Article Text

Abstract
Background Patients with human papillomavirus (HPV+) head and neck squamous cell carcinoma (HNSCC) have superior prognoses compared with patients with HPV− HNSCC and strategies for treatment de-escalation are under investigation for the HPV+ setting. However, the survival advantage associated with HPV is not universal, and a subset of patients with HPV+ HNSCC fail definitive treatment and progress with metastatic/recurrent disease. Currently, no biomarker is available to distinguish aggressive from indolent HPV+ HNSCC. Immune dysfunction facilitates tumorigenesis and is associated with poor treatment response; therefore, we hypothesized that diminished intratumoral immune cell functionality may be attractive biomarkers to identify patients with HPV+ HNSCC at risk for early disease-specific mortality.
Methods This is a retrospective analysis of The Cancer Genome Atlas (TCGA) HPV+ HNSCC cohort.
Results Immunoglobulin J polypeptide (IGJ), uniquely expressed in plasma cells, showed a broad expression range in HPV+ HNSCC. Cox regression model, adjusting for clinical covariates, indicated that IGJ is an independent prognostic biomarker for disease-specific survival (DSS) and overall survival (OS). Patients with low IGJ had a 7.2-fold (p<0.001) increase in risk of disease-specific death with a median DSS of 13 months. Low IGJ showed an area under curve (AUC) of 0.89 with 91.0% sensitivity and 87.6% specificity to identify early disease-specific mortality (defined as DSS ≤12 months). Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis revealed a global dampening of immune pathways in low IGJ tumors.
Conclusions Our work showed that IGJ is a robust and independent prognostic biomarker for disease-specific mortality in HPV+ HNSCC. Patient with HPV+ HNSCC with limited adaptive immune functionality should not be candidates for treatment de-escalation modalities.
- B-Lymphocytes
- Tumor Biomarkers
- Lymphocytes
- Tumor-Infiltrating
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Head and neck squamous cell carcinomas (HNSCCs) are a diverse collection of cancers from distinct anatomical sites in the head and neck region. High risk human papillomaviruses (HPVs) are recognized as an etiological factor, and the incidence of HPV+ HNSCC, primarily located in the oropharynx, has shown a dramatic increase over the past several decades, with this upward trend expected to continue well into the future.1 HPV− HNSCCs are predominantly driven by alcohol consumption and smoking, and do not have a predilection for a particular anatomical site. It is recognized that HPV− and HPV+ HNSCCs are distinct diseases with marked differences in genomic, transcriptomic and immunologic landscapes, clinical presentation and treatment response.2 Patients with HPV+ HNSCC tend to have superior responses and clinical outcomes to standard of care treatment paradigms compared with patients with HPV− HNSCC; therefore, treatment de-escalation strategies are actively being investigated in the clinic.3–5 A subset of patients with HPV+ HNSCC fails definitive treatment and established biomarkers to distinguish this aggressive subgroup are needed and would substantially improve risk stratification for patients with HPV+ HNSCC.
Interrogation of immunologic landscape, particularly T cells, in the context of treatment response and disease progression, is an active area of research for HPV+ HNSCC. Across a spectrum of solid malignancies examined, HPV+ HNSCCs are among the most highly immune cell-infiltrated tumors, with a high preponderance of T cells.6 Although HPV+ HNSCCs have high intratumoral CD8+ T cells, interestingly, these tumors also tend to have a higher degree of immunosuppression.6 HPV+ HNSCC tumors express immune checkpoint receptors, programmed cell death protein 1 (PD-1) and cytotoxic T-lymphocyte-associated protein 4 and, moreover, have high infiltration of regulatory T cells, a negative modulator of other immune cells.6 7 These studies reveal that cytotoxic T cell functionality in HPV+ HNSCC tumors is dynamically regulated through multiple mechanisms. In regards to prognostic impact, elevated CD8+ T cells in the tumor microenvironment (TME) are associated with improved survival in HPV+ HNSCC.8–12 In contrast to T cells, the involvement of B cells in HPV+ HNSCC has not been widely explored. High intratumoral B cell abundance, similar to CD8+ T cell infiltration, is a positive prognostic marker in patients with HPV+ HNSCC.13 This study, further, showed that in HPV+ HNSCC tumors, B cells and CD8+ T cells form cellular aggregates, and the prevalence of this interaction is associated with a higher density of HPV16E6/E7 specific CD8+ T cells in the TME.13
Since adaptive and innate immune dysfunction facilitates tumorigenesis and is associated with clinical outcomes, we assessed if restricted intratumoral immune cell functionality may be used as biomarkers to identify the aggressive HPV+ HNSCC subgroup. In this study, immunoglobulin J polypeptide (IGJ), a plasma cell restricted gene, was revealed as an independent prognostic marker and, robustly predicted early disease-specific mortality in this patient population. Our work supports the exclusion of patients with low IGJ HPV+ HNSCC from treatment de-escalation regimens and, furthermore, increased surveillance and adjuvant therapy in these patients are warranted.
Material and methods
Study cohort
Clinical data were downloaded from the Genomic Data Commons (GDC) data portal from The Cancer Genome Atlas (TCGA) Research Network: http://cancergenome.nih.gov/. Overall survival (OS) and disease-specific survival (DSS) information was obtained from the updated Pan-Cancer dataset.14 HPV annotation was provided by the TCGA and based on analyses of whole genome and exome sequencing datasets. Ninety-six HPV+ cases, consisting of 74 HPV16+, 15 HPV33+, 3 HPV18+, 3 HPV35+ and 1 HPV56+ tumor, were analyzed. Cases with limited clinical data or more than one HPV genotype detected were excluded. Estimating the Proportion of Immune and Cancer cells (EPIC)15 was applied to obtain the fractions of immune cells as described previously.16 Clinical data for HPV− HNSCC, and cervical squamous cell carcinoma and endocervical adenocarcinoma cohorts were downloaded and processed in the same manner for validation.
