT imaging (A–C) revealed significantly improved retention within B16 tumors relative to MC38 tumors, with substantial extracapsular leakage noted in the latter.
Discussion
Since 2016, we have performed over 1500 image-guided intratumoral immunotherapy injections in both clinical trial and standard of care settings at our institution. Our clinical experience has highlighted the fact that while intratumoral delivery of immunotherapeutics is a safe procedure, the variability of intratumoral dispersion, with uncontrolled distribution of the injected drug throughout the tumor volume and leakage into the surrounding tissue, is a substantial challenge. The negative ramifications of off-target delivery include diminished drug efficacy and potentially increased systemic toxicities. In a review of our first 500 intratumoral injections, we found a total adverse event rate of 5.0% related to the injected drug within 24 hours of the injection procedure.4 This finding is not surprising given the degree of intravasation observed in this study. Patients can experience symptoms akin to ‘cytokine storm’ as early as several minutes following the injection. It is not uncommon for patients to develop rigors, shaking chills, tachycardia and hypotension in the procedural suite or recovery area. Major complications (National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTC AE) ≥3) occurred in a small number of patients (2% of patients receiving investigational agents and 4% of patients receiving standard of care talimogene laherparepvec) and included symptoms of sufficient severity to require inpatient admission with close hemodynamic monitoring.
Our clinical experience has highlighted several variables that affect intratumoral drug delivery. These include the stromal composition of the tumor and the organ in which it resides and the design of the injection needle and the rate at which the drug is injected. These challenges are well known to interventional radiologists who are experienced in conventional image-guided injection procedures such as percutaneous ethanol injection, for which specially designed MSHNs are often employed.
As identified in this study, there are substantial advantages to be gained by modifications in needle design. These modifications are particularly useful for patients with firm tumors, suggesting that the contemporary ‘one-size-fits-all’ approach to injections requires re-evaluation. Moreover, current techniques using conventional EHNs are imperfect for several reasons. A singular point for fluid egress from an EHN results in the drug being ejected at fluid pressures over two orders of magnitude greater than physiological fluid pressures. Moreover, the fluid is ejected in a single vector. These factors conspire to cause the drug to be delivered in a high-pressure jet that fractures tumoral tissues and bursts into the surrounding parenchyma. These preclinical studies are corroborated by our clinical experience in which we have witnessed poor tumor localization (online supplemental figure 1). A more apposite needle design is one that lacks an end-hole entirely and instead comprises multiple side holes. In this manner, the injection pressure is distributed across multiple points of fluid egress and in disparate vectors. The MSHN needle used in this study has multiple parallel channels that allow for improved intratumoral distribution and go hand-in-hand with diminished pressure during injection.
Improvements in intratumoral drug distribution are only meaningful if they result in improvements in drug efficacy. Previous simulation-based studies have suggested that even for replicating oncolytic viruses, there is minimal extension of the virus beyond the area of intratumoral deposition and so strategies that broadly distribute the viral vectors throughout the tumor should be employed.13 We sought to experimentally evaluate this hypothesis using a STING agonist. STING agonists have shown great promise in the preclinical setting12 and are being evaluated in numerous clinical trials (eg, NCT03172936). STING is an intracytosolic double-stranded DNA sensor that on activation drives the transcription of interferon-β.12 In addition to its role in the immune response to viruses, STING is a central component in the immune response to tumors. When STING agonists are injected into tumors, their mechanism of action is to alter the phenotype of intratumoral dendritic cells. In order for STING agonists to activate dendritic cells and initiate a type I interferon response, these cells must be exposed to the drug. As these cells comprise a rare minority of the intratumoral cellular milieu, it is reasonable to hypothesize that the STING agonist must be distributed throughout the tumor such that the greatest volume of tumor tissue is exposed to the drug. This approach should maximize the likelihood of the appropriate immune cells encountering the STING agonist and becoming activated. Our scRNAseq data have substantiated this hypothesis. We elected to apply this technology at an early postinjection time point as it would provide the highest resolution evaluation of the direct transcriptional changes due to STING activation. Differences in further downstream effects such as intratumoral interferon concentrations, immune cell infiltration and systemic ramifications could potentially be blunted by the non-linear response of dendritic cells to STING activation. That is, the number of STING-activated dendritic cells could be challenging to predict based on further downstream signals due to positive feedback loops following the release of interferon. Furthermore, STING agonists are both extremely potent and tissue-agnostic, and the off-target delivery of these drugs can lead to the highly morbid cytokine storm syndrome. Therefore, the injection technique, in addition to improving the intratumoral volume of distribution, must also prioritize the suppression of systemic leakage.
