Article Text

Abstract
Background Current immunotherapies including checkpoint blockade therapy have limited success rates in certain types of cancers. Identification of alternative checkpoint molecules for the development of effective strategies for tumor immunotherapy is urgently needed. Immunoglobulin-like transcript 4 (ILT4) is an immunosuppressive molecule expressed in both myeloid innate cells and malignant tumor cells. However, the role of tumor-derived ILT4 in regulating cancer biology and tumor immunity remains unclear.
Methods ILT4 expression in tumor cells and patient samples was determined by real-time PCR, flow cytometry, and immunohistochemistry. T cell senescence induced by tumor was evaluated using multiple markers and assays. Moreover, metabolic enzyme and signaling molecule expression and lipid droplets in tumor cells were determined using real-time PCR, western blot and oil red O staining, respectively. Loss-of-function and gain-of-function strategies were used to identify the causative role of ILT4 in tumor-induced T cell senescence. In addition, breast cancer and melanoma mouse tumor models were performed to demonstrate the role of ILT4 as a checkpoint molecule for tumor immunotherapy.
Results We reported that ILT4 is highly expressed in human tumor cells and tissues, which is negatively associated with clinical outcomes. Furthermore, tumor-derived ILT4/PIR-B (ILT4 ortholog in mouse) is directly involved in induction of cell senescence in naïve/effector T cells mediated by tumor cells in vitro and in vivo. Mechanistically, ILT4/PIR-B increases fatty acid synthesis and lipid accumulation in tumor cells via activation of MAPK ERK1/2 signaling, resulting in promotion of tumor growth and progression, and induction of effector T cell senescence. In addition, blocking tumor-derived PIR-B can reprogram tumor metabolism, prevent senescence development in tumor-specific T cells, and enhance antitumor immunity in both breast cancer and melanoma mouse models.
Conclusions These studies identify a novel mechanism responsible for ILT4-mediated immune suppression in the tumor microenvironment, and prove a novel concept of ILT4 as a critical checkpoint molecule for tumor immunotherapy.
- lymphocytes
- tumor-Infiltrating
- tumor microenvironment
- immunotherapy
- adoptive
- immune evation
- biomarkers
- tumor
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
- lymphocytes
- tumor-Infiltrating
- tumor microenvironment
- immunotherapy
- adoptive
- immune evation
- biomarkers
- tumor
Introduction
It is well established that the suppressive tumor microenvironment (TME) is a significant obstacle for effective antitumor immunity and immunotherapy. Different types of suppressive and/or dysfunctional T cells are the key components in the TME.1 2 Therefore, improving the quality and effector function of tumor-specific T cells is the determinant for successful tumor immunotherapy. Current tumor immunotherapies, including immune checkpoint blockade therapy and adoptive T cell transfer therapy (ACT), have become the most important breakthroughs in the field and obtained promising results in patients with different types of cancer.3–5 The immune checkpoint blockade therapies with anti-CTLA-4 and anti-PD1/PD-L1 antibodies can significantly modulate the pre-existing T cell activity in the TME.3 4 6 However, the clinical effective rates are varied and achieved no more than 40% in solid tumors, whereas it is even lower in tumors harboring a low mutation burden (primary resistance).6 Furthermore, relapses are also observed in approximately one-third of the checkpoint blockade therapy patients after disease control for several months (secondary resistance).7 8 These more recent clinical evidence indicate that anti-PD1/PD-L1/CTLA-4 therapy is insufficient to break the complex immunosuppressive TME in certain types of cancers. Therefore, exploring novel mechanisms responsible for tumor-induced immune tolerance and identification of alternative checkpoint molecules are urgently needed to overcome the immune resistance and maximize the clinical benefit.9
Immunoglobulin-like transcript 4 (ILT4) is an inhibitory molecule of the immunoglobulin superfamily that is mainly expressed in myeloid cells, including monocytes, macrophages, dendritic cells (DCs), and granulocytes.10–12 The ortholog of ILT4 in mouse is paired Ig-like receptor-B (PIR-B).13 ILT4 functions to suppress myeloid cell activation, antigen presentation, and immune response.12 14 15 Increasing evidence suggests that ILT4 is also highly expressed in various tumor cells and stroma cells, such as myeloid-derived suppressor cells (MDSCs) and tumor-associated macrophages (TAMs) in the TME, directly modulating tumor growth, progression and metastasis.16–20 Furthermore, ILT4 expression levels in tumors are negatively associated with clinical outcomes.19 21 However, the molecular processes responsible for ILT4-mediated regulation of tumor behaviors are still unclear. In addition to regulate tumor cells, recent studies have shown that ILT4 can promote tolerogenic DC development, TAM and MDSC polarization, which in turn induce both CD4+ and CD8+ Treg cell development and effector T cell dysfunction and amplify the immunosuppression in the TME.14 20 22 However, whether and how tumor-derived ILT4 directly affects T cell development and function remain unknown.
Malignant tumor cells can use multiple strategies to suppress antitumor immune response and sustain a suppressive TME. Induction of T cell senescence has recently been identified as a novel strategy used by tumor cells to evade immune surveillance.23–28 Different types of human tumor cells, including melanoma, breast, colorectal, ovarian, prostate, head and neck cancers, can directly convert effector T cells into senescent T cells.25 26 Furthermore, tumor-associated Treg cells can also induce responder T cell senescence.23 24 27 28 Importantly, increased senescent T cells have been observed in tumor-infiltrating lymphocytes (TILs) and peripheral blood in patients with various types of cancer.29–31 Senescent T cells express unique phenotypes, including expression of senescence-associated β-galactosidase (SA-β-gal), loss of costimulatory molecules CD27, CD28, upregulation of cell cycle molecules P16, P53, and P21, secretion of proinflammatory and inhibitory cytokines, and possess potent suppressive activity.23–28 Therefore, blockage of effector T cell senescence in the suppressive TME is a critical checkpoint for antitumor immunity and immunotherapy. However, the mechanism(s) responsible for T cell senescence mediated by malignant tumors is incompletely identified. Recent studies have demonstrated tumor-derived cyclic adenosine monophosphate (cAMP) is involved in T cell senescence induction.25 Furthermore, Treg cells can mediate glucose competition with responder effector T cells to promote development of T cell senescence during their crosstalk.27 28 These studies suggest that metabolic regulation is the key for senescent T cell development. Given that ILT4 is highly expressed in tumor cells, whether ILT4 is involved in the molecular processes of tumor-mediated T cell senescence is unclear. An improved understanding of this mechanism will facilitate the development of novel strategies targeting ILT4 for tumor immunotherapy.
To explore the functional role of ILT4 in regulation of tumor biology and antitumor immunity, we identified that ILT4 is a critical immune checkpoint molecule in the TME. We demonstrated that tumor-derived ILT4/PIR-B is responsible for the induction of senescence in naïve/effector T cells mediated by different types of tumor cells in vitro and in vivo. Furthermore, we identified that ILT4/PIR-B increases fatty acid synthesis and lipid accumulation in tumor cells molecularly controlled by MAPK ERK1/2 signaling, resulting in promotion of tumor growth and progression, as well as induction of T cell senescence. In addition, blockage of tumor-derived PIR-B significantly prevents senescence development in tumor-specific T cells and improves antitumor immunity in the breast cancer and therapeutic melanoma mouse tumor models. These studies identify novel mechanism responsible for ILT4-mediated immune suppression in the TME, and provide proof of concept for targeting ILT4 as a checkpoint molecule for enhanced tumor immunotherapy.
Materials and methods
T cells and tumor cell lines
All human and mouse tumor cell lines (lung cancer, breast cancer, melanoma, and prostate cancer cells) were either purchased from American Type Culture Collection (ATCC, Manassas, Virginia, USA) or generated by our laboratory. All the cells were cultured in RPMI-1640 or dullbecco’s modified eagle medium (DMEM) with 10% fetal bovine serum (FBS) (Sigma-Aldrich). Human mammary gland endothelial cell line MCF10A was cultured in DME/F12 supplemented with 2% horse serum, hydrocortisone (1 µg/mL), epidermal growth factor (EGF) (20 ng/mL), cholera toxin (0.1 µg/mL), and insulin (1.2 µg/mL). Mouse mammary gland endothelial cell line FSK4 was maintained in DMEM supplemented with 2% FBS, 1% 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) buffer (1 M), 10 µg/mL insulin and 5 ng/mL EGF. Buffy coats from healthy donors were obtained from the Gulf Coast Regional Blood Center at Houston. Peripheral blood mononuclear cells (PBMCs) were purified from buffy coats using the Ficoll-Paque. Human naïve CD4+ and CD8+ T cells were purified from PBMCs of healthy donors by EasySep enrichment kits (StemCell Technologies). The purity of T cells was >97%, as confirmed by the flow cytometry. Human T cells were maintained in T cell medium containing 10% human AB serum and 50 u/mL recombinant human interleukin-2 (IL-2).
Reverse-transcription PCR and real-time quantitative RT-PCR
Total RNA was extracted from cells or tissues using Trizol reagent (Invitrogen), and cDNA was transcribed using a SuperScript II RT kit (Invitrogen). The mRNA expression of ILT4, PIR-B, or key enzymes related to glycolysis and lipid metabolism in each sample were determined by the real-time quantitative reverse-transcription PCR (qRT-PCR) using specific primers, and mRNA levels in each sample were normalized to the relative quantity of β-actin as previously described.27 28 All experiments were performed in triplicate. The specific primers used are listed as in the online supplemental table 1.
Supplemental material
Flow cytometry analysis
The expression markers on tumor cells or T cells were determined by flow cytometry analysis after surface staining or intracellular staining with anti-human or anti-mouse-specific antibodies conjugated with PE or Alexa Flour488. The human antibodies included anti-ILT4 (clone 287219), anti-CD27 (clone M-T271), anti-CD28 (clone CD28.2), anti-P53 (clone 7F5), anti-P21 (clone 12D1), anti-phospho-ATM, anti-phospho-H2AX, anti-phospho-CHK2, and anti-phospho-53BP1. The mouse antibodies include anti-PIR-B (clone 326414), anti-CD4 (clone RM4-5), anti-CD8 (clone 53–6.7), anti-IFN-γ (clone XMG1.2), anti-granzyme B (clone QA16A02), and anti-perforin (clone S16009A). All these antibodies were purchased from BD Biosciences, Biolegend, R&D system, or Cell Signaling Technology. All stained cells were analyzed on a FACS Calibur flow cytometer (BD Bioscience) and data were analyzed with FlowJo software (Tree Star).
Immunohistochemical staining of ILT4 and quantification method
The cell populations of ILT4+ cells in human cancer and normal tissues (paraffin-embedded) were determined using immunohistochemical staining with the Elivision Plus Polymer Horseradish Peroxidase IHC Kit (Maixin Biotech, Fuzhou, China), as we described previously.32 Immunohistochemical reactions were performed using rabbit polyclonal antibodies against ILT4 at a dilution of 1:20. Controls were performed by incubating slides with the isotype control antibody instead of primary antibodies. ILT4+ cells in tissues were evaluated by two independent pathologists using a computerized image system composed of a cellSens Dimension system equipped on an Olympus U-TV0.63XC microscope (Tokyo, Japan).