B cell and CD8+ T cell activation signatures
Batched-corrected normalized mRNA-seq data were obtained from the supplemental materials of the Pan-Cancer dataset.17 Expression levels of genes represented in the CD8+ T cell and B cell activation signatures were extracted and log-transformed. Three CD8+ T cell activation signatures were used: a cytolytic signature consisting of GZMA, GZMB, GZMH, GZMK, GZMM, CCL5 and PRF1,18 a composite fraction and cytotoxic signature consisting of CD8A, CD8B, GZMA, GZMB and PRF1,19 and a composite effector activity signature consisting of CD8A, GZMA, GZMB, PRF1, IFNG, TBX21, CXCL9 and CXCL10.20 In addition, a B cell cluster signature consisting of IGJ, KIAA0125, TNFRSF17, FCRL5, POU2AF1, CD79A, LOC96610 (BMS1P20), ADAM6, C8orf80 (NUGGC), MEI1, CD79B, PAX5, FCER2, FCRL2, CXCR5, CD19, FCRLA, BLK, FCRL1, MS4A1, CNR2, BANK1 and TNFRSF13B was used.21 Heatmaps were generated using the R package: ComplexHeatmap.22 Unsupervised clustering was performed using the k-means method, starting with 3 initializing centroids and repeating for 100 times to reach a final consensus k-means clustering. Top differentially expressed genes (DEGs) between different functionality groups were analyzed by R package: DESeq223 using the preprocessed RNA-seq data from GDC data portal. Volcano plot was generated by R package: EnhancedVolcano.
BCR segment diversity signature
B cell receptor (BCR) segments were filtered out in the previous batch-corrected mRNA-seq data. Therefore, raw data of mRNA expression levels were downloaded from GDC with the R package: TCGAbiolink.24 BCR segments were selected with gene names starting with ‘IGH’, ‘IGK’ and ‘IGL’, and subsequently genes that were not BCR segments were manually filtered out. A final matrix of 363 BCR segment genes was generated and analyzed. Raw counts were trimmed mean of M-values (TMM) normalized and converted to counts per million (CPM). Log2-transformed CPMs were subjected to unsupervised clustering as described for B cell and CD8+ T cell activation signatures.
Statistical analysis
All statistical analyses were performed using R V.3.5.1. Kaplan-Meier (KM) survival curves were plotted using the R package: survival. Log-rank testing was performed for each group-wise comparison. Univariate and multivariate Cox regression models were used to identify the HRs associated with immune cell fractions as continuous variables and other clinical covariates. Time-dependent receiver operating characteristic (ROC) curves were generated by survivalROC.25 R packages used for plot output were ggplot2. Normality and homoscedasticity were determined using Shapiro-Wilk and Levene’s tests. Student’s t-tests were performed for comparison of two groups if normality was satisfied. Otherwise, non-parametric Mann-Whitney or Kruskal-Wallis tests were used for comparisons of two or more than two groups, respectively.
Results
Immune cell density as a prognostic biomarker in HPV+ HNSCC
EPIC, a deconvolution algorithm, was used to infer the fraction of various immune cells. In the TCGA HPV+ HNSCC cohort, our analysis using EPIC showed that CD4+ and CD8+ T cells were the predominant immune cells infiltrating these tumors (figure 1A). B cells and macrophages were detected in these HPV+ tumors but at a lower density. Next, we determined the association between immune cell fraction and clinical outcomes, DSS and OS. KM survival analyses, using median immune cell fraction to stratify patients into high and low groups, revealed B cell and CD8+ T cell density as prognostic biomarkers in HPV+ HNSCC. High B cell fraction correlated with superior DSS (p=0.002, log rank) and OS (p<0.001, log rank). Similarly, high CD8+ T cell fraction was prognostic for improved DSS (p=0.009, log rank) and OS (p=0.001, log rank).
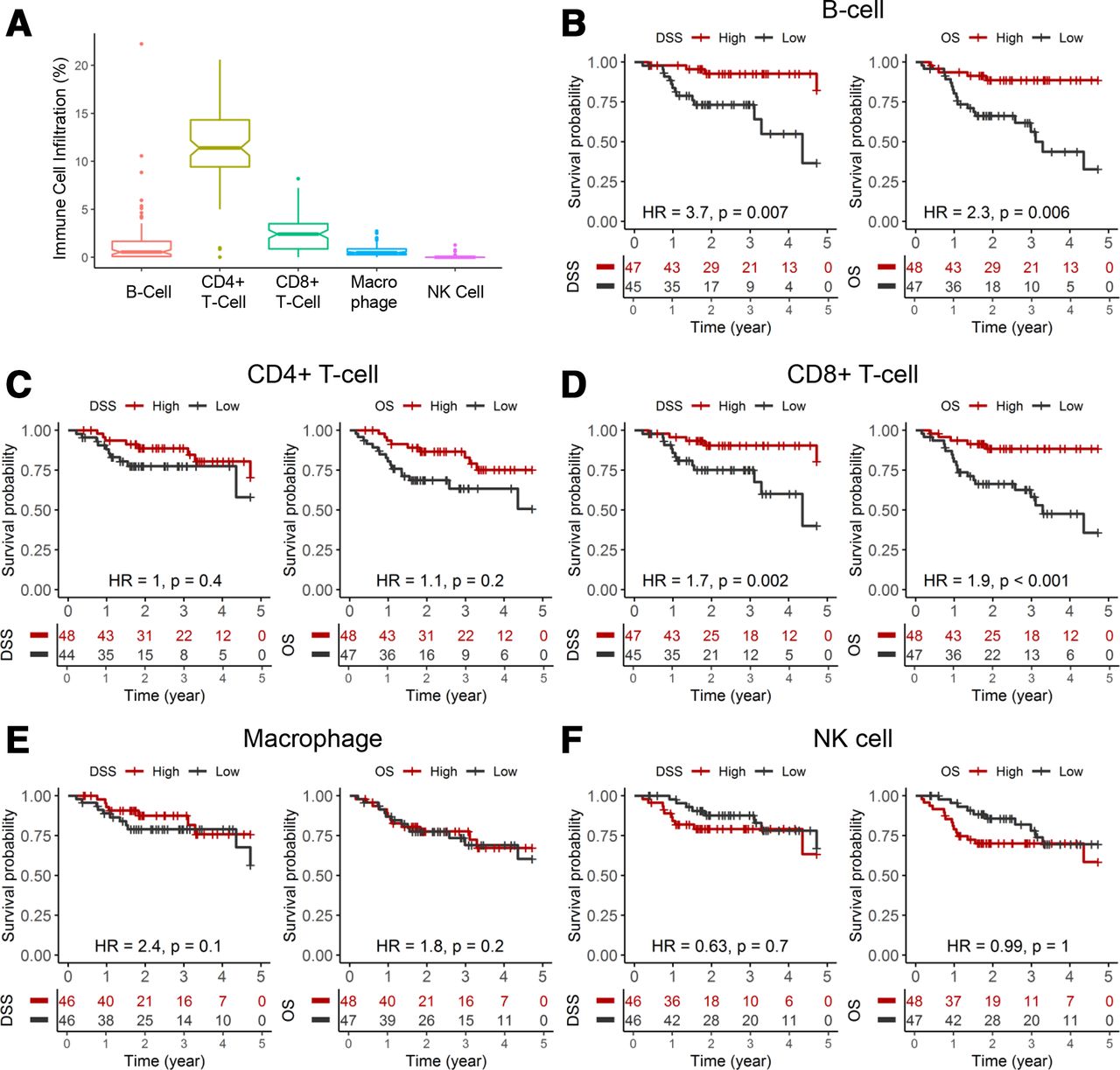
Immune cell density as prognostic biomarkers in HPV+ HNSCC. (A) Fraction of B cell, CD4+ T cell, CD8+ T cell, macrophage and NK cell in HPV+ tumors from the TCGA HNSCC cohort. Immune cell infiltration was inferred from bulk tumor RNA-seq data using a deconvolution algorithm. (B–F) KM analyses for DSS and OS for each immune cell type. HR for each immune cell density was analyzed by univariate Cox regression model for OS and DSS as a continuous value per incremental decrease. DSS, disease-specific survival; HNSCC, head and neck squamous cell carcinoma; HPV, human papillomavirus; KM, Kaplan-Meier; NK, natural killer; OS, overall survival; TCGA, The Cancer Genome Atlas.
