Article Text

Abstract
Background Some sarcomas respond to immune checkpoint inhibition, but predictive biomarkers are unknown. We analyzed tumor DNA methylation profiles in relation to immunological parameters and response to anti-programmed cell death 1 (anti-PD-1) immune checkpoint inhibitor (ICI) therapy in patients with sarcoma.
Patients and methods We retrospectively identified adult patients who had received anti-PD-1 ICI therapy for recurrent sarcoma in two independent centers. We performed (1) blinded radiological response evaluation according to immune response evaluation criteria in solid tumors (iRECIST) ; (2) tumor DNA methylation profiling of >850,000 probes using Infinium MethylationEPIC microarrays; (3) analysis of tumor-infiltrating immune cell subsets (CD3, CD8, CD45RO, FOXP3) and intratumoral expression of immune checkpoint molecules (PD-L1, PD-1, LAG-3) using immunohistochemistry; and (4) evaluation of blood-based systemic inflammation scores (neutrophil-to-lymphocyte ratio, leucocyte-to-lymphocyte ratio, monocyte-to-lymphocyte ratio, platelet-to-lymphocyte ratio). Response to anti-PD-1 ICI therapy was bioinformatically and statistically correlated with DNA methylation profiles and immunological data.
Results 35 patients (median age of 50 (23–81) years; 18 females, 17 males; 27 soft tissue sarcomas; 8 osteosarcomas) were included in this study. The objective response rate to anti-PD-1 ICI therapy was 22.9% with complete responses in 3 out of 35 and partial responses in 5 out of 35 patients. Adjustment of DNA methylation data for tumor-infiltrating immune cells resulted in identification of methylation differences between responders and non-responders to anti-PD-1 ICI. 2453 differentially methylated CpG sites (DMPs; 2043 with decreased and 410 with increased methylation) were identified. Clustering of sarcoma samples based on these DMPs revealed two main clusters: methylation cluster 1 (MC1) consisted of 73% responders and methylation cluster 2 (MC2) contained only non-responders to anti-PD-1 ICI. Median progression-free survival from anti-PD-1 therapy start of MC1 and MC2 patients was 16.5 and 1.9 months, respectively (p=0.001). Median overall survival of these patients was 34.4 and 8.0 months, respectively (p=0.029). The most prominent DNA methylation differences were found in pathways implicated in Rap1 signaling, focal adhesion, adherens junction Phosphoinositide 3-kinase (PI3K)-Akt signaling and extracellular matrix (ECM)–receptor interaction.
Conclusions Our data demonstrate that tumor DNA methylation profiles may serve as a predictive marker for response to anti-PD-1 ICI therapy in sarcoma.
- sarcoma
- biomarkers
- tumor
- biostatistics
- immunotherapy
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Sarcomas are a heterogeneous group of rare mesenchymal cancer entities comprising more than hundred histological and molecular subtypes.1 The treatment of patients with sarcoma necessitates a multidisciplinary approach, involving surgery, radiation and systemic therapy, preferably at expert centers.2 3 In the recurrent setting, therapy options are limited with modest response rates and survival times.4 5 More effective new treatment options are crucially needed; however, due to the rarity of sarcomas, the conduct of clinical trials and efficacy evaluation of new systemic therapies is challenging.
Inhibition of immune checkpoint molecules such as cytotoxic T-lymphocyte antigen 4 (CTLA4), programmed cell death 1 (PD-1) and programmed death ligand 1 (PD-L1) has shown remarkable efficacy in a variety of cancer types. In sarcoma, immune checkpoint inhibitors (ICIs) including the PD-1 inhibitors pembrolizumab and nivolumab have shown evidence of antitumor activity in small studies with response rates ranging from 5% to 40%.6–10 Similar to the situation in other solid cancers, robust biomarkers to predict response to ICI have not been identified in sarcoma so far.8 10 11 Sarcomas are generally characterized as immunologically rather ‘cold’ tumors with limited immune cell infiltration, restricted PD-L1 expression and low tumor mutational burden.12 13 However, the immune composition of the tumor microenvironment in sarcomas is highly variable and has been described to potentially influence the clinical course of patients with sarcoma.12–22
Tumor DNA methylation events are pathophysiologically highly relevant in tumorigenesis and are emerging as novel relevant biomarkers in oncology.23–28 Profiling of these events is increasingly considered for improved classification and differential diagnosis of carcinomas, especially brain tumors and sarcomas.29–34 Moreover, emerging data from several cancer types indicate that tumor DNA methylation profiling may serve as a prognosticator or predictive biomarker of response to specific therapies including chemotherapy, targeted therapy and immunotherapy in several tumor types.23 30 35–37 In sarcoma, the utility of DNA methylation profiling as a predictive marker for response to ICI therapy is so far unknown.
In this study, we performed broad tumor DNA methylation profiling of >850,000 CpG sites in a relatively large multi-institutional series of adult patients with sarcoma treated with anti-PD-1 ICI therapy. We correlated our findings to a comprehensive set of blood-based and tissue-based immunological parameters and therapeutic outcome in order to identify potential biomarkers for selecting patients with sarcoma eligible for anti-PD-1 immunotherapy.
Materials and methods
Patient selection and clinical data collection
We retrospectively identified patients from our hospital record (center 1) and from an independent sarcoma center in Essen, Germany (center 2) according to the following inclusion criteria: age ≥18 years, histological diagnosis of sarcoma, treatment with anti-PD-1 ICI therapy (pembrolizumab or nivolumab) for sarcoma recurrence, measurable disease (at least 10×10 mm) on baseline radiological image prior to initiation of anti-PD-1 ICI therapy, availability of at least one radiological restaging investigation under ICI therapy, availability of tumor tissue for biomarker analyses (online supplemental figure S1, table S1 and S2). Clinical data, including patients’ characteristics, systemic inflammation scores (neutrophil-to-lymphocyte ratio (NLR), leucocyte-to-lymphocyte ratio (LLR), monocyte-to-lymphocyte ratio (MLR), platelet-to-lymphocyte ratio (PLR)) and survival data were collected via retrospective chart review. Response to anti-PD-1 ICI therapy was radiologically assessed by independent radiologists from the institutions according to iRECIST criteria.38 Objective response was defined as complete response (CR) or partial response (PR) as best achieved response to anti-PD-1 ICI therapy. Disease control was defined as CR, PR or stable disease (SD) as best achieved response. The patient cohort was independently treated by multidisciplinary teams according to good clinical practice guidelines.