Lentivirus generation and gene overexpression/knockdown in tumor cells
The methods for generation of recombinant lentivirus carrying target genes and shRNAs have been described previously.33 shRNAs specific for ILT4 (TRCN000057033, TRCN000057034 and TRCN000057035) and PIR-B (TRCN0000067203 and TRCN0000067205), control shRNA and packaging plasmids (psPAX2 and PMD2.G) were purchased from the Sigma-Aldrich. ILT4/PIR-B overexpression plasmids (EX-Z3241-Lv105 and EX-Mm34323-Lv105), control plasmids and HIV expression packaging kit were purchased from GeneCopoeia (Rockville, Maryland, USA) and LV-pReceiver-Lv105/ILT4/PIR-B were constructed following their exact protocols. For lentivirus infection, concentrated lentiviral supernatant with a multiplicity of infection (MOI) of 5–10 in a total volume of 2 mL culture medium was added to the tumor cells growing in 6-well plates containing 5 µg/mL polybrene (Sigma), and then centrifuged at 1000 × g for 1 hour at room temperature. The infected tumor cells were cultured for 48–72 hours and then harvested for subsequent RNA/protein analyses and in vivo injection.
In some experiments, the lentiviral constructs for ILT4/PIR-B shRNA or genes were directly transfected into tumor cells for the studies using X-tremeGENE HP DNA Transfection Reagent (Roche).
Cell growth and proliferation assay
Human or mouse tumor cells infected with lentivirus carrying ILT4 or PIR-B genes or respective shRNAs were cultured in 6-well plates at an initial concentration of 1–2 × 105 cells/well. Cell numbers were counted every 3 days for determination of cell growth. In addition, cell proliferation was evaluated by the 3-(4,5-dimethyl-2-thiazolyl)−2,5-diphenyl-2-H-tetrazolium bromide (MTT) assay. Briefly, 5000 tumor cells/well were seeded into the flat-bottom 96-well plates. After 24 hours of culture, 15 µL of MTT (5 mg/mL) was added and the cells were cultured for an additional 4 hours for detection of MTT absorption at a wavelength of 570 nm.
Adhesion assay
The adhesion of cancer cells was determined as we previously described.34 Flat-bottom 96-well plates were coated with 50 µL/well fibronectin (10 µg/mL, BD Biosciences) overnight at 4°C and blocked with 2% bovine serum albumin (BSA) at 37°C for 2 hours. Tumor cells (1 × 105) were seeded into each well and incubated at 37°C for 45 min. Wash three times with phosphate-buffered saline (PBS) to remove non-adherent cells. The adherent cells on the plates were fixed with 4% formaldehyde for 4 min and stained with 0.5% crystal violet for 15 min. Adherent cells were counted and averaged in 10 fields at high magnification (× 400) with a microscope.
Wound healing and migration assay
Tumor cells were plated in 6-well plates and grew to 80% confluence. A wound area was generated by scraping cells with a 200 µL pipette tip across the entire diameter of the well and extensively rinsed with the medium to remove all cellular debris. The scratches were photographed after additional 0 and 12 hours of culture. The closure was estimated as the wounded area relative to the initial area.34 35
Senescence-associated β-galactosidase staining
Senescence-associated β-galactosidase (SA-β-Gal) activity in senescent T cells was detected as we previously described.23 24 27 Human naïve T cells were activated in anti-CD3 and anti-CD28 (2 μg/mL) precoated 12-well plates, and then cocultured with tumor cells. After 24 hours of coculture, the T cells were separated and cultured for additional 3 days in medium of RPMI-1640 containing 50 u/mL IL-2 and 10% FBS, and then stained for SA-β-Gal.
For some experiments, the cocultured T cells were determined for SA-β-Gal expression after the tumor cells pretreated with the MAPK inhibitor U0126 (10 µM) (Calbiochemistry), or FASN inhibitor C75 (5 µM) (Cayman Chemical Company), anti-ILT4 antibody (500 ng/mL), or anti-HLA-G antibody (10 µg/mL) (R&D System). In addition, tumor cells were infected with lentivirus carrying ILT4 gene or ILT4-specific shRNAs to overexpress ILT4 or knockdown ILT4 expression, and then cocultured with T cells and SA-β-Gal expression in T cells were determined.
Western blotting analysis
Whole cell lysates of ILT4-overexpressed/downregulated tumor cells were prepared for western blotting. Western blots were developed with chemiluminescent substrate (KPL, Maryland, USA). The antibodies used in western blotting are as follows: anti-ILT4 (Abgent, San Diego, California, USA), anti-PIR-B (Abiocode, Belgrade, Serbia), anti-ERK, anti-phospho-ERK (Thr180/Tyr182), anti-P38, anti-phospho-P38 (Thr180/Tyr182), anti-JNK, anti-phospho-JNK (Thr183/Tyr185), and anti-GAPDH rabbit polyclonal antibodies (Cell Signaling Technology, Danvers, Massachusetts, USA).
Oil red O staining
The treated different types of tumor cells were washed in PBS (pH 7.2), fixed with 4% formaldehyde for 30 min, and treated with 60% isopropanol for 2–5 min. Cells were further stained with freshly prepared Oil Red O staining solution (Sigma-Aldrich) in isopropanol (60%) for 5 min, followed by quick rinse with isopropanol (60%). The stained tumor cells were then washed with H2O thoroughly and lipid droplets in tumor cells were evaluated using a light microscope.
Analysis of prognostic significance of ILT4 in lung and breast cancers
The online tool of KM-plotter (http://kmplot.com/) database was used to analyze ILT4-based survival in patients with non-small cell lung cancer (NSCLC). A total of 1926 and 982 patients were included for overall survival (OS) and progress-free survival (PFS) analyses, respectively. Patients in each cohort were divided by ‘auto selected best cut-off’ and all other parameters were at default settings. The HR with 95% CIs and log-rank p values were displayed online. Another independent mRNA expression dataset with clinical parameters (GSE1456) was obtained from the Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/). GSE1456 was based on GPL96 and GPL97 (Affymetrix Human Genome U133A and U133B Array), and included 159 patients with breast cancer. The series matrix file data of the datasets were downloaded and normalized by R package ‘limma’. The expression data of ILT4 were extracted and the survival data were integrated for survival analysis. Patients with breast cancer were divided into two groups according to ILT4 expression with the cut-off value of 6.37. The relationship between ILT4 expression and patient survival was verified by Kaplan-Meier survival and the log-rank (Mantel-Cox) test.36
In vivo studies
NOD/SCID IL2Rγnull mice (NSG) (Stock No. 005557), C57BL/6 mice (B6, Stock No. 000664) and B6.Cg-Thy1a/Cy Tg (TcraTcrb) 8Rest/J mice (Pmel-1 TCR transgene mice, Stock No. 005023) were purchased from The Jackson Laboratory and maintained in the institutional animal facility. All animal studies have been approved by the Institutional Animal Care Committee at Saint Louis University (Protocol No. 2411).
Mouse tumor growth models
E0771 tumor cells (2 × 105 cells/mouse) infected with lentivirus carrying LV-PIR-B, LV-PIR-B-shRNA, or LV-control vector were subcutaneously injected into female C57BL/6 or NSG mice (6–8 weeks old). Five mice were included in each group. Tumor size was measured with calipers every 2–3 days. Tumor size was calculated based on two-dimensional measurements. At the end of experiments, the mice were sacrificed and tumors were isolated and weighed. The fresh tumor tissues from NSG mice were prepared for real-time qPCR or western blotting analyses as described above. Furthermore, in the immunocompetent C57BL/6 mice, lymphocytes from the blood, spleen, lymph nodes, and tumors were also isolated using Ficoll-Paque for subsequent SA-β-Gal expression and flow cytometry analyses.
Mouse T cell adoptive transfer therapy models
C57BL/6 mice (6-week to 8-week-old female) were injected with B16F0 tumor cells (2 × 105 cells/mouse) infected with lentivirus carrying LV-PIR-B-shRNA or LV-control-shRNA. Splenocytes from Pmel-1 TCR/Thy1.1 transgenic mice were prepared and activated in the presence of plate-coated anti-CD3 (2 µg/mL) and anti-CD28 (1 µg/mL) antibodies for 6–9 days. The activated Pmel-1 T cells (2 × 106 cells/mouse) were adoptively transferred into B16F0-bearing mice at day 9 (tumor diameter reached a size of 5–6 mm) post tumor inoculation. The B16F0 tumor-bearing mice were radiated with a non-myeloablative dose (500 cGy) using an XRAD 320 irradiator (Precision X-RAY) to induce lymphopenia 1 day before T cell adoptive transfer. Five mice were included in each group. Tumor size was measured with calipers every 3 days and tumor size was calculated based on two-dimensional measurements. At the end of experiments, the mice were sacrificed and tumors were isolated and weighed. Furthermore, blood, spleens, and tumors were harvested and the transferred CD8+ T cells from different organs and tumor tissues were isolated by antibody-coated microbeads (StemCell Technologies) for subsequent SA-β-Gal staining, and phenotypic and functional analyses.
Statistical analysis
Statistical analysis was performed with GraphPad Prism5 software. Data are expressed as mean ± SD. One-way analysis of variance (ANOVA) was employed for multiple-group comparisons. The paired Student’s t-test was used for a single comparison between two groups, and the non-parametric t-test was also employed if the sample size was too small and unfit for Gaussian distribution.