Univariate analysis indicated that B cell fraction (HR: 2.3, 95% CI: 1.3 to 4.2; p=0.006) and CD8+ T cell fraction (HR: 1.9, 95% CI: 1.4 to 2.6; p<0.001) had similar impact on OS, whereas B cell fraction (HR: 3.7, 95% CI: 1.4 to 9.5; p=0.007) showed a greater impact on DSS compared with CD8+ T cell fraction (HR: 1.7, 95% CI: 1.2 to 2.5; p=0.002) (figure 1B,D). Densities of other immune cells, CD4+ T cell, macrophage and natural killer cell, did not demonstrate a significant correlation to these clinical endpoints. Multivariate Cox regression models, controlling for key clinical covariates, showed that the association between B cell or CD8+ T cell density to DSS and OS is dampened (online supplemental tables 1 and 2). Clinicopathologic variables, such as anatomical site, HPV genotype, T stage and nodal status, showed limited independent prognostic impact.
Supplemental material
CD8+ T cell and B cell functionality signatures are prognostic markers in HPV+ HNSCC
Since immune cell functionality may be compromised in the TME, immune cell activation state, rather than density, may better represent the intratumoral immunologic landscape. CD8+ T cell functionality signatures have been developed by various groups independently; some genes are unique, while other genes, namely, granzymes and perforin, are common to multiple signatures.18–20 We applied three CD8+ T cell functionality (cytotoxic, cytolytic, and effector) signatures to the TCGA HPV+ HNSCC dataset, and unsupervised clustering separated this cohort into three groups, low, medium and high CD8+ T cell functionality, for each signature analyzed (figure 2).

CD8+ T cell activation signatures as prognostic biomarkers in HPV+ HNSCC. (A-C Unsupervised clustering of CD8+ T cell activation signatures shown in the top section. Expression (log2(normalized RSEM+1)) of genes representative of three different CD8+ T cell activation signatures was clustered via unsupervised k-means method. KM analyses for DSS and OS shown in the bottom section. DSS, disease-specific survival; HPV, human papillomavirus; HNSCC, head and neck squamous cell carcinoma; KM, Kaplan-Meier; OS, overall survival; RSEM, RNA-Seq by Expectation Maximization.
KM survival analyses showed each CD8+ T cell functionality signature was prognostic for DSS and OS; cytolytic signature (DSS: p=0.008, log rank; OS: p=0.002, log rank) outperformed cytotoxic (DSS: p=0.03, log rank; OS: p=0.003, log rank) and effector (DSS: p=0.08, log rank; OS: p=0.02, log rank) signatures. Clearly, for all three signatures, patients with high CD8+ T cell activation have better prognosis than those with medium and low activation. A multivariate Cox regression model, including key clinical covariates, showed that the top performing CD8+ T cell functionality signature, cytolytic, was an independent prognostic biomarker for OS (HR: 5.0, 95% CI: 1.1 to 1.23; p=0.041) and trending toward significance for DSS (HR: 4.6, 95% CI: 0.96 to 23; p=0.057) (table 1).
Multivariate Cox regression model of OS and DSS for CD8+ T cell cytolytic signature
Similar to the CD8+ T cell functionality data, unsupervised clustering using a B cell activation signature21 segregated the HPV+ HNSCC cohort into three groups: high, medium and low (figure 3A). High B cell activation patients showed the best prognosis and had 5 year DSS and OS of 97% and 91%, respectively (figure 3B). Patients in the medium B cell activation group showed intermediate prognosis and patients with low B cell activation had the worst prognosis with 5-year DSS of 22% and 5-year OS of 26%. Multivariate Cox regression, adjusted for clinical covariates, revealed that B cell activation is an independent prognostic marker (table 2). Patients with HPV+ HNSCC in the low B cell activation group had a 12-fold (95% CI: 3.4 to 39; p<0.001) increase in risk of disease-specific death and 4.4-fold (95% CI: 1.8 to 11; p=0.001) increase in risk of overall death. B cell activation signature outperformed, in a bivariate regression model, the most robust CD8+ T cell functionality signature, cytolytic, as a prognostic biomarker for DSS (figure 4A). ROC analyses showed that B cell activation had an area under curve (AUC) of 0.94 (100% sensitivity and 86.4% specificity) to predict early disease-specific mortality (defined as DSS ≤12 months) (figure 4B). B cell activation was significantly better than the three CD8+ T cell functionality signatures (p<0.001, DeLong’s test) (figure 4C).