Supplemental material
Determination of tumorous immune cell infiltration and immune checkpoint expression by immunohistochemistry (IHC)
Immune cell subsets and immune checkpoint molecules were immunohistochemically stained using the Ventana BenchMark Special Stains System (Ventana Medical Systems, Roche, Oro Valley, Arizona, USA) and semiautomatically analyzed with the Definiens Tissue Studio software (Definiens, Carlsbad, California, USA). Used antibodies and dilution protocols are summarized in online supplemental table S3. Density of immune cell subsets was specified by absolute numbers of positively stained cells per square millimeter (mm2) tumor.
DNA extraction and sodium bisulfite treatment
Genomic DNA (gDNA) was extracted from 3 to 5×10 µm slices of formalin-fixed, paraffin-embedded (FFPE) sarcoma specimens that were collected before ICI therapy start and contained at least 70% tumor cells using the Maxwell FFPE Plus Kit (Promega, Madison, Wisconsin, USA). Five hundred nanograms of isolated gDNA was first sodium bisulfite converted using the EZ DNA Methylation Kit (Zymo Research, Irvine, California, USA) and then repaired using the Infinium HD FFPE DNA Restore Kit (Illumina, San Diego, California, USA) according to the manufacturer’s recommendations.
Genome-wide DNA methylation analysis
DNA methylation profiling of more than 850,000 CpG sites was performed using Infinium MethylationEPIC BeadChip microarrays (Illumina, San Diego, California, USA). In brief, sodium bisulfite–treated gDNA was amplified, enzymatically fragmented and hybridized to a microarray as recommended by the manufacturers. After washing and staining, microarrays were analyzed on an iScan device (Illumina, San Diego, California, USA). Raw .idat files were imported into R (V.R 3.6.1, R Foundation for Statistical Computing, Vienna, Austria) using the ChAMP package for initial quality control, beta-value extraction and calculation of differential DNA methylation.39 40 Probes with a detection p value >0.01 in one or more samples, with a bead count <3 in at least 5% of samples, non-CpG probes, probes with single nucleotide polymorphisms (SNPs) and, probes that align to multiple locations and sex chromosome-specific probes were removed from subsequent analyses. Beta values represent the percentage of methylation for each CpG site on the MethylationEPIC BeadChip. These values range from 0 (completely unmethylated) to 1 (completely methylated) and are calculated as: intensity of the methylated signal (M)/(intensity of the unmethylated signal (U)+intensity of the methylated signal (M)+100). Data normalization was performed by the SWAN41 algorithm. Singular value decomposition (SVD) was used to determine variation within our dataset associated with center of sample origin, sex, microarray batch, sarcoma type and immune cell subsets determined by IHC. Significant variation was adjusted using ComBat. Differential methylation between groups was defined as |beta value difference| >0.1 and adjusted p value (Benjamini-Hochberg method, FDR)<0.1. Gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses were performed using the WebGestalt tool.42 Raw data were deposited at ArrayExpress database.
Assessment of copy number variations (CNVs)
CNVs were analyzed using the .idat files from EPIC microarrays and the R package conumee. In brief, multiple linear regression was used to normalize data from each tumor sample to a normal control sample cohort yielding the linear combination of controls which most closely fits the intensities of the tumor sample. In a next step, the log2 ratio of probe intensities of the tumor sample versus the combination of control samples was calculated. Then, probes were merged within predefined genomic bins and intensity values were shifted to minimize the median absolute deviation of all bins to zero to determine the copy number neutral state. The genome is segmented into regions of the same copy number state using the circular binary segmentation algorithm.43