Results
Enriched ILT4 expression in tumor cells is associated with poor outcomes in patients
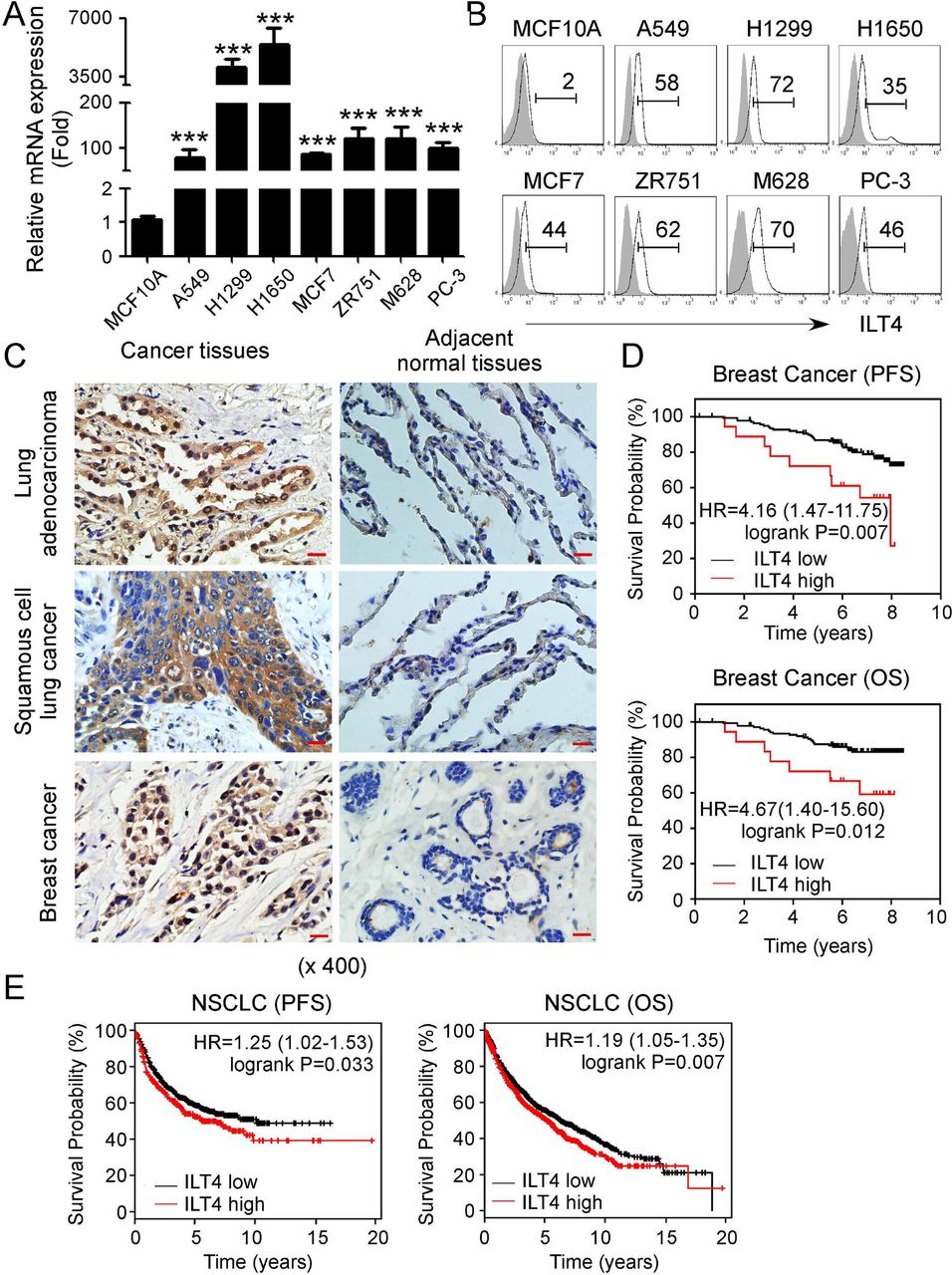
Increased ILT4 expression has been identified in certain types of cancers.19 21 37 38 To determine whether ILT4 is a general prognosis biomarker for malignant tumors, we first detected ILT4 gene expression in different human tumor cell lines, including lung cancer (A549, H1299 and H1650), breast cancer (MCF7 and ZR751), melanoma M628, and prostate cancer PC-3 cells. We found high expression of ILT4 mRNA in all the tumor cell lines compared with that in the normal epithelial cells (MCF10A) (figure 1A). The gene expression results of ILT4 were further confirmed in its protein expression levels in those tumor cell lines (figure 1B). We also detected PIR-B (ILT4 ortholog) expression levels in mouse lung cancer (MLE12 and LL/2), breast cancer (E0771) and melanoma (B16F0) cell lines. Consistent with ILT4 expression in human cancer cells, both gene and protein expressions of PIR-B were also abundant in mouse tumor cells (online supplemental figure 1A,B). To further determine ILT4 expression in the TME, we collected tumor tissues from patients with lung cancer and breast cancer and performed histochemical staining. Our results demonstrated that ILT4 was strongly expressed in the tumor tissues rather than in adjacent normal tissues in both tumor types (figure 1C). These results clearly indicate an accumulated expression of ILT4 in tumors.

Upregulated immunoglobulin-like transcript 4 (ILT4) expression in human tumors predicts poor patient survival. (A, B) Gene and protein expression levels of ILT4 were upregulated in different human cancer cell lines using real-time quantitative PCR (qPCR) and flow cytometry analyses. Tumor cell lines included non-small cell lung cancer (NSCLC) (A549, H1299, H1650), breast cancer (MCF7, ZR751), melanoma (M628), and prostate cancer (PC-3). The mammary gland endothelial cell line (MCF10A) was included as a control. mRNA levels in each cell line were normalized to the relative quantity of β-actin expression and then adjusted to ILT4 level in MCF10A cells (served as 1). Results shown in the histogram are mean ± SD from three independent experiments. ***p<0.001 compared with MCF10A cells (in A). Protein levels in cell lines were determined using the flow cytometry analysis (in B). (C) ILT4 expression was highly increased in NSCLC and breast cancer tissues but rare in the adjacent normal tissues analyzed by the immunohistochemical staining. The positive staining was displayed as brown granules and mainly observed in the membrane and cytoplasm of tumor cells. Scale bar: 20 µm. (D, E) ILT4 expression levels in human breast cancer (in D) and NSCLC (in E) tissues were negatively associated with patient progress-free survival (PFS) and overall survival (OS). Transcriptional expression levels of ILT4 and patient survival information in patients with breast cancer and NSCLC were obtained from the GEO database. One hundred fifty-nine patients with breast cancer from GSE1456 dataset were included for PFS and OS analysis with the cut-off value of 6.37. While the sample sizes for pooled PFS and OS analysis in patients with NSCLC were 1926 and 982, respectively, and the best cut-off values were auto-selected by the online tool of KM-plotter database. The relationships between ILT4 and patient survivals were further verified by the Kaplan-Meier survival analysis and the log-rank (Mantel-Cox) test.
To explore the role of ILT4/PIR-B in the pathogenesis of tumor development, we determined the effects of PIR-B gene expression levels (overexpression or knockdown) on mouse tumor cell proliferation, adhesion and migration of breast cancer and melanoma. The efficiencies of overexpression and knockdown of PIR-B in mouse cell lines were determined using infection of lentivirus carrying PIR-B-shRNA, control shRNA, PIR-B or control vector at the MOI of 5–10 for 48 hours (online supplemental figure 2A,B). Furthermore, knockdown of PIR-B gene expression with the lentivirus-based shRNAs dramatically inhibited tumor proliferation and growth of both E0771 and B16F0 tumor cells (online supplemental figure 2C). In contrast, overexpression of PIR-B gene in tumor cells significantly promoted the tumor cell growth during the culture. Furthermore, knockdown of PIR-B gene expression strongly inhibited the abilities of tumor cell adhesion and migration of E0771 and B16F0 tumor cells, whereas overexpression of PIR-B in tumor cells promoted those biological behaviors (online supplemental figure 2D,E). These results suggest that ILT4 is a critical molecule to drive tumor growth and progression.
We further retrospectively analyzed the correlations between ILT4 expressions in tumor tissues and clinical outcomes in patients with cancer using the Gene Expression Omnibus (GEO) database. We observed that high ILT4 expression indicated shortened PFS and OS in patients with breast cancer and NSCLC (figure 1D and E). Collectively, enriched ILT4 expression in tumor cells promotes tumor growth and biologic functions, and predicts poor outcomes of patients with cancer.
Tumor-derived ILT4 is responsible for the induction of T cell senescence
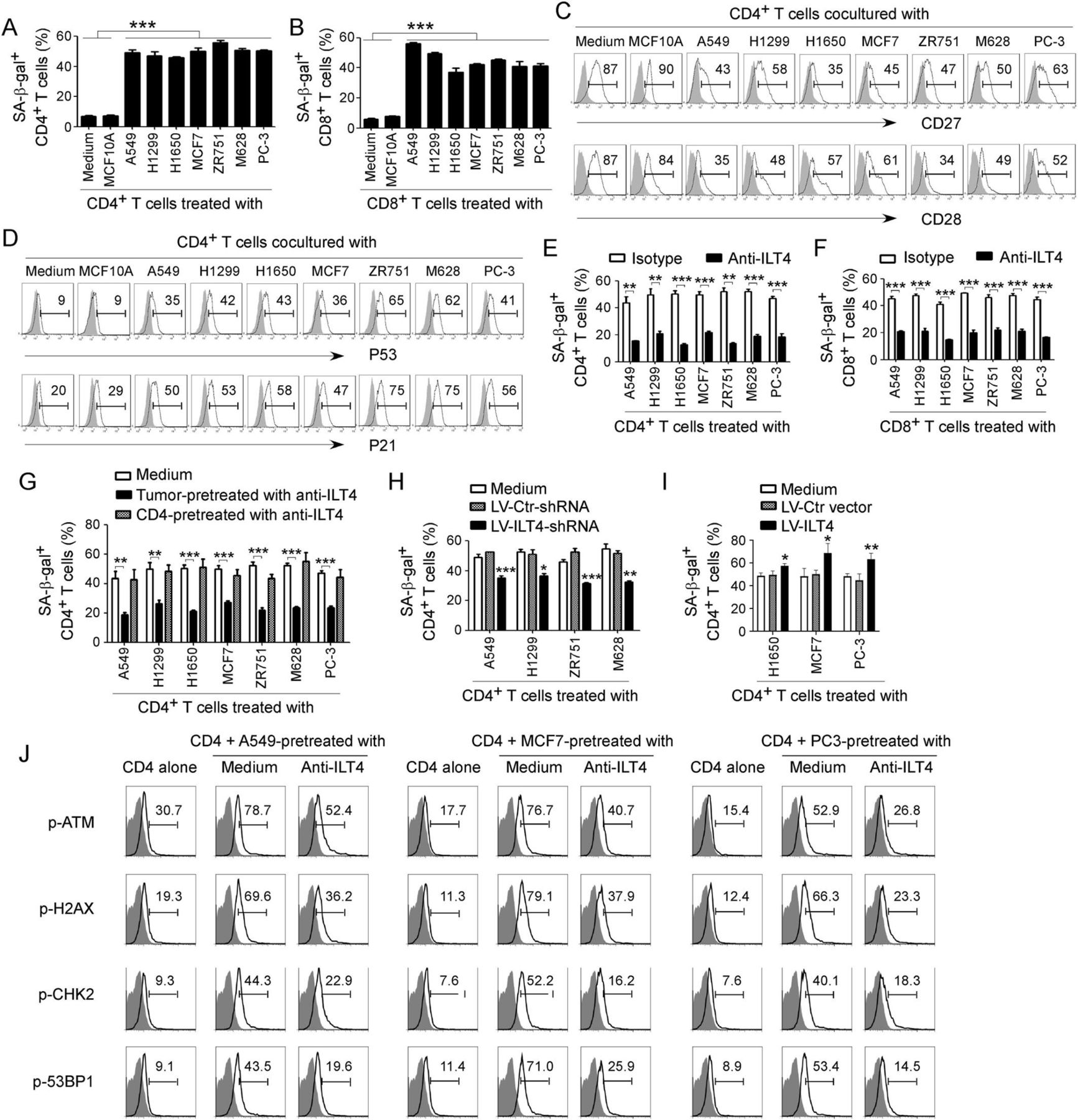
Recent studies have identified that induction of T cell senescence mediated by malignant tumors is a novel mechanism for tumor immune escape.25 39 We then determined the generality of T cell senescence induced by different types of tumor cells. We found that coculture with different tumor cells, including lung, breast, melanoma and prostate cancers, significantly promoted the development of senescent CD4+ and CD8+ T cell populations in cocultures (figure 2A and B and online supplemental figure 3A). Furthermore, coculture with different types of tumor cells dramatically downregulated expression of costimulatory molecules CD27 and CD28, and increased the expression of cell cycle regulatory molecules P53 and P21 in senescent T cells (figure 2C and D and online supplemental figure 3B,C).23 27 28 Recent studies have shown that increased expression of ILT4 in the TME is associated with the reduced TILs in patients with cancer, suggesting that ILT4 could be an immune inhibitory molecule.16 19 22 37 40 Therefore, we reasoned that tumor-derived ILT4 could be involved in senescence induction in T cells mediated by tumor cells.25 26 To test this possibility, we determined whether the development of T cell senescence can be prevented if we block ILT4 activity in the cocultures. We observed that neutralization of ILT4 significantly decreased both senescent CD4+ and CD8+ T cell populations and prevented the loss of CD27 and CD28 expression in senescent T cells mediated by tumor cells (figure 2E and F and online supplemental figure 3D,E).