Multivariate Cox regression model of OS and DSS for B cell activation signature
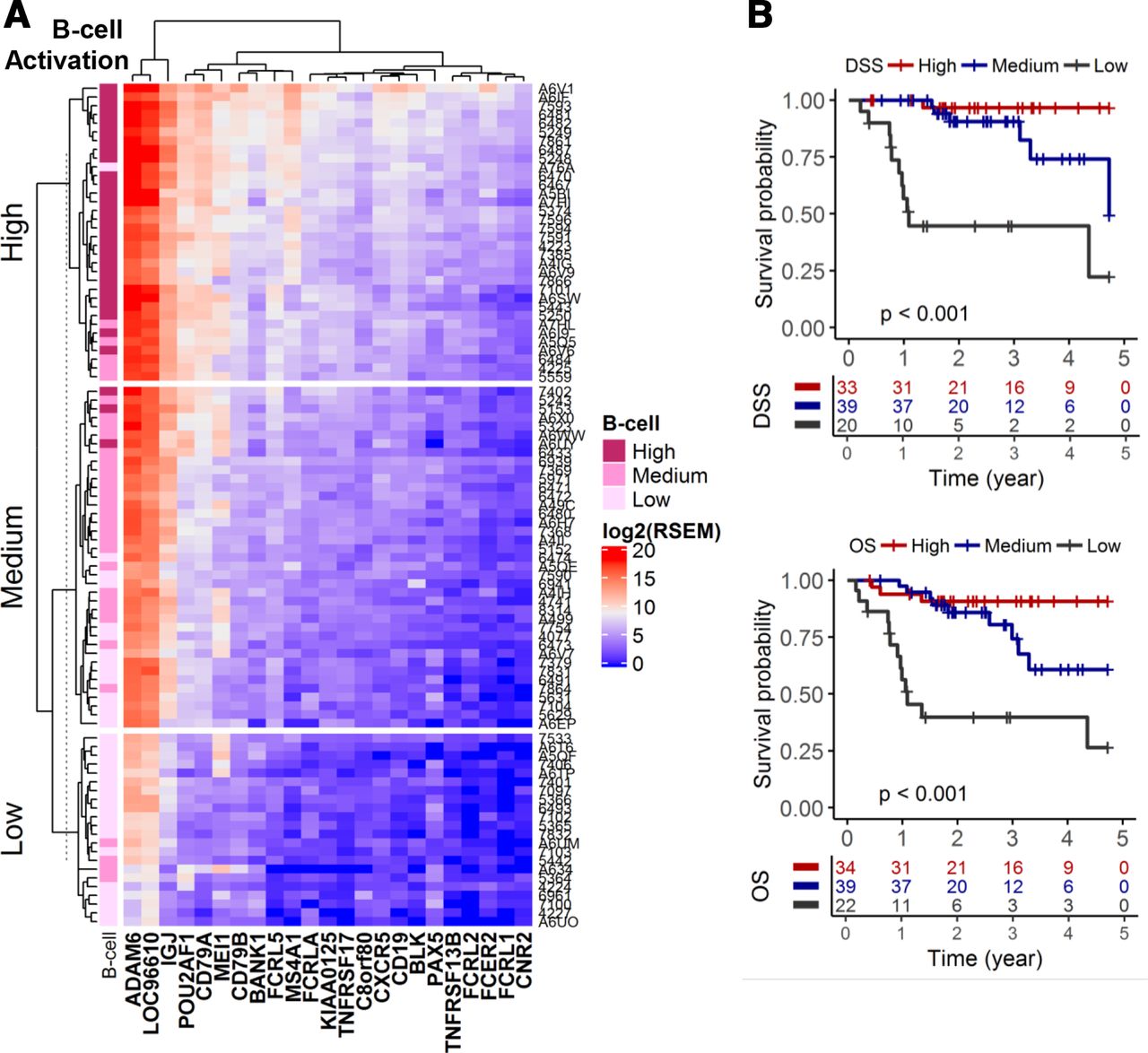
B cell activation signature as a prognostic biomarker in HPV+ HNSCC. (A) Unsupervised clustering of a B cell activation signature. Expression (log2(normalized RSEM+1)) of genes representative of a B cell activation signature was clustered via unsupervised k-means method. (B) KM analyses for DSS and OS. DSS, disease-specific survival; HPV, human papillomavirus; HNSCC, head and neck squamous cell carcinoma; KM, Kaplan-Meier; OS, overall survival; RSEM, RNA-Seq by Expectation Maximization.

B cell activation signature outperforms CD8+ T cell functionality signatures to predict early disease-specific mortality in HPV+ HNSCC. (A) Multivariate Cox regression models of B cell activation and CD8+ T cell cytolytic signatures for DSS and OS. (B) ROC curves for B cell activation and CD8+ T cell functionality signatures to predict early disease-specific mortality (DSS ≤12 months). (C) DeLong’s test to compare ROCs for B cell activation and CD8+ T cell functionality signatures. AUC, area under curve; DSS, disease-specific survival; FTR, false positive rate; HNSCC, head and neck squamous cell carcinoma; OS, overall survival; ROC, receiver operating characteristic; TPR, true positive rate.
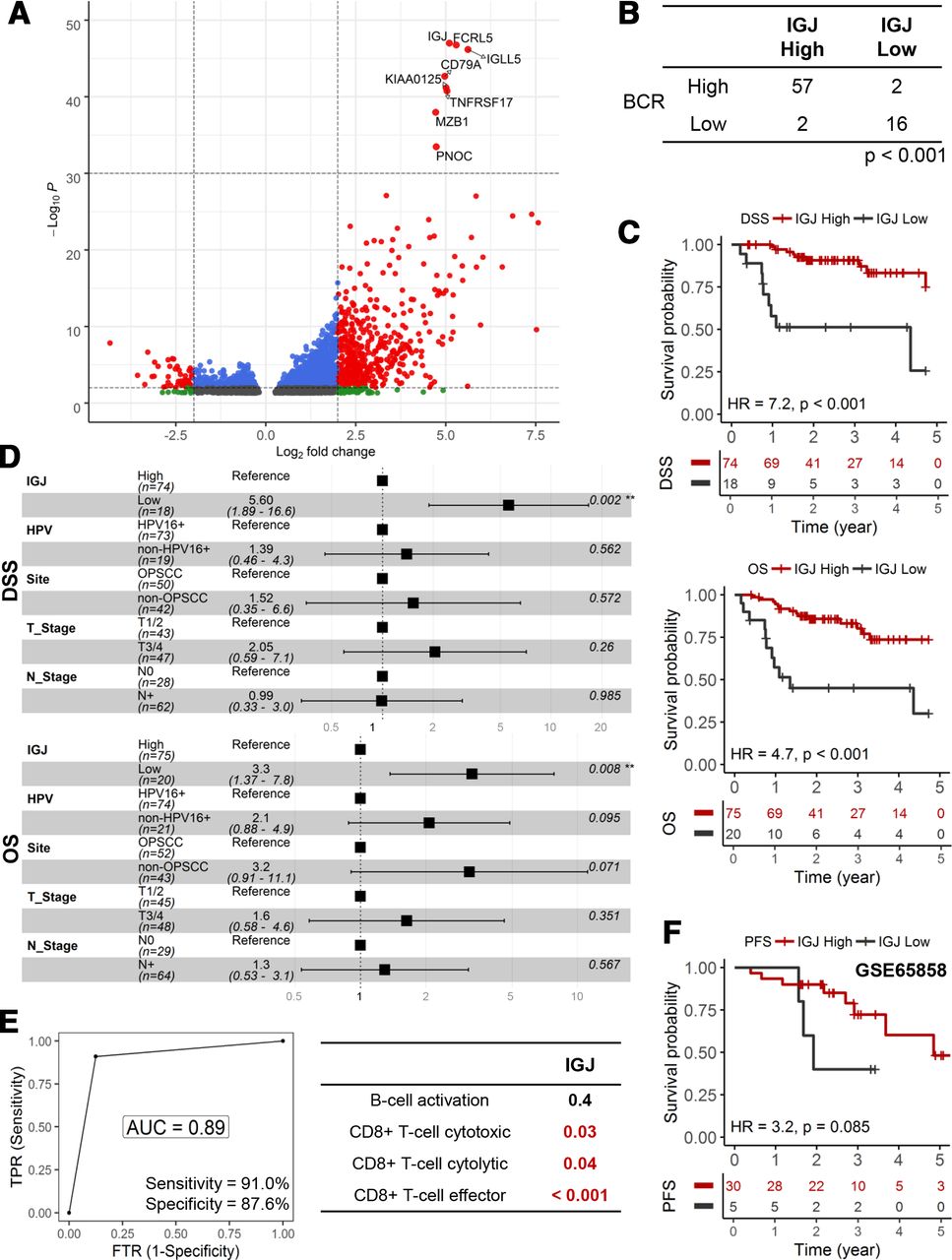
IGJ predicts early disease-specific mortality in HPV+ HNSCC
We performed further analyses to illuminate genomic and transcriptomic features that may be unique for the low B cell functionality tumors. Genomic characteristics, total mutational burden, aneuploidy and mutant allele tumor heterogeneity,26 were similar between high/medium and low B cell activation tumors (online supplemental figure 1). In figure 5A, volcano plot showed that a number of genes were differentially regulated between high/medium and low B cell activation tumors; more genes were upregulated (3303 genes, adjusted p<0.05) than downregulated (1436 genes, adjusted p<0.05) in high/medium B cell activation tumors. We focused our subsequent analyses on IGJ since it was the top ranked gene based on adjusted p value. BCR diversity, a measure of active adaptive immunity activity, was correlated with IGJ expression (p<0.001, Χ2) (figure 5B and online supplemental figure 2). Low IGJ was significantly associated with inferior DSS and OS in HPV+ HNSCC (figure 5C). These correlations were maintained after adjusting