Statistical analysis
Progression-free survival (PFS) and overall survival (OS) from anti-PD-1 ICI therapy start were calculated from first application of therapy until disease progression or death. Patients who neither progressed nor died by the data cut-off were censored at their last follow-up in the hospital. Differences between groups were calculated using the Mann-Whitney U test and the Kruskal-Wallis test. A two-tailed p value ≤0.05 was considered to indicate statistical significance. Correction for multiple testing was performed using the Bonferroni correction method. Statistical analysis was performed with Statistical Package for the Social Sciences (SPSS) V.23.0 software (SPSS) and standard R functions (V.R 3.6.1, R Foundation for Statistical Computing, Vienna, Austria).
Results
Patients’ characteristics
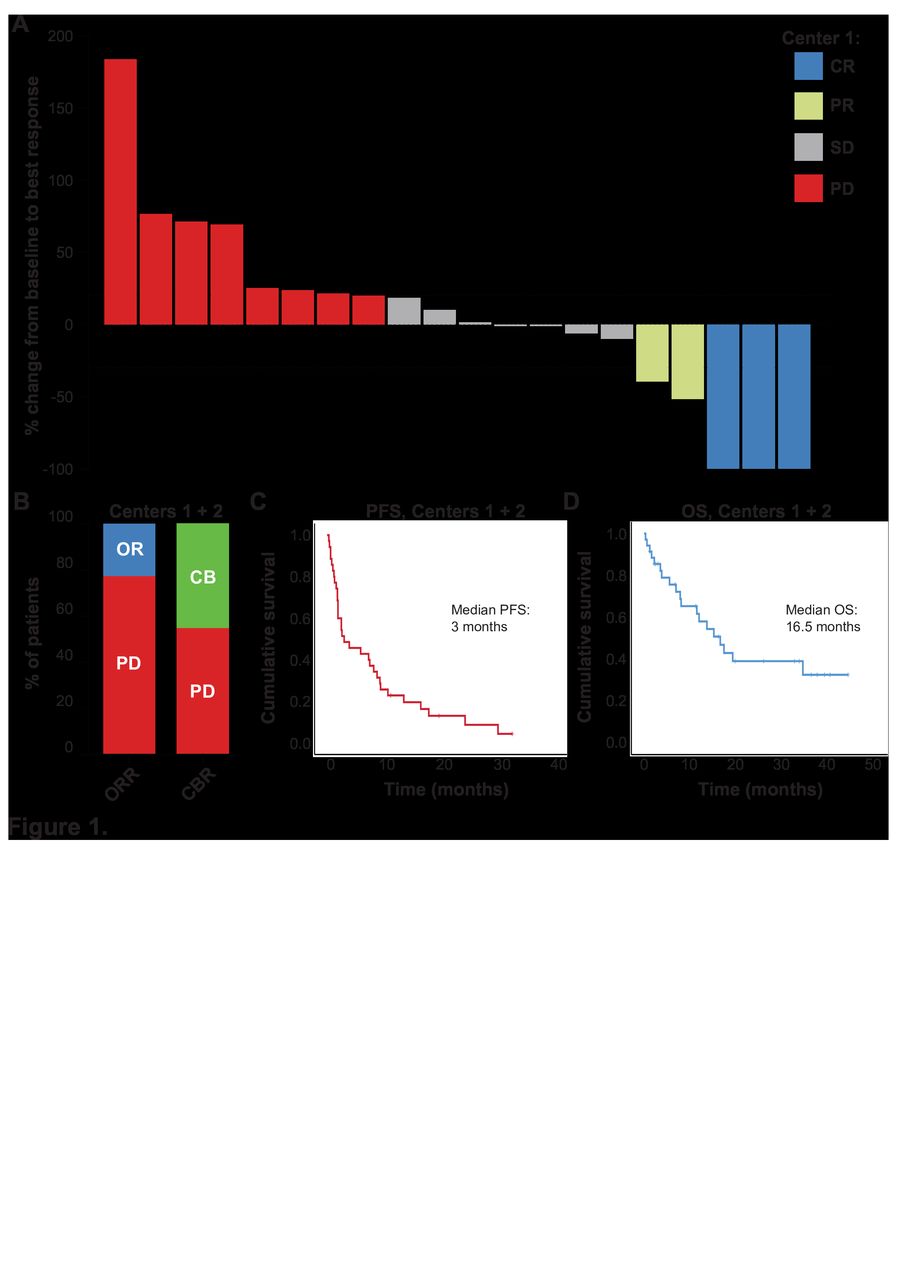
Thirty-five patients (median age of 50 (23–81) years; 18 females, 17 males; 27 (77.1%) soft tissue sarcomas; 8 (22.9%) osteosarcomas) were included in our study. Median number of prior systemic therapies was 3 (0–9). Thirty patients (85.7%) were treated with pembrolizumab and five patients (14.3%) were treated with nivolumab. Median number of anti-PD-1 ICI therapy applications was 4.5 (1–42). Three out of 35 (8.6%) patients achieved a complete response and 5 out of 35 (14.3%) patients a PR during anti-PD-1 therapy, resulting in an objective response rate of 22.9%. Eight out of 35 (22.9%) patients achieved an SD as best response, resulting in a clinical benefit rate of 45.7% (figure 1A,B). Median PFS and OS from anti-PD-1 ICI start were 3.0 (0.3–32.6) and 16.5 (0.3–44.2) months, respectively (figure 1C,D). Patients’ characteristics are summarized in table 1 and online supplemental tables S1 and S2.

Efficacy of anti-programmed cell death 1 (anti-PD-1) immune checkpoint inhibitor (ICI) therapy in patients with sarcoma. (A) Waterfall plot of % change of tumor burden from anti-PD-1 ICI therapy start until best response from center 1 patients. The dashed lines indicate the margins for PD (+20% change of tumor burden) and PR (−30% change of tumor burden). (B) Objective response rate (ORR) and clinical benefit rate (CBR) as % of patients (both centers). (C) Progression-free survival (PFS) and (D) overall survival (OS) of patients with sarcoma from anti-PD-1 ICI therapy start in months (both centers). Vertical lines indicate censored patients at last follow-up. CB, clinical benefit; OR, objective response; PD, progressive disease; PD-1, programmed cell death 1.
Patients’ characteristics
Correlation of immune cell density and systemic inflammation with response to anti-PD-1 ICI therapy
PD-L1 staining was available in 20 out of 35 patients. Three out of 20 (15%) patients had programmed cell death ligand 1 (PD-L1) expressing tumor cells (>1% PD-L1 expression). Density of tumor-infiltrating immune cell subsets (CD3+tumor-infiltrating lymphocytes (TILs), CD8+TILs, CD45RO+TILs, FOXP3+TILs, PD-1+TILs, LAG3+TILs) and systemic inflammation scores (NLR, LLR, PLR, MLR) are summarized in table 2. There was no correlation of PD-L1 expression, density of investigated immune cell subsets or systemic inflammation scores with response to anti-PD-1 ICI therapy (Mann-Whitney U test, p>0.05; online supplemental figure S2).