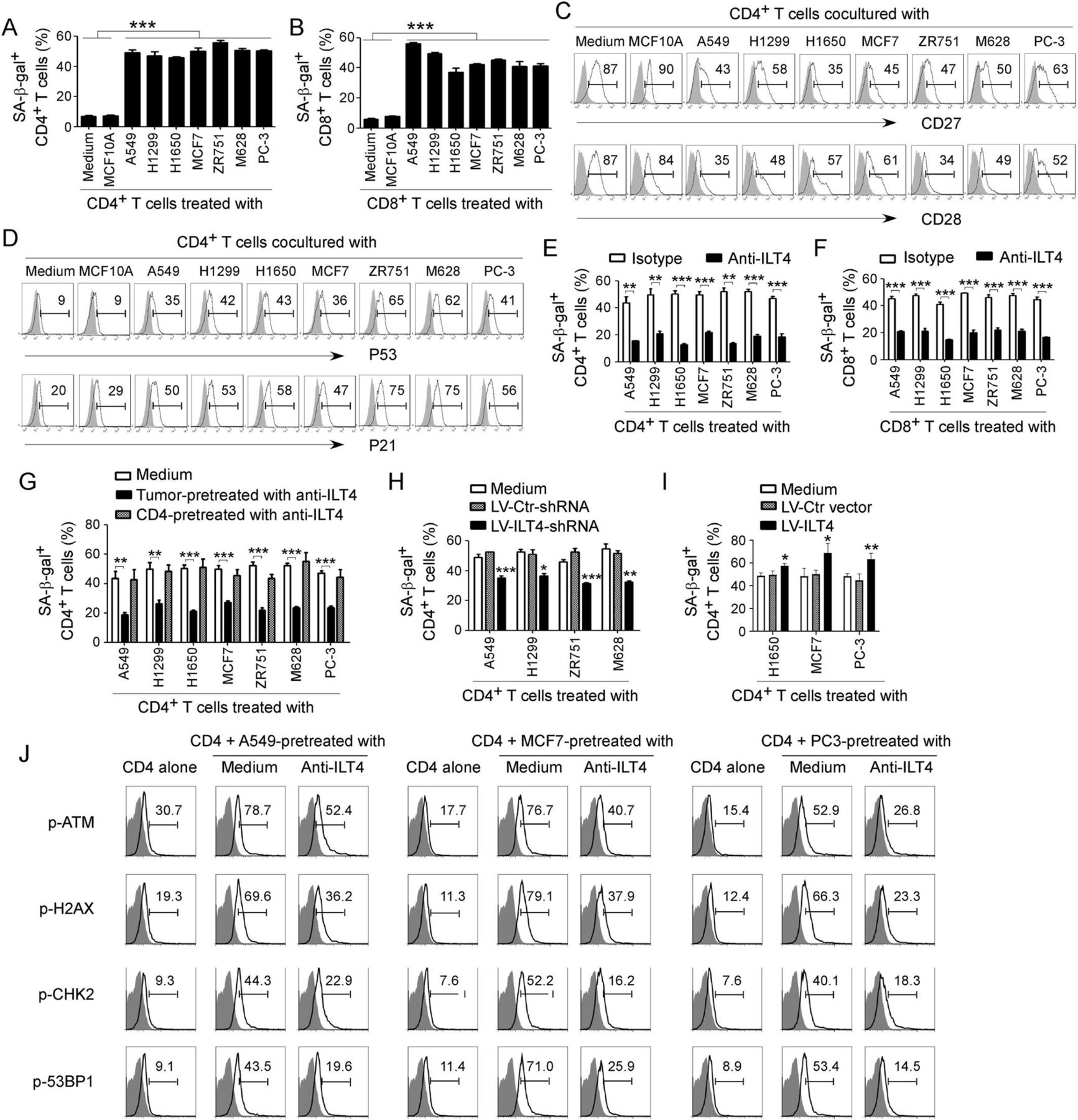
Tumor-derived immunoglobulin-like transcript 4 (ILT4) is responsible for T cell senescence. (A, B) Coculture with different tumor cells significantly increased SA-β-Gal+ T cell populations in cocultured CD4+ (in A) and CD8+ (in B) T cells. However, T cells cocultured with the breast epithelial cell line MCF10A had no or little senescence-associated β-galactosidase (SA-β-Gal) expression. Anti-CD3 pre-activated naïve T cells were cocultured with tumor cells or control MCF10A cells at a ratio of 1:1 for 24 hours. Cocultured CD4+ and CD8+ T cells were then separated and cultured for additional 3 days for SA-β-Gal activity analyses. Tumor cell lines included non-small cell lung cancer (NSCLC) (A549, H1299 and H1650), breast cancer (MCF7 and ZR751), melanoma (M628) and prostate cancer (PC-3). Results shown in the histograms are mean ± SD from three independent experiments. ***p<0.001. (C, D) Tumor cell treatment decreased expression of costimulatory molecules CD27 and CD28 (in C) but upregulated expression of cell cycle regulatory molecules P53 and P21 (in D) in senescent T cells. Cell treatment and procedure were the same as in (A). Expression levels of CD27, CD28, P53, and P21 in cocultured T cells were analyzed by the flow cytometry analyses. (E, F) Blockade of ILT4 through an anti-ILT4 neutralizing antibody remarkably prevented tumor-induced senescence in both CD4+ (in E) and CD8+ (in F) T cells. The cell coculture procedures are identical to (A) and (B). Anti-ILT4 neutralizing antibody (500 ng/mL) or isotype control antibody were included in the coculture system. Results shown in the histogram are mean ± SD from three independent experiments. **p<0.01 and ***p<0.001. (G) Tumor cells but not T cells pretreated with anti-ILT4 antibody decreased SA-β-Gal+ cell populations in cocultured CD4+ T cells. Different tumor cells or anti-CD3 pre-activated naïve CD4+ T cells were pretreated with anti-ILT4 antibody (500 ng/mL) for 2 hours, and then cocultured with untreated T cells or tumor cells, respectively, as described in (A). Results shown in the histogram are mean ± SD from three independent experiments. **p<0.01 and ***p<0.001. (H) Knockdown of ILT4 in tumor cells prevented T cell senescence induced by tumor cells. Tumor cell lines (A549, H1299, ZR751 and M628) with ILT4 high expression were infected with lentivirus carrying ILT4 shRNA or control shRNA at the multiplicity of infection (MOI) of 5–10 for 48 hours and then cocultured with T cells as described in (A) and (B). SA-β-Gal+ cell populations in cocultured CD4+ T cells were determined. Results shown in the histogram are mean ± SD from three independent experiments. *p<0.05, **p<0.01, and ***p<0.001, compared with the control shRNA (LV-Ctr-shRNA) group. (I) Overexpression of ILT4 in tumor cells promoted T cell senescence induced by tumor cells. Tumor cell lines (H1650, MCF7, and PC-3) with ILT4 low expression were infected with lentivirus carrying ILT4 or control vector at the MOI of 5–10 for 48 hours and then cocultured with T cells as described in (A) and (B). SA-β-Gal+ cell populations in cocultured CD4+ T cells were determined. Results shown in the histogram are mean ± SD from three independent experiments. *p<0.05 and **p<0.01, compared with the control vector group. (J) Blockade of ILT4 prevented tumor-induced DNA damage response in CD4+ T cells. Different tumor cells were pretreated with anti-ILT4 neutralizing antibody (500 ng/mL) for 2 hours, then cocultured with anti-CD3 pre-activated naïve CD4+ T cells at the ratio of 1:1 for 24 hours. Cocultured CD4+ T cells were separated and cultured for additional 3 days. Phosphorylation levels of ATM, H2AX, CHK2, and 53BP1 in cocultured T cells were analyzed by the flow cytometry analyses.
To elucidate the sources of ILT4 responsible for T cell senescence, we pretreated the tumor or T cells with anti-ILT4 neutralizing antibody, and then cocultured with T cells or tumor cells, respectively. We observed that only pretreatment of tumor cells but not T cells with the neutralizing antibody can prevent induction of T cell senescence mediated by tumor cells (figure 2G). To further explore the functional role of ILT4 in tumor-induced T cell senescence, we downregulated or overexpressed ILT4 gene expression in tumor cells and then determined its effect on T cell senescence induction. Tumor cell lines infected with lentivirus carrying ILT4 shRNA or ILT4 gene at the MOI of 5–10 for 48 hours showed significant downregulation or upregulation of ILT4 gene expression levels in human tumor cells, respectively (online supplemental figure 3F,G). In addition, we observed that knockdown of ILT4 expression in tumor cells (ILT4 protein high expression) with lentivirus-based shRNAs significantly prevented tumor-induced T cell senescence (figure 2H and online supplementary figure 3H). In contrast, overexpression of ILT4 in tumor cells (ILT4 protein low expression) promoted tumor-induced T cell senescence (figure 2I and online supplementary figure 3I). The results clearly suggest that tumor-derived ILT4 is the key molecule involved in tumor-induced T cell senescence.
Our studies have shown that initiation of ATM-associated DNA damage is the cause for T cell senescence and dysfunction induced by both human Treg cells and tumor cells.25 27 We therefore determined whether ILT4 regulates ATM-associated DNA damage responses, resulting in T cell senescence induced by tumor cells. We observed that different types of tumor cells significantly induced activation and phosphorylation of DNA damage molecules in responder CD4+ T cells, including ATM, H2AX, 53BP1, and CHK2 (figure 2J). Furthermore, functional blockage of ILT4 in tumor cells with the neutralizing antibody can markedly prevent phosphorylated activation of DNA damage molecules in senescent CD4+ T cells mediated by tumor cells (figure 2J). These results identify the mechanistic relationships among the ILT4 regulation, DNA damage initiation, and cell senescence in T cells during the crosstalks between tumors and T cells.