for clinical covariates in a multivariate Cox regression; low IGJ patients had a 3.3-fold (95% CI 1.37 to 7.8; p=0.008) increase in risk of death and 5.60-fold (95% CI: 1.89 to 16.6; p=0.002) increase in risk for disease-specific death (figure 5D). IGJ was as robust as the 23-gene, B cell activation signature to predict early disease-specific mortality and showed an AUC of 0.89 with 91.0% sensitivity and 87.6% specificity (figure 5E). Moreover, IGJ outperformed the three CD8+ T cell functionality signatures. In the GSE65858 HNSCC cohort, patients with HPV+ with low IGJ tend to present with disease progression earlier than patients with high IGJ; median progression-free survival (PFS) was 23 months and 58 months for patients with low and high IGJ, respectively (figure 5F). These findings trended (p=0.085, log-rank) but did not reach significance, likely due to limited sample size and clinical events. Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis showed that low IGJ tumors have a global diminishment in immune cell pathways (BCR signaling pathway, T cell receptor signaling pathway, natural killer cell mediated cytotoxicity, and antigen processing and presentation) and enrichment in metabolic and DNA repair pathways (online supplemental figure 3). Additionally, the prognostic utility of IGJ was shown in an HPV+ cervical carcinoma cohort indicating that the connection between B cell dysfunction and prognosis may generally apply to HPV-driven malignancies (online supplemental figure 4).
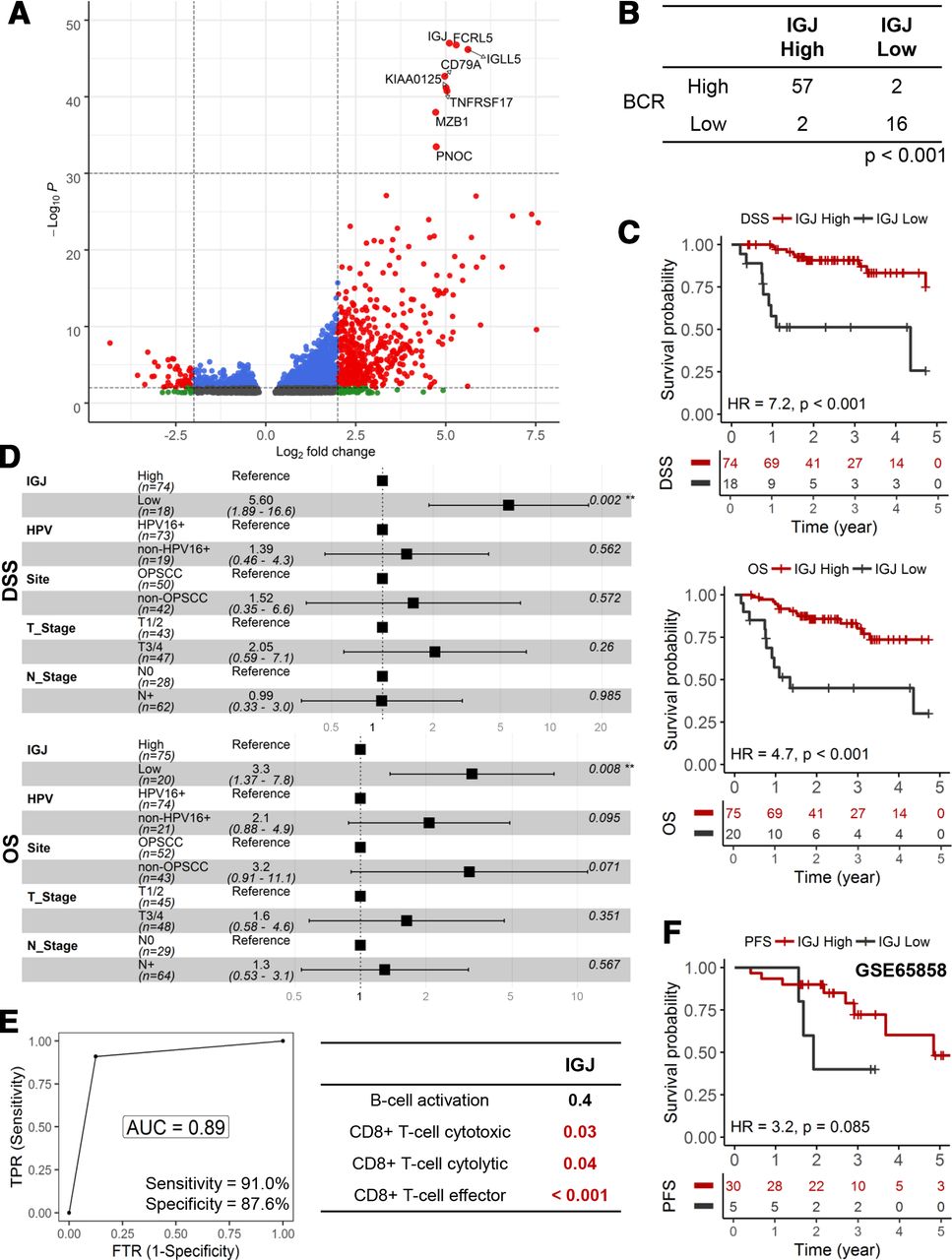
IGJ predicts early disease-specific mortality in HPV+ HNSCC. (A) Volcano plot to depict differentially gene expression. The top eight genes ranked according to adjusted p value formed a discrete cluster. (B) Association between IGJ and BCR diversity. (C) KM analyses for DSS and OS. (D) HRs from multivariate Cox regression models of IGJ and clinical covariates for DSS and OS. (E) ROC curve analysis of IGJ to predict early disease-specific mortality (DSS ≤12 months). DeLong’s test was used to compare ROCs. (F) KM analysis for PFS in the GSE65858 HNSCC cohort. AUC, area under curve; BCR, B-cell receptor; DSS, disease-specific survial; FTR, false positive rate; HNSCC, head and neck squamous cell carcinoma; IGJ, immunoglobulin J polypeptide; KM, Kaplan-Meier; OS, overall survival; PFS, progression-free survival; ROC, receiver operating characteristic; TPR, true positive rate.