There has been a recent proliferation of biomaterials with improved localization and sustained release properties that are amenable to minimally invasive, image-guided interventions such as intratumoral injections.14 Shear stress-thinning hydrogels represent a class of biomaterials with tunable viscosities that are well-suited for such applications. When forced through a needle, their viscosity decreases, allowing for injection; once they are delivered into the body, the undergo a gelation process and re-acquire their pre-injection resistance to flow. Furthermore, hydrogels can be functionalized to serve as sustained release depots for a variety of drugs, including immunotherapeutics.15 We have previously demonstrated that STING-loaded MDP hydrogels can have dramatic effects on tumor size reduction and survival in murine models of oral cancer.10 We likewise found in this study that the MDP hydrogel is very effective at intratumoral retention and tumor necrosis, although the subsequent immune response may be targeted to the hydrogel itself rather than to tumor antigens since the MDP is primarily composed of cationic amino acids. Variations in hydrogel composition, however, can potentially overcome this limitation to combine the benefits of hydrogel-based intratumoral deposition with the pharmacokinetic advantages of sustained drug release.16 17
While variations on injection needle design and drug delivery platform are fertile grounds for innovation and experimentation, manipulation of the immunosuppressive tumor stroma into which the immunotherapy is to be delivered presents fewer options. Several non-invasive and minimally invasive interventions have been shown to reduce tumor interstitial pressure and could therefore potentially be used to modify the tumor’s mechanical properties to render it more amenable to intratumoral drug delivery. For example, external beam radiation has long been known to reduce tumor interstitial pressure18; this is likewise true for hyperthermia.19 Thus, beyond the potential immune simulating effects of these adjuvant interventions, there may be additional benefit from their impact on tumor biomechanics.
Intratumoral immunotherapy is a burgeoning paradigm in immuno-oncology with the potential to substantially increase the impact of immunotherapy across the cancer spectrum. While a tremendous degree of creativity has been applied towards the development of novel intratumoral immunotherapy agents, it is also critical to ensure that these therapies are delivered into the target lesions accurately. Innovations in injection technique, drug formulation and tumor microenvironmental modifications will be instrumental to achieve this goal.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Twitter @AnaingMD, @DrSapnaPatel
Contributors Conception and study design: NM, SZM, DL, JH, SY, PP, PH, RAS. Development of methodology: NM, MW, KD, CD, AW, RA, SZM, KMcH, RM, AT, AN, SP, DL, JH, SY. Data acquisition: NM, MW, KD, CD, RA, DL, JH, SY, RAS. Analysis and interpretation of data: NM, SZM, KMcH, RM, AT, AN, SP, SY, PP, PH, RAS. Manuscript preparation: NM, SZM, RAS. Study supervision: NM, RAS. All authors read and approved the final manuscript.
Funding This work was supported by funds from the Society for Interventional Radiology Foundation, the Radiological Society of North America Research and Education Foundation, NIH/NIBIB (R21 EB026089-01A1) and NIH/NIDRC (R21 DE027794). This research was in part performed in the Flow Cytometry & Cellular Imaging Core Facility and Advanced Technology Genomics Core, which are supported in part by the National Institutes of Health through MD Anderson's Cancer Center Support Grant CA016672. This work was also funded in part by the The Welch Research Fund C-1557.
Competing interests None declared.
Patient consent for publication Not required.
Ethics approval All mice and rats were maintained under specific pathogen-free conditions according to the American Association for Accreditation of Laboratory Animal Care (AAALAC) standards. All experimental research on mice was done in accordance with protocols approved by MD Anderson Cancer Center.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available on reasonable request. All data relevant to the study are included in the article or uploaded as supplementary information.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.