HLA-G is an important ligand for ILT4 to induce T cell senescence
Increasing evidence suggests that crosstalk between ILT4 and specific ligands is required for its unique biological functions.22 HLA-G is an important ligand for ILT4, which is abundant in tumor cells and participates in immunosuppression and increases migration and metastasis of tumor cells.41 42 HLA-G is also expressed in T cells and involves T cell suppressive function.43 44 Therefore, we reasoned that ILT4-HLA-G ligation is required for tumor-induced T cell senescence. We found that HLA-G is highly expressed in naïve, activated, and senescent CD4+ and CD8+ T cells (figure 3A). Furthermore, HLA-G is also expressed in different types of tumor cells (figure 3B). To identify the role of HLA-G in tumor-derived ILT4-mediated T cell senescence, we added the neutralizing antibody against HLA-G activity and then determined whether we can prevent T cell senescence induced by tumor cells. Our studies clearly showed that neutralization of HLA-G significantly prevented T cell senescence induced by different types of cancer cells (figure 3C). We then determined whether tumor-derived HLA-G or T cell-derived HLA-G is responsible for ILT4-mediated T cell senescence. We pretreated T cells or tumor cells with anti-HLA-G neutralizing antibody, and then cocultured with tumor cells or T cells, respectively. We found that pretreatment of either tumor cells or T cells with the neutralizing antibody can prevent induction of T cell senescence mediated by tumor cells, suggesting that tumor-derived ILT4 might regulate both tumor and T cells for the induction of T cell senescence (figure 3D and E). In addition, pretreatment of T cells with the HLA-G neutralizing antibody dramatically suppressed the tumor-induced activation and phosphorylation of ATM-associated DNA damage molecules in responder CD4+ T cells, including ATM, H2AX, CHK2, and 53BP1 (figure 3F). These studies collectively suggest that ILT4-HLA-G ligation is critical for controlling tumor cell-induced T cell senescence in the TME.

HLA-G is important for tumor-derived immunoglobulin-like transcript 4 (ILT4)-induced T cell senescence. (A) HLA-G is expressed in naïve, activated and senescent CD4+ and CD8+ T cells. Naïve CD4+ and CD8+ T cells were purified from peripheral blood mononuclear cells (PBMCs) of healthy donors. Activated T cells were obtained from naïve T cells cultured in the plate-coated anti-CD3/CD28 (2 µg/mL) for 24 hours. Senescent T cells were induced from pre-activated naïve CD4+/CD8+ T cells cocultured with human lung cancer A549 cells as described in figure 2A. HLA-G expression was determined by intracellular staining with the flow cytometry analysis. (B) HLA-G expression in human non-small cell lung cancer (NSCLC) (A549, H1299 and H1650), breast cancer (MCF7 and ZR751), melanoma (M628) and prostate cancer (PC-3) cell lines determined by surface staining with the flow cytometry analysis. (C) Neutralization of HLA-G significantly prevented tumor-induced CD4+ T cell senescence. Anti-CD3/CD28 pre-activated naïve CD4+ T cells were cocultured with different tumor cells at the ratio of 1:1 for 24 hours in the presence of 10 µg/mL anti-HLA-G neutralizing antibody or isotype antibody in the coculture system. CD4+ T cells were then separated and senescence-associated β-galactosidase (SA-β-Gal) activity was determined as described in figure 2. Results shown in the histogram are mean ± SD from three independent experiments. **p<0.01 and ***p<0.001. (D, E) Blockade of HLA-G either in CD4+ T cells (in D) or tumor cells (in E) decreased tumor-induced CD4+ T cell senescence. Anti-CD3 pre-activated CD4+ T cells or tumor cells were pretreated with 10 µg/mL anti-HLA-G neutralizing antibody for 2 hours, and then cocultured with respective untreated tumor or T cells, as described in figure 2. T cell senescence was determined by SA-β-Gal staining. Results shown in the histogram are mean ± SD from three independent experiments. *p<0.05, **p<0.01 and ***p<0.001. (F) Neutralization of HLA-G significantly prevented tumor-induced DNA damage response in CD4+ T cells. Anti-CD3 pre-activated CD4+ T cells were treated anti-HLA-G neutralizing antibody and then cocultured with different tumor cells as described in (D). Phosphorylation levels of ATM, H2AX, CHK2, and 53BP1 in cocultured T cells were analyzed with the flow cytometry analyses.
We next explored whether ILT4 expression status controls HLA-G expression in tumor cells. We found that knockdown of ILT4 in lung (A549 and H1299) and breast cancer (ZR751) cell lines with high ILT4 expression significantly decreased surface expression but not intracellular expression of HLA-G in these tumor cells (online supplemental figure 4A,B). In addition, overexpression of ILT4 in melanoma (M628), breast cancer (MCF7) and prostate cancer (PC-3) cell lines also promoted surface expression but not intracellular expression levels of HLA-G in the tumor cells (online supplemental figure 4C,D).
ILT4 promotes lipid metabolism in tumor cells, resulting in T cell senescence
Recent studies have shown that tumor cells require increased energy metabolism to support their rapid growth and progression, which causes nutrient deprivation or toxic metabolite accumulation that in turn restrict T cell function.45 46 Furthermore, we have identified that tumor-derived cAMP is directly involved in T cell senescence induction mediated by tumor cells.25 These studies prompted us to determine whether ILT4 can regulate tumor cell metabolism and subsequently induce T cell senescence. We determined the changes of the key metabolic enzyme genes involving glucose and lipid metabolism in tumor cells after neutralization of ILT4 using real-time quantitative PCR analyses.28 Those molecules include glucose transporters Glut1 and Glut3, glycolysis-related enzymes hexokinase 2 (HK2), glucose-6-phosphate isomerase (GPI), phosphofructokinase 1 (PFK1), triosephosphate isomerase 1 (TPI1), enolase 1 (ENO1), pyruvate kinase muscle 2 (PKM2), as well as lipid-related enzymes 3-hydroxy-3-methyl-glutaryl-CoA reductase (HMGCR), 3-hydroxy-3-methylglutaryl-CoA synthase 1 (HMGCS1), squalene monooxygenase (SQLE), isopentenyl-diphosphate delta isomerase 1 (IDI1), carnitine palmitoyltransferase I (CPT-1), fatty acid synthase (FASN) and acetyl-CoA carboxylases 1 (ACC1).28 We found that mRNA expression levels of the enzymes involved in glucose and cholesterol metabolism were varied in different tumor cells induced by ILT4 neutralization and we did not obtain consistent alteration results (online supplemental figure 5A). However, ILT4 neutralization in tumor cells significantly downregulated gene expression of limiting enzymes ACC1 and FASN in de novo fatty acid synthesis (figure 4A). We further used the gain-of-function and loss-of-function strategies and demonstrated that knockdown of ILT4 with shRNAs in tumor cells significantly decreased ACC1 and FASN expression in tumor cells (figure 4B). In contrast, overexpression of ILT4 in tumor cells markedly increased ACC1 and FASN expression in tumor cells (figure 4C). We next determined whether PIR-B has a similar role as ILT4 in regulation of lipid metabolism in mouse tumor cells. However, unlike ILT4, knockdown of PIR-B in B16F0 cells significantly decreased gene expressions in enzymes involved in both fatty acid and cholesterol metabolism (online supplemental figure 5B). Furthermore, overexpression of PIR-B in E0771 cells increased the gene expression of those enzymes (online supplemental figure 5C).

Tumor-derived immunoglobulin-like transcript 4 (ILT4) promotes T cell senescence via upregulation of tumor lipid metabolism. (A) Neutralization of ILT4 significantly reduced gene expression of key enzymes for fatty acid synthesis (ACC1 and FASN) in tumor cells. Human tumor cell lines were treated with anti-ILT4 neutralizing antibody (500 ng/mL) or isotype control antibody for 48 hours, and mRNA expression levels of ACC1 and FASN were determined by real-time quantitative PCR (qPCR). The expression of ACC1 and FASN genes was normalized to β-actin and adjusted to the levels in corresponding isotype antibody-treated cells (served as 1). Results shown are mean ± SD from at least three independent experiments. *p<0.05, **p<0.01 and ***p<0.001, compared with the cells treated with isotype antibody. (B) Knockdown of ILT4 in tumor cells downregulated expression levels of ACC1 and FASN by tumor cells. Tumor cell lines (A549, ZR751 and M628) were infected with lentivirus carrying ILT4 shRNA or control shRNA at the multiplicity of infection (MOI) of 5–10 for 48 hours and then mRNA expression of ACC1 and FASN were analyzed by real-time qPCR. The expression levels of ACC1 and FASN gene were normalized to β-actin and adjusted to the level in respective control group (served as 1). Results shown in the histogram are mean ± SD from at least three independent experiments. *p<0.05, compared with the control shRNA (LV-Ctr-shRNA)-transfected cells. (C) Overexpression of ILT4 in tumor cells upregulated expression levels of ACC1 and FASN by tumor cells. Tumor cell lines (H1650, MCF7, and M628) were infected with lentivirus carrying ILT4 or control vector at the MOI of 5–10 for 48 hours and then mRNA expression of ACC1 and FASN were analyzed by real-time qPCR as described in (B). *p<0.05 and **p<0.01, compared with the respective cells infected with lentivirus carrying control vector. (D, E) Neutralization of ILT4 significantly reduced lipid droplet (LD) formation in tumor cells. Different types of tumor cell lines were treated with anti-ILT4 neutralizing antibody (500 ng/mL) or isotype control antibody for 48 hours, and then stained for Oil red O. For cell lines with high LD accumulation (H1299, H1650, MCF7 and PC-3), positive cells were counted based on the cells with LDs of more than 1/3 cytoplasm. Results in (D) showed the typical images of tumor cells with the Oil red O staining. Results shown in (E) are mean ± SD from three independent experiments. *p<0.05, **p<0.01 and ***p<0.001. (F) Knockdown of ILT4 in tumor cells decreased LDs in tumor cells. Tumor cell lines were infected with lentivirus carrying ILT4 shRNA or control shRNA at the MOI of 5–10 for 72 hours and then stained for Oil red O. Results shown are mean ± SD from three independent experiments. *p<0.05, **p<0.01 and ***p<0.001, compared with the control shRNA (LV-Ctr-shRNA)-transfected cells. (G) Overexpression of ILT4 in tumor cells promoted LD accumulation in tumor cells. Tumor cell lines (H1650, MCF7, and M628) were infected with lentivirus carrying ILT4 at the MOI of 5–10 for 72 hours and then stained for Oil red O. Results shown are mean ± SD from three independent experiments. **p<0.01 and ***p<0.001, compared with the respective cells infected with lentivirus carrying control vector. (H, I) Inhibition of FASN activity in tumor cells reversed ILT4-induced CD4+ (in H) and CD8+ (in I) T cell senescence. Tumor cells were pretreated with FASN inhibitor C75 (5 µM) for 24 hours, and then infected with lentivirus carrying ILT4 for 48 hours in the presence of C75. The transfected cells were further cocultured with pre-activated T cells and T cell senescence were determined as described in figure 2. Results shown are mean ± SD from three independent experiments. ***p<0.001, compared with the LV-Ctr vector-transfected cells. ###p<0.001, compared with the corresponding LV-ILT4-transfected cells.
Our studies clearly suggest that tumor-derived ILT4 facilitates fatty acid biosynthesis in tumor cells, which is a key component for lipid droplets (LDs) that are lipid-rich cytoplasmic organelles directly regulating inflammation and cancer.47 48 Therefore, we determined whether ILT4 can regulate LD formation in tumor cells using the Oil red O staining. As expected, we observed significant accumulation of Oil red O+ tumor cell populations in different types of tumor cell lines (figure 4D and E). However, neutralization of ILT4 markedly reduced LD accumulation in tumor cells, suggesting that ILT4 promotes lipid storage in tumor cells (figure 4D and E). Furthermore, knockdown of ILT4 with shRNAs or overexpression of ILT4 in different tumor cell lines dramatically decreased or increased LD formation and accumulation in tumor cells, respectively (figure 4F and G). To further dissect the causative relationship between ILT4-induced upregulation of tumor fatty acid synthesis and induction of T cell senescence, we blocked FASN activity in tumor cells with a pharmacological inhibitor C75 and then determined ILT4 overexpression-induced T cell senescence. Consistent with previous results shown in figure 2, overexpression of ILT4 significantly promoted tumor-induced T cell senescence. However, blockage of fatty acid synthesis with C75 dramatically prevented ILT4-induced promotion of T cell senescence in both CD4+ and CD8+ T cells mediated by tumor cells (figure 4H and I). In addition, blockage of fatty acid synthesis significantly prevented ILT4 overexpression-induced tumor cell proliferation and adhesion abilities (online supplemental figure 5D-F). Collectively, our results clearly indicate that tumor-derived ILT4 promotes lipid metabolism of tumor cells, which is responsible for tumor-induced T cell senescence.