Inflammatory characteristics
DNA methylation profiling and correlation with response to anti-PD-1 ICI therapy
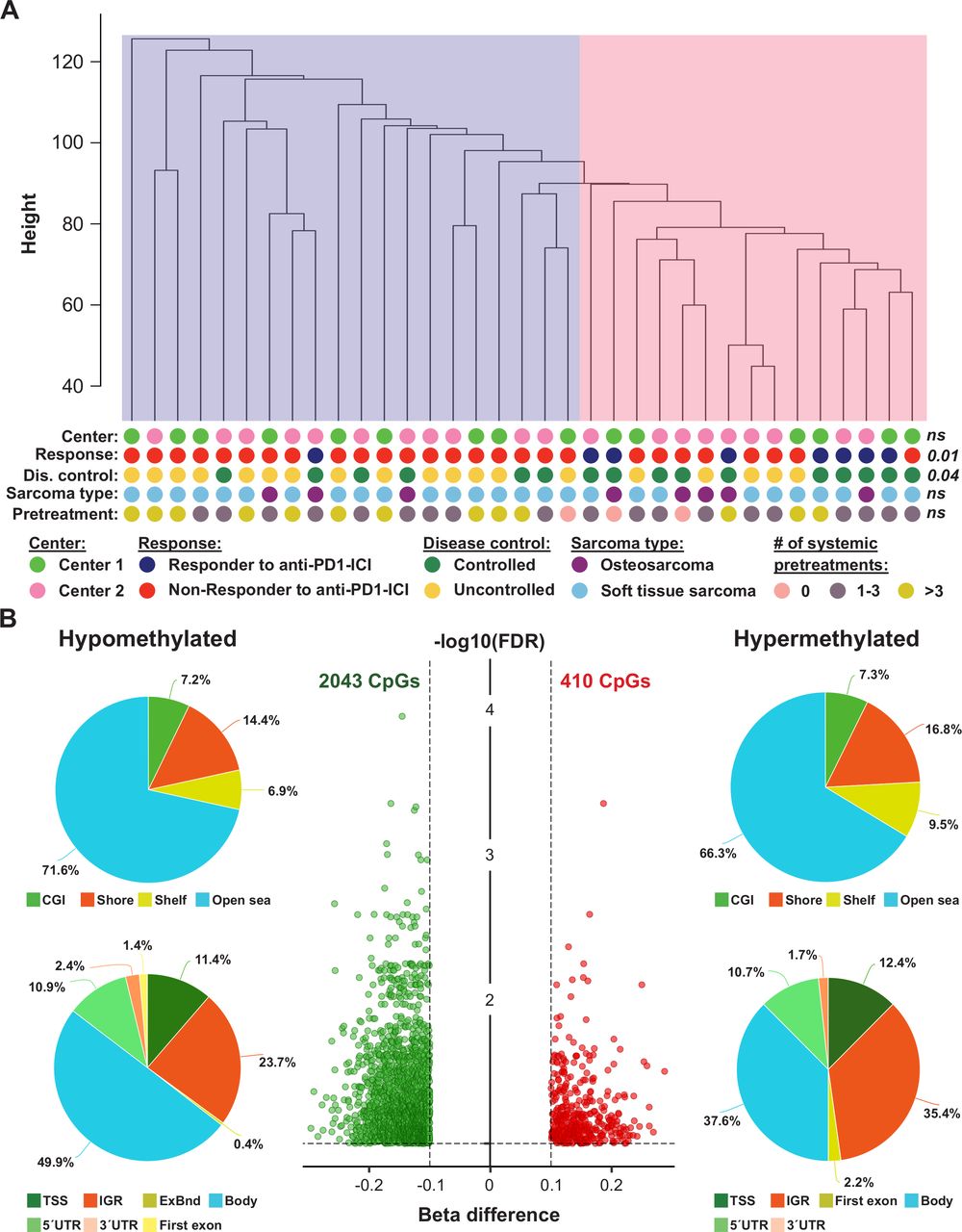
To determine if the methylome of primary tumors from patients with sarcoma who had an objective response to anti-PD-1 ICI (n=8) differs from the methylome of patients with sarcoma who did not achieve an objective response to anti-PD-1 ICI (n=27), we performed Illumina methylation EPIC microarray analyses. Overall, 704,003 probes remained for further statistical evaluation after quality and sex chromosome filtering. SVD analysis indicated that a significant variation of DNA methylation within the whole study cohort was associated with CD3+/CD8+ immune cell content, center of sample origin and sex (online supplemental figure S3A). In addition, although data were normalized, a strong variation within our dataset was seen in beta value density (online supplemental figure S3B). This variation was reduced after adjusting methylation data for the factors mentioned above (online supplemental figure S3C). Unsupervised hierarchical clustering of the whole study population based on 704,003 adjusted beta values was performed to test for DNA methylation heterogeneity within the cohort. Two subclusters were identified: one cluster containing only 1 responder and 19 non-responders and one clusters containing 7 responders and 8 non-responders to anti-PD-1 ICI (p=0.011; figure 2A). This clustering was independent of sample origin (center 1 or 2) and sarcoma type (osteosarcoma or soft tissue sarcoma).
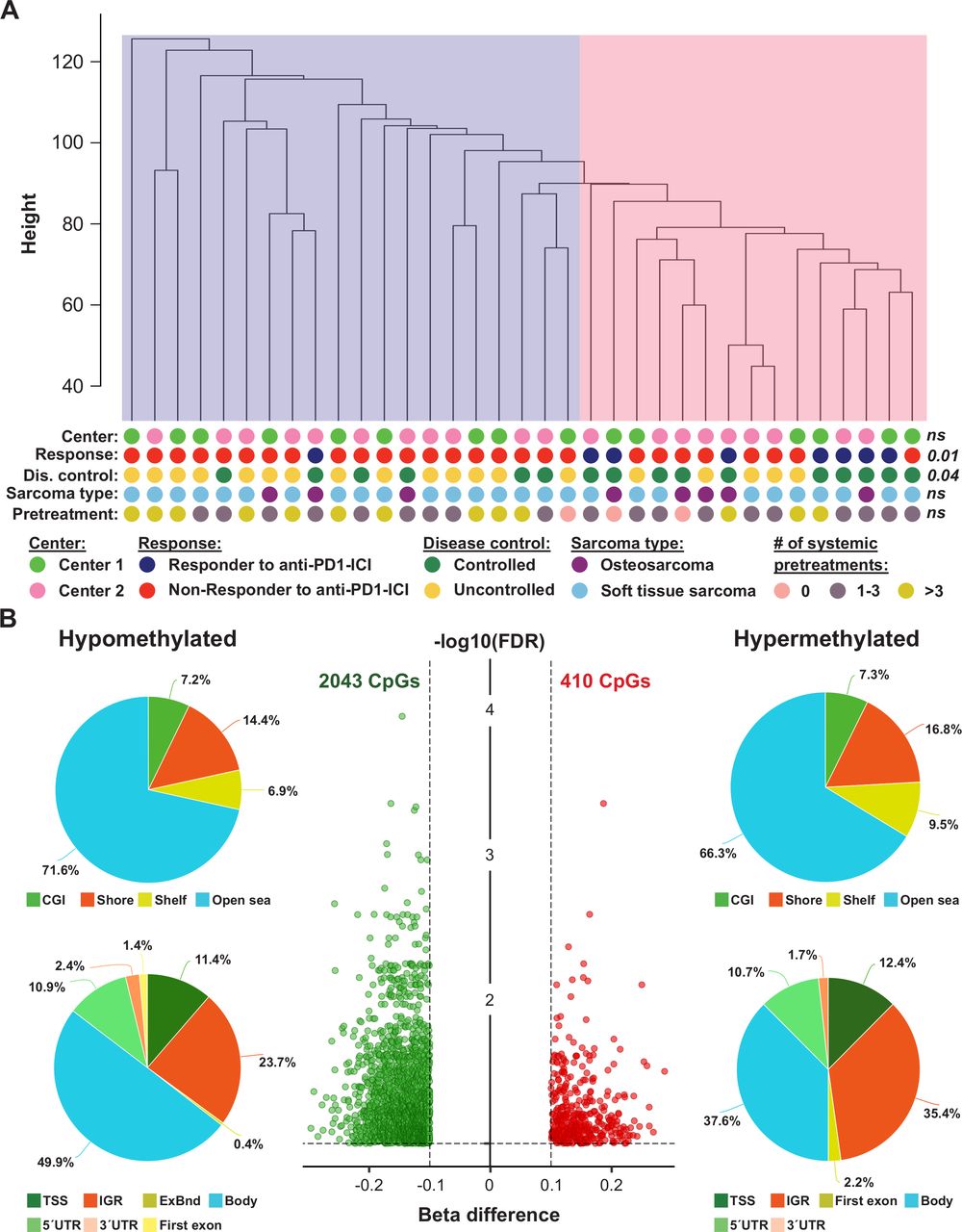
Exploratory analysis of EPIC DNA methylation data of tumors from anti-programmed cell death 1 (anti-PD-1) immune checkpoint inhibitor (ICI)-treated patients with sarcoma. (A) Unsupervised hierarchical clustering of 35 patients with sarcoma based on beta values of 704,003 probes. The pink cluster is significantly enriched for responders (dark blue dots) to anti-PD-1 ICI, while the blue cluster contains only one responder to anti-PD-1 ICI. P values were calculated using Fisher’s exact tests; ns, not significant. (B) The volcano plot (middle panel) illustrates differentially methylated CpG sites (DMPs; green, hypomethylated; red, hypermethylated) between non-responders and responders to anti-PD-1 ICI. Each dot represents a unique DMP. The location of these DMPs relative to CpG islands and genomic features are shown in the upper and lower pie charts, respectively.