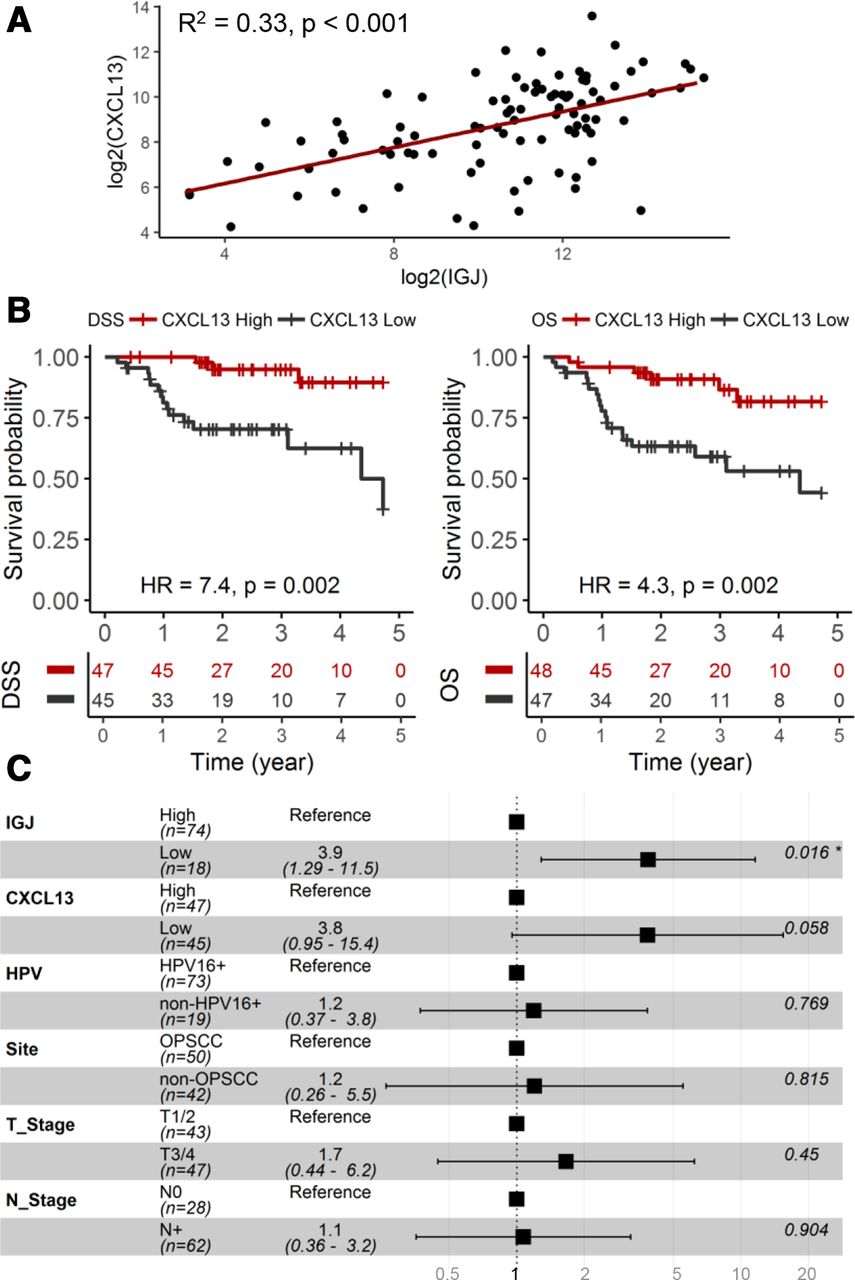
Chemokine C-X-C motif ligand 13 (CXCL13) is associated with IGJ and a prognostic marker in HPV+ HNSCC
Tertiary lymphoid structures (TLSs) form in tumors due to persistent inflammation and exist in different maturation states, including germinal center formation, to elicit an adaptive antitumor immune response. Chemokine C-X-C motif ligand 13 (CXCL13) mediates the recruitment of B-cells and facilitates the formation of TLSs in tumors.27–29 TLSs consist of various immune cells, including memory B cells and plasma cells; therefore, we determined if CXCL13, a marker for TLSs, is associated with IGJ and has utility as a prognostic biomarker in HPV+ HNSCC. As shown in figure 6A, CXCL13 expression correlated with IGJ expression in these tumors (p<0.001). Patients with low CXCL13 showed inferior DSS (p=0.002) and OS (p=0.002) compared with patients with high CXCL13 (figure 6B). IGJ, but not CXCL13, was shown to be an independent prognostic biomarker for DSS and OS in a multivariate model, including CXCL13, IGJ and clinical covariates (figure 6C).