ILT4 controls tumor fatty acid synthesis and T cell senescence via activation of ERK1/2 signaling
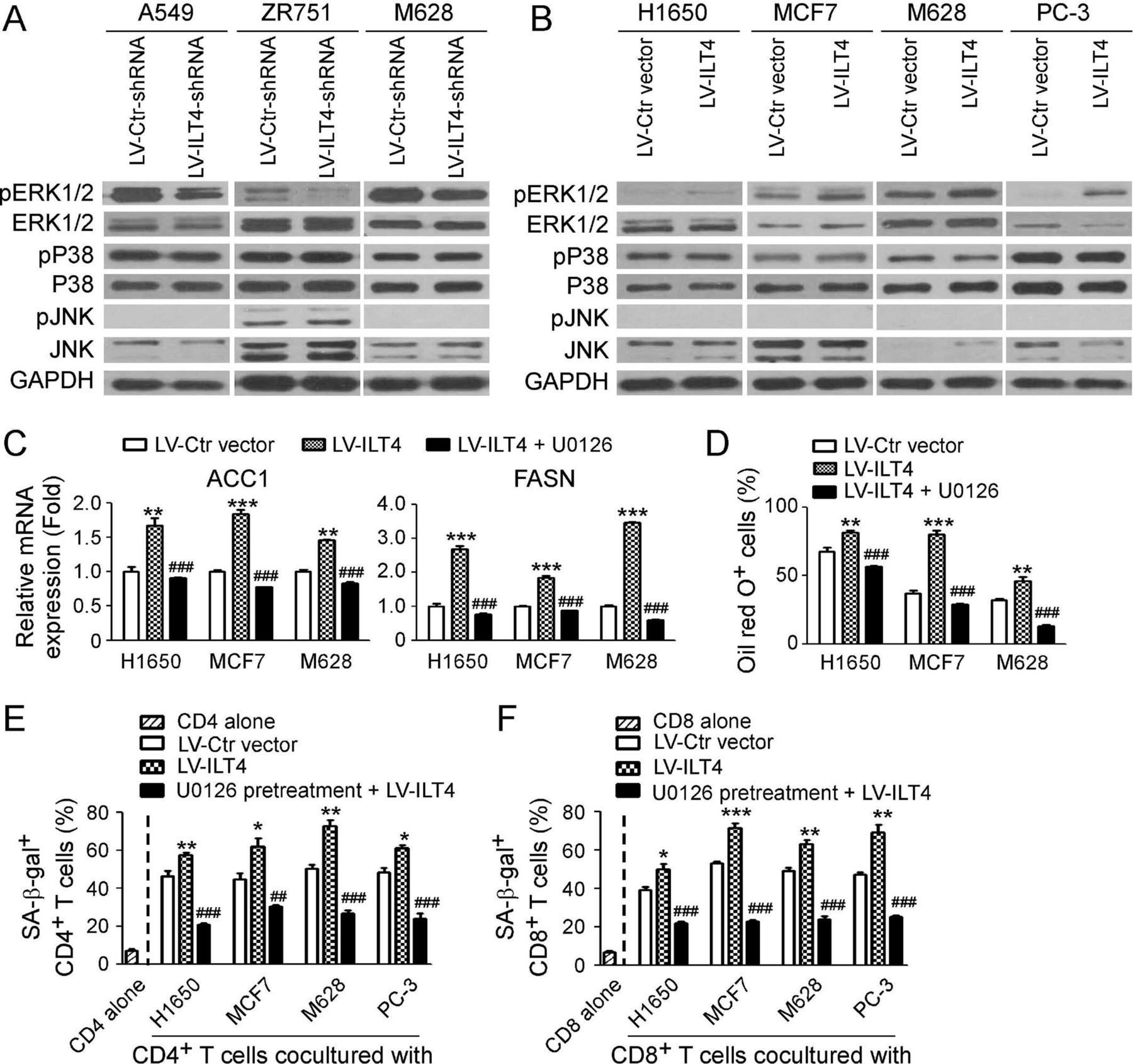
We next explored the downstream signaling in tumor cells regulated by ILT4 to promote lipid metabolism and induce T cell senescence. Previous studies have shown that ILT4 in tumor cells or DCs can potentially activate ERK1/2, AKT-mTOR, NF-κB or CaMK signaling to perform its biological functions.22 49 Therefore, these signaling pathways were priorities for our study. We set two panels of tumor cell lines with knockdown or overexpression of ILT4, respectively. As shown in figure 5A, knockdown of ILT4 in A549, ZR751 and M628 tumor cells with shRNAs significantly downregulated phosphorylation of ERK1/2 but not P38 and JNK. Furthermore, overexpression of ILT4 in H1650, MCF7, M628 and PC-3 tumor cells dramatically promoted phosphorylation of ERK1/2 but not P38 and JNK (figure 5B). Notably, knockdown or overexpression of ILT4 did not alter activation and expression of p-AKT and p-mTOR levels (data not shown). We then functionally blocked ERK1/2 signaling with specific pharmacological inhibitor U0126 and further confirmed whether ERK1/2 signaling is involved in control of ILT4-promoted lipid metabolism of tumor cells and T cell senescence. We found that overexpression of ILT4 significantly increased gene expressions of ACC1 and FASN, and promoted LD formation in H1650, MCF7 and M628 tumor cells. However, pretreatment of tumor cells with U0126 dramatically prevented ILT4-induced promotion of ACC1 and FASN gene expression and LD accumulation in the tumor cells (figure 5C and D). In addition, blockage of ERK1/2 signaling with U0126 in tumor cells markedly decreased both senescent CD4+ and CD8+ T cell populations induced by the ILT4-overexpressed tumor cells (figure 5E and F). Our studies clearly demonstrate that ERK signaling in tumor cells is critical for regulation of ILT4-mediated lipid metabolism promotion in tumor cells and cell senescence induction in responder T cells.
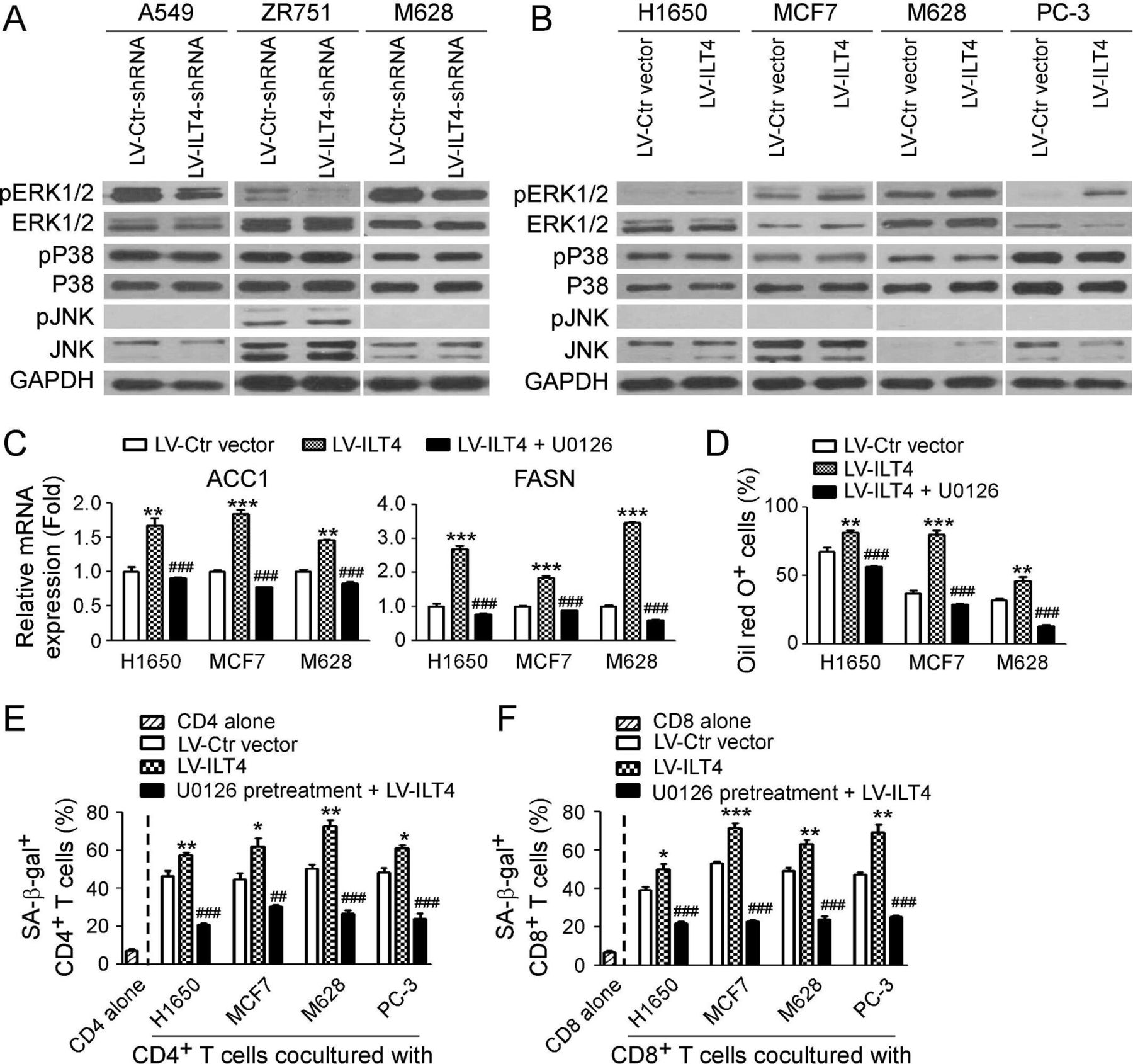
Immunoglobulin-like transcript 4 (ILT4) controls fatty acid synthesis and T cell senescence via ERK1/2 signaling pathway. (A) Knockdown of ILT4 expression decreased the phosphorylation of ERK1/2 but not P38 or JNK in tumor cells. Tumor cells were transfected with LV-ILT4-shRNA or LV-Ctr-shRNA for 72 hours, and the whole cell lysates were prepared for analysis of phosphorylated and total ERK1/2, P38, and JNK protein expression by the western blot. (B) Overexpression of ILT4 promoted activation and phosphorylation of ERK1/2 but not P38 or JNK in tumor cells. Tumor cells were transfected with lentivirus-based ILT4 or control vector for 72 hours, and the whole cell lysates were prepared for analysis of phosphorylated and total ERK1/2, P38, and JNK protein expression by the western blot. (C, D) Inhibition of ERK signaling restored ILT4-induced upregulation of ACC1 and FASN gene expression (in C) and accumulation of lipid droplets (LDs) (in D) in tumor cells. Different types of tumor cells were pretreated with ERK inhibitor U0126 (10 µM) for 24 hours, and then transfected with lentivirus-based LV-ILT4 or control LV-Ctr vector for 72 hours in the presence of 10 μM U0126. The gene expression levels of ACC1 and FASN were evaluated using the real-time qPCR (in C). LD accumulation was determined by the Oil red O staining (in D). Results shown in (C) and (D) are mean ± SD from three independent experiments. **p<0.01 and ***p<0.001, compared with cells transfected with the LV-Ctr vector. ###p<0.001, compared with the cells transfected with the LV-ILT4. (E, F) U0126 pretreatment in tumor cells prevented ILT4-induced CD4+ (in E) and CD8+ (in F) T cell senescence. Different types of tumor cells were pretreated with ERK inhibitor U0126 (10 µM) for 24 hours and then infected with lentivirus carrying ILT4 or control vector for 72 hours in the presence of U0126 (10 µM). Treated tumor cells were further cocultured with pre-activated T cells as described in figure 2, and SA-β-Gal activity in T cells was determined. Results shown in the histogram are mean ± SD from three independent experiments. *p<0.05, **p<0.01 and ***p<0.001, compared with the respective LV-Ctr vector group. ##p<0.01 and ###p<0.001, compared with the respective LV-ILT4 group.