Differential methylation analysis
In a next step, we performed an in-depth differential methylation analysis between responders and non-responders to anti-PD-1 therapy. In total, 2453 CpG sites (DMP; 0.3% of the CpG sites analyzed) were differentially methylated between the two patient groups. While methylation of 2043 DMPs was decreased, methylation of 410 DMPs was increased (figure 2B). A detailed description of these DMPs is shown in online supplemental table S4. When we mapped the DMPs to their genomic location within, around or between CpG islands, we found an over-representation of hypomethylated non-CpG island DMPs (71.6%). The frequency of hypermethylated non-CpG island DMPs was only slightly lower (66.3%, figure 2B). In addition, the frequencies of hypomethylated and hypermethylated CpG island-associated DMPs were 7.2% and 7.3% for CGIs, 14.4% and 16.8% for shore regions (2 kb regions flanking CpG islands), and 6.9% and 9.5% for shelve regions (2 kb regions of flanking shore regions), respectively. The locations of hypomethylated DMPs in the context of gene-associated regions were 49.9% in gene bodies, 23.7% in intergenic regions (IGR), 11.4% in transcriptional start sites, 10.9% in 5′ UTRs, 2.4% in 3′ UTRs, 1.4% in first exons and 0.4% in exon boundaries. Again, these values are very similar for hypermethylated DMPs with the exception of a higher IGR frequency and a lower gene body frequency. Chromosomal distribution of hypomethylated and hypermethylated DMPs was relatively even, as shown in online supplemental figure S4.
Supplemental material
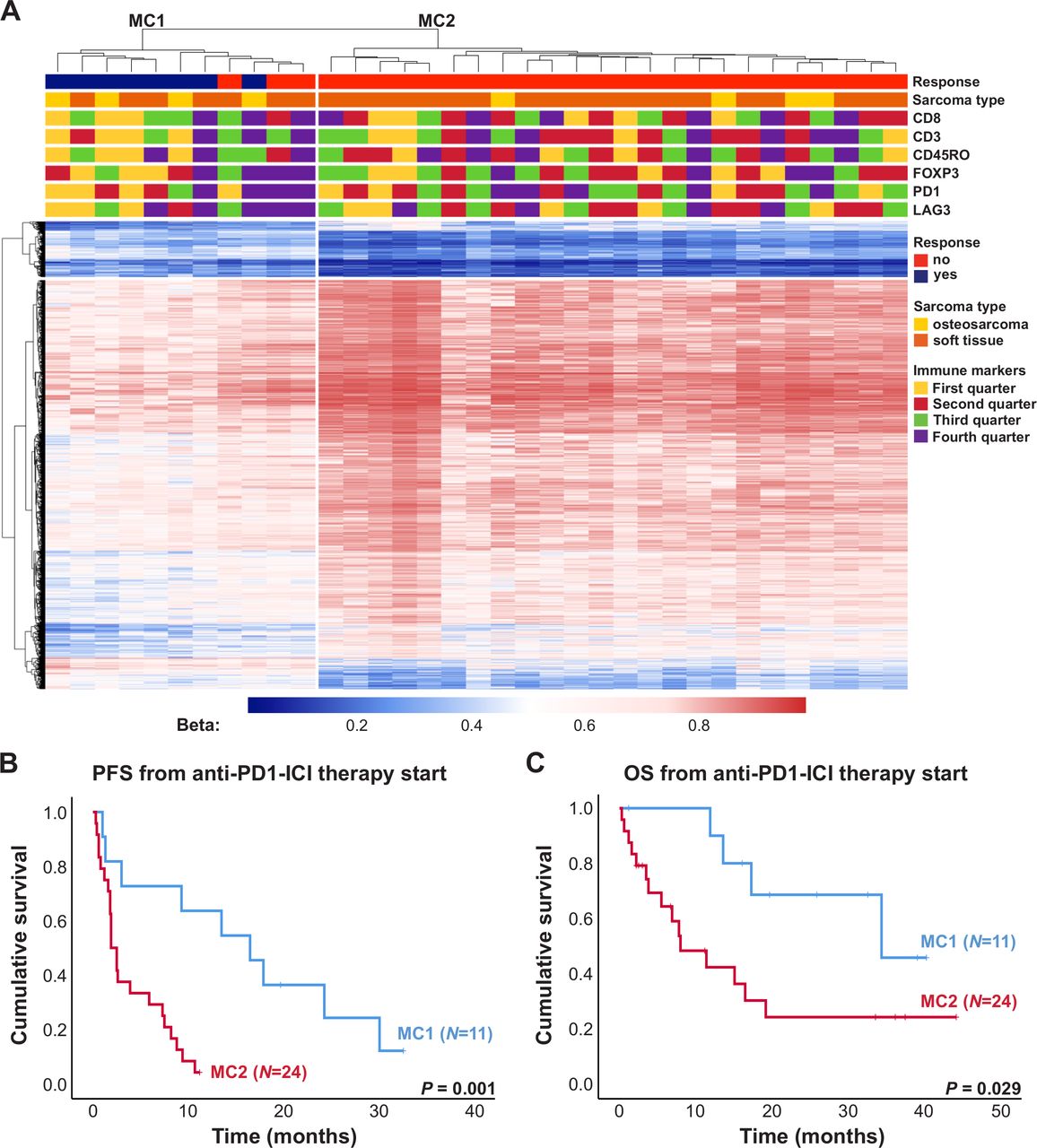
Clustering of sarcoma samples based on the 2453 DMPs revealed two main clusters, of which one consisted of 73% responders to anti-PD-1 ICI (MC1) and the other one contained only non-responders (MC2, figure 3A). This clustering was only associated with response to anti-PD-1 ICI and no association with sarcoma subtype or immune cell infiltration was seen. Median PFS from anti-PD-1 therapy start of patients included in MC1 and MC2 was 16.5 and 1.9 months, respectively (figure 3B, p=0.001). Median OS from anti-PD-1 therapy start of these patients was 34.4 and 8.0 months, respectively (figure 3C, p=0.029).