CXCL13 is associated with IGJ and a prognostic biomarker in HPV+ HNSCC. (A) Association between CXCL13 and IGJ expression. (B) KM analyses for DSS and OS. (C) HRs from multivariate Cox regression model of CXCL13, IGJ and clinical covariates for DSS. CXCL13, Chemokine C-X-C motif ligand 13; DSS, disease-specific survival; HPV, human papillomavirus; HNSCC, head and neck squamous cell carcinoma; IGJ, immunoglobulin J polypeptide; KM, Kaplan-Meier; N stage, node stage; OS, overall survival; T stage, tumor stage.
IGJ is associated with PD-1 in HPV+ HNSCC
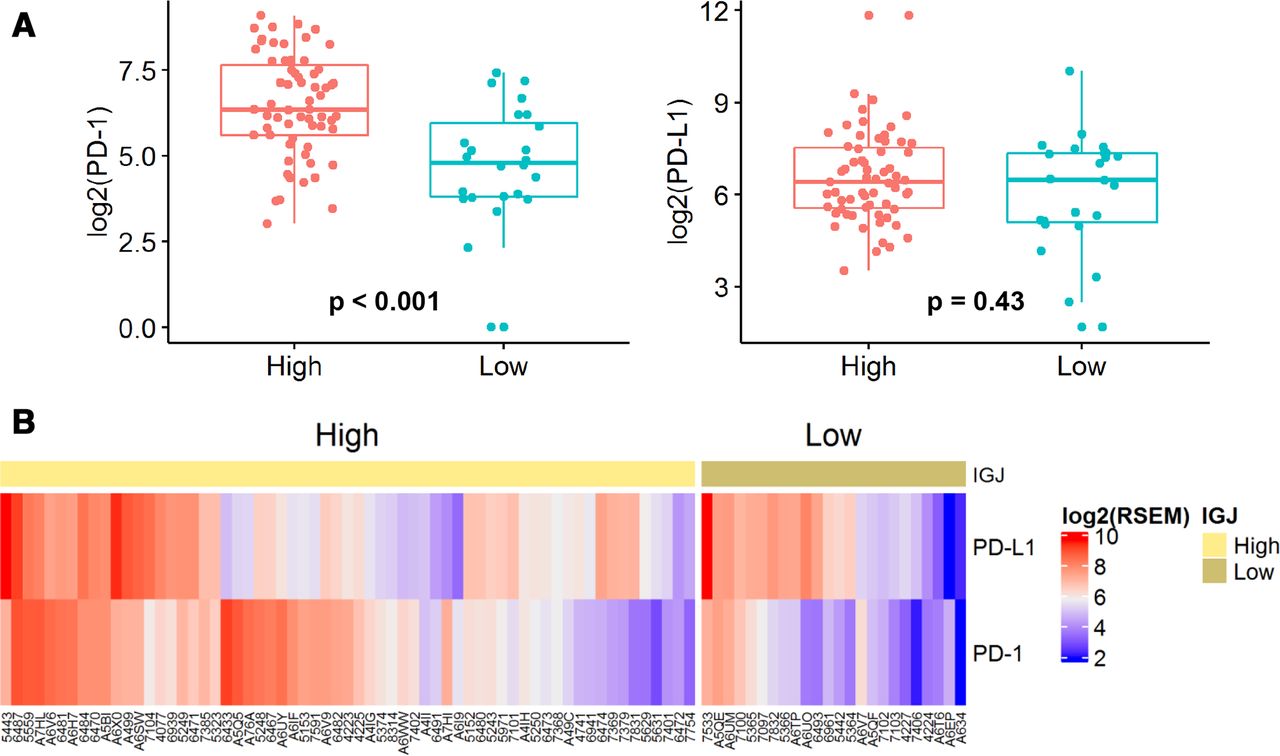
Anti-PD-1 immunotherapies, nivolumab and pembrolizumab, are approved for recurrent/metastatic (R/M) HNSCC. Clinical benefit of these two checkpoint inhibitors is limited to ~15%–20% of patients and this response rate is similar between HPV+ and HPV− R/M HNSCC.30 31 Better understanding of the PD-1 and PD-L1 landscapes in HPV+ HNSCC may reveal patients who are more likely to respond to PD-1 blockade. Analysis of the HPV+ HNSCC cohort showed that the mean PD-1 expression was lower in low IGJ tumors than in high IGJ tumors, whereas PD-L1 was similar between both groups (figure 7).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
IGJ is associated with PD-1 in HPV+ HNSCC. (A) PD-1 and PD-L1 expression. (B) Heatmap for PD-1 and PD-L1 expression in patients with low and high IGJ HPV+ HNSCC. HPV, human papillomavirus; HNSCC, head and neck squamous cell carcinoma; IGJ, immunoglobulin J polypeptide; PD-1, programmed cell death protein 1.
Discussion
HPV+ HNSCCs predominantly arise in the oropharynx since high risk HPV has a predilection to infect basal cells that reside in the tonsillar crypt. Patients with HPV+ oropharyngeal squamous cell carcinoma (OPSCC) have higher response rates to standard of care treatment, cisplatin-based chemoradiation, resulting in superior prognosis compared with patients with HPV− OPSCC.3–5 Patients with HPV+ OPSCC have high cure rates but are often burdened with long-term treatment-associated morbidities. These clinical observations have prompted investigation of the feasibility of treatment de-escalation strategies to maintain oncologic control with lower toxicities in the HPV+ OPSCC setting.
De-escalation by substituting cisplatin for a molecularly targeted agent, anti-epidermal growth factor receptor (EGFR) antibody cetuximab, showed disappointing results in two randomized, multicenter phase 3 clinical trials. In RTOG 1016, patients with HPV+ OPSCC treated with cetuximab +RT had inferior DSS and OS compared with cisplatin +RT.32 Another trial, De-ESCALaTE HPV, limited to patients with low risk (non-smokers or lifetime smoking history of <10 pack-years) HPV+ OPSCC, had comparable results and showed inferiority of cetuximab +RT to cisplatin +RT.33 Surprisingly, in both studies, severe toxicities were similar between cisplatin and cetuximab treatment arms.32 33 Another strategy, reduction in RT dose, has been assessed in phase 2 trials and showed promising results compared with historical standard RT dose controls; however, randomized trials are still needed to fully vet this approach.34 35 A presumption in these clinical trials is that all patients with HPV+ OPSCC are candidates for treatment de-escalation, but this is not the case since early treatment failures are seen in a subset of patients with HPV+ OPSCC. As treatment de-escalation continues to be explored in the HPV+ OPSCC population, biomarkers are needed to appropriately select patients who will benefit and also to limit the number of patients who will be severely under treated.