Tumor-derived PIR-B promotes tumor growth and induces T cell senescence in vivo
Our current in vitro studies have indicated that tumor-derived ILT4 is the cause of T cell senescence. In addition, ILT4 can also promote tumor growth, adhesion and transmigration. We next explored whether ILT4 is critical for controlling tumor development and T cell senescence in vivo. We first used the mouse E0771 breast cancer model in immunocompetent C57BL/6 mice to determine how PIR-B in tumor cells (human ILT4 homolog) affects tumor development and T cell functions.35 E0771 tumor cells (2 × 105 cells/mouse) infected with lentivirus carrying PIR-B gene (overexpression), shRNAs against PIR-B (knockdown), or control vector (control) were injected subcutaneously into female C57BL/6 mice. Tumor growth was evaluated. At the end of the experiments, tumors were isolated from the sacrificed mice and weighed. Furthermore, T cells in tumor-bearing mice were purified and recovered from different groups and organs at the end of experiments, and analyzed for the effects on T cell functions and senescence induction.
E0771 tumor cells infected with lentivirus carrying LV-Ctr vector grew progressively in mice. However, knockdown of PIR-B expression in tumor cells significantly inhibited E0771 tumor growth. In contrast, overexpression of PIR-B markedly accelerated tumor growth in vivo (figure 6A). Furthermore, tumor sizes collected from the E0771 with PIR-B knockdown group (LV-PIR-B-shRNA) on day 24 post inoculation were significantly smaller than those in control vector group of E0771 tumor, while tumor sizes from the E0771 cells with PIR-B overexpression group (LV-PIR-B) were much larger than those in control vector infection group (figure 6B). In addition, we obtained consistent results of the average tumor weights from the three groups (figure 6C). We confirmed the molecular and functional changes of recovered T cells in vivo from different organs and tumors. Consistent with the results shown in in vitro studies, we observed significantly increased senescent T cell (SA-β-Gal+) populations in T cells obtained from blood and TILs in tumor-bearing mice compared with blood T cells from normal tumor-free mice (figure 6D). Furthermore, overexpression of PIR-B in tumor cells significantly promoted induction of senescent T cells, while knockdown of PIR-B markedly prevented T cell senescence (figure 6D). These results clearly suggest that tumor cells can induce T cell senescence in vivo, and PIR-B on tumor cells is responsible for tumor-induced T cell senescence.25 We also accessed whether PIR-B expression levels affect T cell numbers in tumor tissues. We did not observe changes of total T cell numbers in TILs from three tumor-bearing groups regardless of PIR-B expression (figure 6E). However, knockdown of PIR-B in tumor cells increased fraction of CD8+ T cells rather than CD4+ T cells in the TILs in tumor-bearing mice (figure 6F). In addition to the effect on T cell fractions, we observed that knockdown of PIR-B in tumor cells significantly augmented T cell effector functions, showing increased IFN-γ+ populations in both tumor-infiltrating CD4+ and CD8+ T cells, and granzyme B+ and perforin+ CD8+ T cell populations in TILs (figure 6G and H). In contrast, overexpression of PIR-B in tumor cells markedly suppressed production of effector molecules IFN-γ, granzyme B and perforin in T cells (figure 6G and H). Notably, we did not observe dramatic alterations of IL-17-producing CD4+ T cell and FoxP3+CD4+ T cell populations in blood and TILs of tumor-bearing mice induced by either knockdown or overexpression of PIR-B in tumors (data not shown). These results collectively suggest that ILT4/PIR-B controls T cell fate and functions in the TME.

Tumor-derived PIR-B promotes tumor growth and induces T cell senescence in vivo. (A) Knockdown of PIR-B gene in E0771 breast cancer cells markedly inhibited transplanted cancer growth in immunocompetent C57BL/6 mice. However, overexpression of PIR-B in E0771 promoted tumor growth. Mouse breast cancer cell line E0771 (2 × 105 cells/mouse) infected with lentivirus carrying PIR-B gene or shRNA against PIR-B were subcutaneously injected into female C57BL/6 mice. Tumor size was measured every 3 days and presented as mean ± SD (n=5 mice/group). (B) Representative tumor images obtained from the indicated groups at the endpoint of the experiments (day 24). (C) E0771 breast cancer cells with PIR-B knockdown had decreased tumor weights, but cancer cells overexpressing PIR-B had increased tumor weights. Results shown are mean ± SD of tumor weights from indicated groups at the endpoint of the experiments (day 24) (n=4 mice/group). **p<0.01 and ***p<0.001, compared with the LV-Ctr vector group. (D) Knockdown of PIR-B gene in E0771 breast cancer significantly prevented T cell senescence in blood and tumor-infiltrating lymphocytes (TILs). In contrast, overexpression of PIR-B in tumor cells remarkably induced T cell senescence. Lymphocytes were separated from blood and tumor tissues, and SA-β-Gal expression determined. Results are presented as mean ± SD (n=4 mice/group). *p<0.05 and ***p<0.001, compared with the LV-Ctr vector group. (E) Knockdown or overexpression of PIR-B in E0771 did not alter lymphocyte infiltration in tumor tissues. Tumor tissues from different groups were isolated and the absolute numbers of TILs in each mouse were counted and divided by the tumor volume. Data shown are mean ± SD from different groups (n=4 mice/group). (F) Knockdown of PIR-B gene in E0771 tumor cells increased CD8+ T cell populations but not CD4+ T cells in TILs. However, overexpression of PIR-B gene in tumor cells did not alter CD4+ and CD8+ T cell subsets in TILs. TILs were separated from tumor tissues of each group and fractions of T cell subsets were evaluated by the flow cytometry analysis. Results shown are mean ± SD from different groups (n=4 mice/group). **p<0.01, compared with the LV-Ctr vector group. (G, H) Knockdown of PIR-B gene in E0771 breast cancer significantly increased IFN-γ+ cell populations in both tumor-infiltrating CD4+ and CD8+ T cells (in G), and granzyme B+ and perforin+ cell populations in tumor-infiltrating CD8+ T cells (in H). In contrast, overexpression of PIR-B in tumor cells dramatically decreased those populations in tumor-infiltrating CD4+ and CD8+ T cells. TILs were separated from tumor tissues, and different T cell subsets/populations were analyzed by the flow cytometry. Data shown are mean ± SD from different groups (n=4 mice/group). *p<0.05 and **p<0.01, compared with the LV-Ctr vector group. (I–K) Knockdown of PIR-B gene in E0771 cells inhibited tumor growth and decreased tumor sizes and weights in immunodeficient NSG mice. However, overexpression of PIR-B gene in tumor cells promoted tumor growth and increased tumor sizes and weights in NSG mice. Cell preparation, cell injection numbers and procedures were identical to descriptions in (A). Tumor sizes were measured every 3 days (in I). Mice were sacrificed and tumors in different groups were separated and weighed (in J and K) at day 22 after tumor injection. Data shown in (I) and (K) are mean ± SD from different groups (n=4 mice/group). **p<0.01 and ***p<0.001, compared with the LV-Ctr vector group (in K). (L) PIR-B knockdown in tumor cells downregulated the gene expression levels of key enzymes involved in lipid metabolism, while PIR-B overexpression enhanced their expression levels in tumor cells from NSG mice. Total RNAs were extracted from fresh tumor tissues of each tumor-bearing NSG mouse and gene expression levels of each enzyme were determined by the real-time quantitative PCR analysis. All the data are expressed as mean ± SD from different groups (n=4 mice/group). *p<0.05, **p<0.01 and ***p<0.001, compared with the LV-Ctr vector group.
To further dissect the importance of tumor-derived PIR-B-mediated regulation of T cells on tumor growth, we next performed complementary in vivo studies, using E0771 cells in humanized NSG mouse model, and explored how PIR-B directly affects tumorigenesis in vivo. We observed that overexpression or knockdown of PIR-B expression in E0771 cells can also increase or inhibit tumor growth in NSG mice, respectively (figure 6I–6K). However, differences in E0771 tumor growth and sizes between overexpression and knockdown of PIR-B groups with control vector group were much smaller than those of E0771 tumor in immunocompetent C57BL/6 mice (figure 6A–6C), suggesting that immune system is regulated by PIR-B and critical for control of tumor growth in vivo. In addition, we observed a remarkable increase or decrease in gene expression levels of the key enzymes associated with fatty acid and cholesterol metabolism in the isolated tumors from respective PIR-B overexpression or knockdown groups. These results further confirmed that PIR-B directly regulates tumor lipid metabolism (figure 6L). Collectively, these studies indicate that PIR-B is a critical inhibitory molecule for tumor immunity, and knockdown of PIR-B could be an effective strategy to suppress tumor cell metabolism and reverse T cell senescence and dysfunction for tumor treatment.