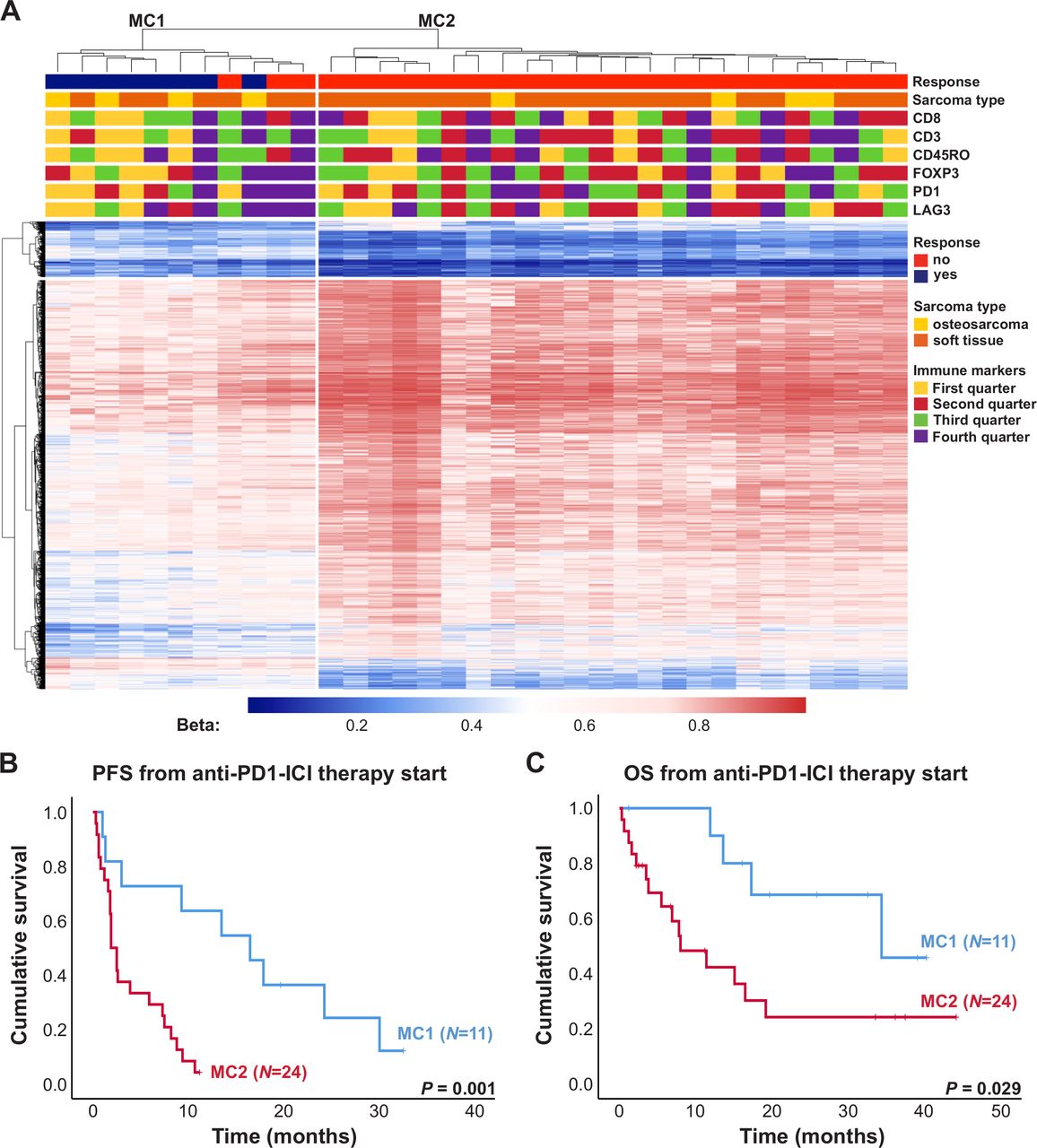
Impact of DNA methylation on progression-free survival (PFS) and overall survival (OS) of patients with sarcoma treated with anti-programmed cell death 1 (anti-PD-1) immune checkpoint inhibitor (ICI). (A) Heatmap summarizing methylation of 2453 DMPs in 35 sarcoma samples. Patients’ characteristics including response to anti-PD-1 ICI, sarcoma subtype as well as expression of immune markers are displayed according to the color scheme shown at the right-hand side of the figure. Each row represents a unique CpG site and each column represents a unique patient sample. Heatmap colors reflect beta values representing the degree of methylation from low (blue) to high (red). No centering/scaling of beta values was performed. Immune markers were divided into first, second, third and fourth quarter. (B) PFS and OS (C) of patients with sarcoma from anti-PD-1 ICI therapy start in months. Blue and red lines indicate patients included in the methylation clusters MC1 and MC2, respectively. Vertical lines indicate censored patients at last follow-up. P values were calculated using log-rank tests.