Features of the tumor immune microenvironment, in particular immune cell functionality, are attractive candidates as prognostic markers in virally induced, neoantigen-rich carcinomas. B cell functionality signature consists of 23 genes and further analysis showed that 8 of these genes had the greatest differential expression between low and high/medium B cell activation tumors. We focused our subsequent work on IGJ, a gene which is uniquely expressed in plasma cells, since it was the top ranked DEG. Cox regression analyses, adjusting for clinical covariates, including anatomic site, revealed IGJ as an independent prognostic marker. HPV+ HNSCC with low IGJ had a 7.9-fold increase (p<0.001) in risk of disease-specific death. Dramatically, median DSS and OS was only 13 months for patients with HPV+ HNSCC with low IGJ expression. IGJ performed as well as the B cell activation signature and more importantly, significantly outperformed the three CD8+ T cell functionality signatures to predict early disease-specific mortality in HPV+ HNSCC. Single marker IGJ offers a key advantage over the B cell activation signature as a clinical assay; IGJ has a broad signal range and thus, amendable to transition to an inexpensive and easy to perform immunohistochemistry (IHC) test. In contrast, B cell activation signature requires unsupervised hierarchical clustering to classify tumors into distinct B cell functionality states; this approach is infeasible at the individual patient level. Based on our findings, we argue that patients with HPV+ HNSCC with low IGJ tumors are poor candidates for treatment de-escalation paradigms, and require increased surveillance and adjuvant therapy.
Our results in HPV+ HNSCC were not, generally, applicable to HPV– HNSCC. CD8+ T cell functionality signatures were not prognostic biomarkers in the TCGA HPV– HNSCC cohort (online supplemental figure 5). B cell activation correlated with OS (p=0.03) and DSS (p=0.05) in HPV– HNSCC but these associations were not as robust as in HPV+ HNSCC (online supplemental figure 6). Low IGJ was associated with inferior OS (p=0.02) but not DSS (p=0.2) (online supplemental figure 7a). In the GSE65858 HNSCC cohort, low IGJ was not associated with poor PFS (p=0.5) (online supplemental figure 7B). Compared with HPV+ HNSCC tumors, infiltration of various types of immune cells was poor in HPV– HNSCC tumors (online supplemental figure 8), which may explain the lack of prognostic impact of immune cell functionality signatures in this patient population. Consistent with our data, B cell density, based on computational deconvolution and traditional immunohistochemistry, was reported to be higher in HPV+ than in HPV– HNSCC.36 Additionally, analysis of fresh HNSCC tumors, 27 HPV– and 11 HPV+, using multiparameter flow cytometry revealed that CD19+/CD20+ B cells are 6.6-fold higher in HPV+ than in HPV– tumors.37 These results were not surprising since HPV+ carcinomas have an intrinsic and persistent source of viral antigens to illicit an antitumor adaptive immunity response. Although HPV+ HNSCCs share a common etiologic mediator, intratumoral B cell functionality was not uniform in this patient population but stochastic. Similarly, another group reported that HPV viral antigens, E6 and E7, generate a robust B cell response in patients with HPV+ HNSCC but seropositivity was not universal.38 It is unclear why some HPV+ HNSCC seroconvert while this activity is limited in others. We speculate that this differential B cell responsiveness may be due to each patient’s intrinsic capacity to generate an adaptive immune response against HPV+ cells; this ability may be dependent on genetic and/or lifestyle factors, such as smoking. Alternatively, the dynamic process of immune cell-HPV+ tumor cell co-evolution during the tumorigenesis program may reshape HPV+ tumor cells toward an immunosuppressive phenotype, through autonomous and non-autonomous mechanisms, to thwart B cell infiltration and activation.
Anti-PD-1 blockade, with pembrolizumab and nivolumab, has become standard of care options for R/M HNSCC, in both the HPV– and HPV+ settings. Data from pivotal clinical trials showed that the clinical benefits from these biologics are restricted to a minority of patients with HNSCC. Expression of PD-L1, the ligand to PD-1, as a predictive biomarker to PD-1 blockade was explored using two different metrics: ≥1% tumor cell positivity in a minimum of 100 tumor cells for nivolumab and ≥1 combined positive score (CPS; number of PD-L1 positive tumor cells, lymphocytes and macrophage/total number of cells×100) for pembrolizumab. In the Checkmate141 trial for platinum-refractory R/M HNSCC, post-trial analysis, restricted to patients with ≥1% PD-L1 expression, showed a survival advantage with nivolumab compared with standard treatment; OS of 8.7 months and 4.8 months for nivolumab and control arms, respectively.30 Analysis of KEYNOTE-048 demonstrated a median survival of 12.3 months for pembrolizumab compared with 10.3 months for the standard treatment arm in the subgroup of patients with CPS ≥1.31 PD-L1 immunohistochemical approaches have shown value to identify responder to anti-PD-1 treatment and, in fact, pembrolizumab was approved as first-line, single agent therapy for patients with R/M HNSCC whose tumors have CPS ≥1.
PD-1 expression is not limited to CD8+ T cells but also found in other immune cells. In fact, PD-1 is found on the surface of B cells and functions to negatively regulate B cell activation and proliferation.39 Our data showed that patients with HPV+ HNSCC who experience early disease-specific mortality have tumors with poor B cell functionality and, furthermore, these tumors tend to have low PD-1 expression. We speculate that PD-1 blockade in the low IGJ, patient population with HPV+ HNSCC may likely have limited therapeutic efficacy since PD-1/PD-L1-mediated exhaustion does not appear be the primary mechanism of immune cell, either B cell or CD8+ T cell, dysfunction.
In summary, our data showed that IGJ is an independent prognostic biomarker and, more importantly, is robust to predict early disease-specific mortality in HPV+ HNSCC. We argue that patients with HPV+ HNSCC with limited B cell functionality should not be candidates for treatment de-escalation modalities. Further work needs to be prioritized to validate the utility of IGJ as a predictor of treatment response in HPV+ HNSCC.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors SG for acquisition of data, data analysis, data interpretation, statistical analysis, participating in study design and manuscript preparation. WQO for study design, data interpretation and manuscript preparation. TNT for study design and manuscript preparation. QP for study design, data interpretation, manuscript preparation and study supervision. All authors read and approved the final manuscript.
Funding This study was funded by grants from National Institutes of Health R01CA193590 and R01DE023555.
Competing interests None declared.
Patient consent for publication Not required.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available in a public, open access repository. The Cancer Genome Atlas's head and neck squamous cell carcinoma dataset can be downloaded http://cancergenome.nih.gov/.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.