Functional implication of differentially methylated genes
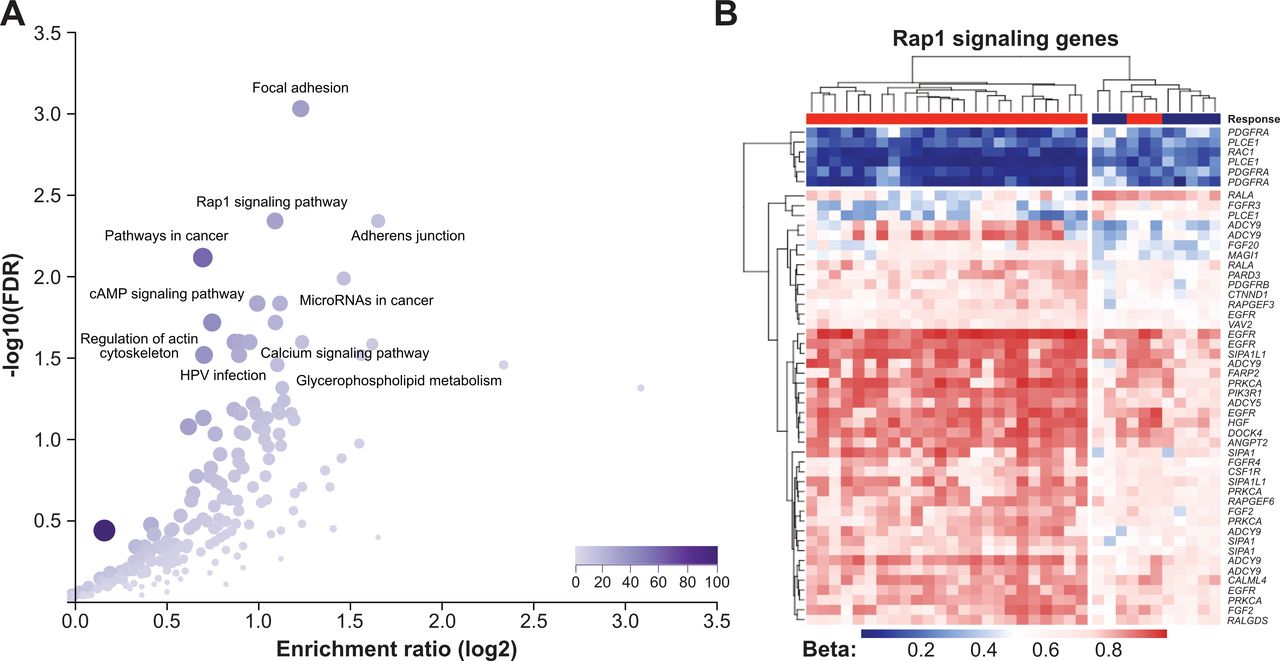
To investigate the potential biological relevance of differential methylation between responders and non-responders to anti-PD-1 therapy, the 2453 DMPs were annotated to 1360 genes, which were used for further pathway enrichment analyses. Among KEGG annotated pathways, the top five hits were focal adhesion (hsa04510, FDR=0.0009), adherens junction (hsa4520, FDR=0.005), Rap1 signaling pathway (hsa04015, FDR=0.005), pathways in cancer (hsa05200, FDR=0.008) and ECM–receptor interaction (hsa04512, 0.01) (figure 4A, online supplemental table S5). The methylation pattern of Rap1 signaling pathway members in our sarcoma patient cohort is shown in figure 4B. GO enrichment analysis revealed a statistically significant over-representation of genes mainly involved in developmental processes, cell adhesion and cell migration (online supplemental table S6). DMPs of another cancer-related pathway, PI3K/AKT, and its members affected by differential methylation are illustrated in online supplemental figure S5.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Functional implication of differentially methylated CpG sites (DMPs) between responders and non-responders to anti-programmed cell death 1 (anti-PD-1) immune checkpoint inhibitor (ICI) therapy. (A) Results from Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis using WebGestalt software (left panel). Each dot represents a unique pathway. (B) Heatmap summarizing methylation of Rap1 signaling genes in 35 sarcoma samples. Genes which appear repeatedly in the heatmap were affected by multiple DMPs. Heatmap colors reflect beta values representing the degree of methylation from low (blue) to high (red). No centering/scaling of beta values was performed.
CNV in sarcomas
Because sarcomas are characterized by high levels of CNVs,23 we were also interested if these genetic alterations differ between responders and non-responders to anti-PD-1 therapy. The data obtained from EPIC methylation microarrays of the 35 patients with sarcoma described above were used for these analyses. As shown in online supplemental figures S6 and S7, CNVs were detected at various extent in the tumor samples. While some tumors showed no or only a small number of CNVs, others were affected by many CNVs. However, when we compared the CNV pattern of responders and non-responders to anti-PD-1 therapy, no distinct difference between these two patient groups was seen indicating that CNVs are not suitable to distinguish between responders and non-responders to anti-PD-1 ICI therapy in patients with sarcoma.
Discussion
Anti-PD-1 ICI with pembrolizumab or nivolumab is a promising treatment that seems to benefit some patients with advanced sarcomas. In our current multicenter series of 35 adult patients with sarcoma treated with pembrolizumab or nivolumab monotherapy as salvage treatment for recurrent disease we observed favorable outcome data with an objective response rate of 22.9%, median PFS of 3 months and median OS of 16.5 months. Anti-PD-1 therapy was well tolerated in our patients treated in the real-world setting and no new safety concerns were identified. Thus, our experience is in line with prior reports and provides further support for considering anti-PD-1 ICI as treatment option in individual patients with sarcoma. However, better biomarkers to select patients most likely to derive a benefit from this therapy are urgently needed.
The results from the correlative studies in our cohort introduce DNA methylation profiling as a novel candidate biomarker for selecting sarcoma patient potentially benefiting from anti-PD-1 ICI therapy. Interestingly, PD-L1 expression on tumor cells was not associated with response to ICI treatment in the cohort from center 1. In line, previous studies also failed to observe an association of ICI response and PD-L1 expression.44 Moreover, also dense TIL infiltration as a marker of inflammatory tumor microenvironment activation was not associated with response. Therefore, ‘classic’ predictive biomarkers might not be suitable in sarcoma. Indeed, PD-L1 and/or TILs density are far away from perfect predictive biomarkers as they fail to reliably predict ICI response in various other solid cancer types like lung cancer and head and neck cancer patients.45 46 Here, we investigated DNA methylation as a potential predictive biomarker for ICI response. Differential DNA methylation analysis led to a separation of patients achieving an objective radiological response from patients not achieving an objective response to anti-PD-1 ICI. A similar predictive role of microarray DNA methylation profiles for response to PD-1 inhibition has recently been reported for stage IV non-small cell lung cancer, thus underlining the potential value of this approach.47 Importantly, in our study on sarcomas, the effect was detectable only after bioinformatically adjusting the DNA methylation data for the content of tumor-infiltrating immune cells. Thus, our findings indicate that reliable response prediction in the clinical setting may necessitate integrated multimodal biomarker analysis to account for the heterogeneous composition of the tumorous microenvironment in sarcoma and potentially also other tumors.
Pathway enrichment analyses showed that the genes most prominently affected by differential methylation are involved in Rap1 signaling, focal adhesion, adherens junction, pathways in cancer and ECM–receptor interaction. These pathways have not been extensively investigated in sarcoma and further studies are needed to elucidate the association with patient outcome observed in our study. However, some of these factors have known functions in immune response, and based on our findings, aberrant DNA methylation may influence sensitivity of immune checkpoint inhibition. Rap1 is important for a range of diverse cellular functions including control of cell–matrix and cell–cell adhesion via activation of integrins and other adhesion molecules.48 It has also been demonstrated to play crucial roles in immune response during the formation of the immunological synapse and to interact with immune checkpoint molecules such as CTLA-4.48 49 Our data indicate that functional investigations should focus on these pathways and their potential role in determining immune response of sarcoma.
Several limitations of our study have to be acknowledged for correct interpretation of our results. Owing to the rarity of sarcomas, the sample size of our cohort is limited, although it represents a comparatively large multi-institutional series of homogeneously treated and well-defined patients. In addition, the retrospective design precludes definite conclusions. However, we introduced clear and strict inclusion criteria, performed a comprehensive evaluation of clinical and tissue-based parameters and performed blinded radiological review of all baseline and restaging images in order to maximize the information value and quality of data derived from this investigation. Still, it needs to be stressed that further studies with more patients, prospective designs and adequate statistical power are needed to substantiate our findings.
In conclusion, we demonstrate that tumor DNA methylation profiling may serve as a predictive marker for response to anti-PD-1 ICI therapy in sarcoma, especially when performed in conjunction with analysis of tumor-infiltrating immune cell composition. Further studies are needed to validate our findings and elucidate the pathobiological basis of our hypothesis-generating observations.
Acknowledgments
This study was performed within the PhD thesis of AS with the title “Immune monitoring in cancer patients” in the N790 doctoral program at the Medical University Vienna, Austria. The financial support by the Austrian Federal Ministry for Digital and Economic Affairs, the National Foundation for Research, Technology and Development and the Christian Doppler Research Association is gratefully acknowledged.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
GH and TB contributed equally.
Contributors Data analyses and experimental analyses: AS, AB, RH, ET, KF, TH, ST, WL, LM, GA, GH, and TB. Central radiological assessment: JF and IN. Statistical analysis: AS, GH, and AB. Manuscript writing: AS, AB, GH, MP, and TB. Intellectual revision and approval of manuscript: AS, AB, RH, ET, KF, JF, IN, WL, GA, LM, SB, H-US, MP, GH, and TB.
Funding The study was supported by the research budget of the Medical University of Vienna, an unrestricted research grant by Hoffmann La Roche, the Austrian Federal Ministry for Digital and Economic Affairs, the National Foundation for Research, Technology and Development and the Christian Doppler Research Association.
Competing interests AS has received travel support from PharmaMar. AB has research support from Daiichi Sankyo and Roche; honoraria for lectures, consultation or advisory board participation from Roche Bristol-Meyers Squibb, Merck, Daiichi Sankyo; as well as travel support from Roche, Amgen, Daiichi Sankyo and AbbVie. MP has received honoraria for lectures, consultation or advisory board participation from the following for-profit companies: Bayer, Bristol-Myers Squibb, Novartis, Gerson Lehrman Group (GLG), CMC Contrast, GlaxoSmithKline, Mundipharma, Roche, BMJ Journals, MedMedia, Astra Zeneca, AbbVie, Lilly, Medahead, Daiichi Sankyo, Sanofi, Merck Sharp & Dome, Tocagen. The following for-profit companies have supported clinical trials and contracted research conducted by MP with payments made to his institution: Böhringer-Ingelheim, Bristol-Myers Squibb, Roche, Daiichi Sankyo, Merck Sharp & Dome, Novocure, GlaxoSmithKline, AbbVie. TB reports personal fees from Roche (lecture fee), personal fees from Amgen (lecture fee, advisory board), personal fees from Bayer (lecture fee, advisory board), personal fees from Novartis (lecture fee, advisory board), personal fees from PharmaMar (lecture fee, advisory board), personal fees from Eisai (lecture fee, advisory board), personal fees from Eli Lilly (lecture fee, advisory board), outside the submitted work. RH is supported by Clinician Scientist Program of the University Medicine Essen Clinician Scientist Academy (UMEA) sponsored by faculty of medicine and Deutsche Forschungsgemeinschaft (DFG). RH has received travel grants from Lilly, Novartis and PharmaMar, as well as fees from Lilly outside of the submitted work. SB reports personal fees from Deciphera, grants from Incyte, grants and personal fees from Blueprint Medicines, personal fees from Lilly, grants and personal fees from Novartis, personal fees from Daichii-Sankyo, personal fees from Plexxikon, personal fees from Exelixis, personal fees from Bayer, other from Pfizer, during the conduct of the study; personal fees from Pharmamar, personal fees from Lilly, personal fees from Roche, personal fees from GSK, outside the submitted work. All other authors report no conflicts of interest concerning this specific publication.
Patient consent for publication Not required.
Ethics approval This project was approved by the ethics committee of the Medical University of Vienna (EK 2195/2019) and by the ethics committee of the Medical Faculty, University of Duisburg Essen (20-9558-BO).
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available upon reasonable request. All data relevant to the study are included in the article or uploaded as supplementary information. Relevant additional data can be shared on request to the authors.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.