Reversal of tumor-specific effector T cell senescence by inhibiting PIR-B enhances antitumor immunity in vivo in the adoptive transfer therapy model
We further investigated the potential translation of this novel concept that inhibition of ILT4/PIR-B in tumor cells could be an effective and novel strategy for tumor immunotherapy. We used the well-established B16 melanoma and Pmel (gp100-specific) TCR transgenic (Tg) mouse models to test our hypothesis.50 B16F0 tumor cells (2 × 105 cells/mouse) infected with lentivirus carrying shRNA against PIR-B (knockdown), or control scrambled shRNA, were injected subcutaneously into C57BL/6 mice. Pre-activated gp100-specific CD8+ T cells were adoptively transferred into B16F0-bearing mice through intravenous injection at day 9 after tumor cell inoculation (tumor size over 5 × 5 mm). Tumor growth was evaluated. Furthermore, the adoptively transferred gp100-specific CD8+ T cells were purified and recovered from different groups and organs at the end of experiments, and analyzed for the effects of the PIR-B manipulation on T cell senescence and function. We observed that B16F0 tumor cells grew very fast in mice. When tumor-specific Pmel-1 T cells were adoptively transferred into mice, tumor growth was inhibited. Knockdown of PIR-B in B16F0 tumor cells with shRNAs dramatically promoted the inhibition of tumor growth mediated by gp100-specific CD8+ T cells (figure 7A). Furthermore, tumor sizes collected from the PIR-B knockdown tumor group (B16F0-LV-PIR-B-shRNA) were much smaller than those in the T cell therapy groups with B16F0 tumor alone and B16F0 tumor cells transfected with control shRNAs at day 39 post tumor inoculation (figure 7B). In addition, the average tumor weights obtained from the B16F0-LV-PIR-B-shRNA group were much lower than that from the other two control groups (figure 7C). These results suggest that knockdown of PIR-B in tumor cells enhances antitumor ability mediated by tumor-specific effector T cells. We then determined senescence induction and functional alterations in T cells obtained from different groups. Consistent with the findings in our previous report, we observed that increased senescent T cell populations in adoptively transferred Pmel T cells were induced in B16F0 tumor-bearing mice.25 Furthermore, knockdown of PIR-B in tumor cells significantly prevented senescence induction in transferred Pmel T cells in blood and tumor tissues (figure 7D). In addition, knockdown of PIR-B in tumor cells markedly increased IFN-γ production from Pmel T cells in blood and tumor tissues (figure 7E). These in vivo studies strongly suggest that blockage of PIR-B/ILT4 can prevent T cell senescence and enhance subsequent antitumor immune responses, which is a novel and effective strategy for tumor immunotherapy.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
PIR-B blockade enhances antitumor efficacy mediated by the adoptively transferred tumor-specific CD8+ T cells in vivo. (A–C) PIR-B knockdown showed enhanced antitumor immunity mediated by the adoptively transferred CD8+ T cells. Mouse melanoma cell line B16F0 cells (2 × 105 cells/mouse) infected with/without lentivirus carrying shRNA against PIR-B or control shRNA were subcutaneously injected into 6–8 week C57BL/6 mice. When the tumor diameter reached 5–6 mm, all the mice were intravenously injected with anti-CD3/anti-CD28 pre-activated Pmel CD8+ T cells (2 × 106 cells/mouse) after irradiation with a dose of 500 cGy. Tumors sizes were measured every 3 days (in A). When mice were euthanized, tumors in each group were isolated and weighed (in B and C). All the data are presented as mean ± SD (n=5 mice/group). **p<0.01, compared with the LV-Ctr-shRNA group (in C). (D) Knockdown of PIR-B in tumor cells significantly prevented senescence induction in the adoptively transferred CD8+ T cells in blood and TILs from tumor-bearing mice. The transferred Pmel CD8+ T cells were isolated from blood and tumor tissues, and stained for SA-β-Gal. Data shown are mean ± SD from different groups (n=4 mice/group). ***p<0.001, compared with the LV-Ctr-shRNA group. ###p<0.001, compared with the Pmel T only group. (E) Knockdown of PIR-B in tumor cells enhanced IFN-γ-producing cell populations in adoptively transferred CD8+ T cells in the blood and TILs. IFN-γ+CD8+ T cell populations in blood and TILs were determined by the flow cytometry analysis. Results shown are mean ± SD from different groups (n=4 mice/group). ***p<0.001, compared with the LV-Ctr-shRNA group.
Discussion
Current immunotherapies, including checkpoint blockade therapy and adoptive T cell transfer therapy, has dramatically changed therapeutic paradigm for patients with cancer and resulted in encouraging results.3–5 However, these immunotherapeutic strategies exert limited clinical benefits in patients with solid tumors due to the potent suppressive TME with dysfunctional T cells.1 2 Therefore, identification of alternative novel checkpoint molecules is urgently needed for the development of novel and effective immunotherapies to overcome the tumor intrinsic and extrinsic immune resistance in solid tumors.3–5 51 52 Our previous and current studies have clearly demonstrated that ILT4 is a potential checkpoint molecule for tumor immunotherapy.22 In this study, we further identified that tumor-derived ILT4 is involved in induction of effector T cell senescence and dysfunction mediated by tumor cells. Furthermore, we demonstrated that ILT4 in tumor cells promote fatty acid synthesis and lipid accumulation in tumor cells controlled by MAPK ERK1/2 signaling, which is responsible for T cell senescence mediated by tumor cells. In addition, our in vivo studies using both breast cancer and melanoma tumor models indicated that blockage of ILT4 can significantly prevent senescence in tumor-specific T cells and enhance antitumor efficacy for tumor immunotherapy.
Studies from our group and others have demonstrated that ILT4 is highly expressed in different types of malignant cancers in both hematopoietic and solid tumors in addition to the myeloid cells, which is positively associated with advanced disease stages and poor survivals in patients with cancer.19 21 ILT4 directly regulates cancer cell proliferation, differentiation, invasion, metastasis and epithelial-mesenchymal transformation.16 18 19 37 38 53 54 These results indicate that ILT4 is an important driver for tumor development and progression and could be a prognosis biomarker for patients. However, the molecular mechanism responsible for ILT4-mediated cancer promotion is still unclear. In this study, we discovered that ILT4 selectively regulates tumor cell lipid metabolism rather than glucose metabolism. Increasing studies suggest that glycolysis is critical for cancer proliferation, growth and biological behaviors.55–57 However, using loss-of-function and gain-of-function strategies, we found that ILT4 in tumor cells significantly promote fatty acid synthesis and lipid accumulation in tumor cells, resulting in tumor proliferation, growth and migration. These novel results further suggest that both glucose and lipid metabolism are important and required for cancer cell biological functions, and that targeting ILT4 could be an effective strategy to reprogram tumor cell lipid metabolism for cancer treatment. Indeed, our in vivo studies have demonstrated that tumor-derived ILT4/PIR-B promotes tumor growth in vivo, and knockdown of ILT4 significantly suppresses tumor development and progression.
In addition to regulate tumor metabolism and functions, our current studies identify a novel mechanism that tumor-derived ILT4 is responsible for tumor-mediated induction of T cell senescence. ILT4 was initially identified in innate immune cells, such as DCs and macrophages, regulating innate immune cell functions and promoting development of tolerogenic DCs and MDSCs.14 22 58 59 Increasing evidence suggests that ILT4 is a potent suppressive molecule and ILT4 in tolerogenic DCs can promote the development of both CD4+ and CD8+ Treg cells.14 22 58 59 However, very limited information is known about whether ILT4 directly affect T cell development and function. More recent study suggests that LILRB4 (ILT3) expression on AML leukemia cells can mediate T cell suppression and tumor infiltration.60 However, whether and how tumor-derived ILT4 can affect the effector T cells in the suppressive TME is unknown. We have demonstrated that different types of tumor cells can induce effector/naïve T cell senescence.25 Furthermore, studies from our and other groups showed accumulation of senescent T cells in the suppressive TME from patients with cancer, and senescence is a critical dysfunctional state for tumor-infiltrating T cells.23–26 29–31 However, the molecular mechanism responsible for tumor-induced T cell senescence remains incompletely understood. Our current studies have made significant progress in understanding these challenging issues in the field. We first identified that tumor-derived ILT4 is a key molecule involved in induction of DNA damage response and senescence in T cells mediated by different types of tumor cells in vitro and in vivo. We then showed that ILT4 promotes lipid metabolism and increases lipid accumulation in tumor cells, which promotes T cell senescence mediated by tumor cells. Studies from other groups have shown that lipid accumulation in tumor cells and stroma cells including DCs and MDSCs suppressed T cell activation and immune responses, suggesting that the lipid metabolic process in tumor cells can reprogram the functional state of T cells.61 62 In addition, we identified that ERK1/2 signaling molecularly controls ILT4-mediated promotion of lipid metabolism and tumor-induced T cell senescence. Importantly, our in vivo studies have demonstrated that PIR-B blockade can prevent tumor-specific T cell senescence in the TME and enhance their antitumor efficacy in mouse tumor models. Collectively, our current studies identify the causative links among the ILT4 signaling, tumor cell metabolism and functions, and T cell senescence. Our previous studies showed that tumor-derived cAMP can activate LCK/cAMP/PKA/CREB signaling in T cells and induce T cell senescence.25 Therefore, future studies will focus on the identification of the mechanistic relationship between ILT4-mediated lipid metabolism, cAMP production, and T cell senescence. Notably, our studies have also identified that HLA-G may be involved in regulation of ILT4-mediated induction of T cell senescence. It is established that HLA-G is one of the important ligands for ILT4.42 43 Our current studies have clearly showed that HLA-G is highly expressed on both T cells and tumor cells, and blockage of HLA-G can prevent T cell DNA damage response and senescence mediated by tumor cells. However, how the crosstalk between tumor-derived ILT4 and HLA-G in both tumor and T cells promotes the development of T cell senescence is still unknown in the current study. An improved understanding of these molecular processes will facilitate the development of novel strategies for tumor immunotherapy.
Our studies have also brought a novel concept that ILT4 is an important checkpoint for tumor immunotherapy.22 Although current checkpoint blockade therapies using anti-CTLA-4, PD1 and PD-L1 have resulted in promising benefits in patients with cancer, only around 30% patients respond to these therapeutics.2–4 These clinical trial results prompt us to explore other potential tolerogenic mechanisms utilized by malignant tumors and further identify alternative checkpoint molecules for tumor immunotherapy. Our recent studies have suggested that induction of T cell senescence is a novel mechanism for tumor immune escape, and blockage of effector T cell senescence in the TME should be an emerging target for tumor immunotherapy.23–28 Our previous and current studies have clearly shown that tumor-derived ILT4 is a driver for tumor growth and proliferation, invasion and progression.19 37 Our current studies further identified that tumor-derived ILT4 is responsible for induction of effector/naïve T cell senescence mediated by tumor cells. In addition, studies from other groups have shown that ILT4/PIR-B can promote MDSCs, TAM and Treg cell development in the TME.16–20 All these studies collectively indicate that ILT4 is an important checkpoint molecule controlling both tumor cells and immune cells in the suppressive TME. In support of this assertion, our in vivo studies using the breast cancer E0771 model showed that knockdown/blockage of PIR-B in tumor cells can significantly prevent T cell senescence and suppress tumor growth and progression. In addition, our in vivo therapeutic studies demonstrated that combination of ILT4 blockage and adoptive transfer T cell therapy can dramatically enhance antitumor efficacy mediated by adoptively transferred tumor-specific T cells. ACT, especially CAR-T therapy, has achieved an immense success in the treatment of hematologic malignancies.63 64 However, it is difficult to translate into solid tumors because of the suppressive TME that presents a major barrier for its successful clinical application.1 2 Our recent and current studies also suggested that tumor cells can convert transferred tumor-specific T cells to become senescent T cells, resulting in T cell dysfunction and impaired antitumor immunity.25 26 In this study, using melanoma B16 and gp100-specific TCR Tg T cell models, we discovered that the endogenous blockade of tumor-derived PIR-B (ILT4) remarkably prevents the adoptively transferred CD8+ T cell senescence, enhances their antitumor efficacy, and suppresses tumor growth. These results further suggest that ILT4 blockade is an effective strategy to reverse T cell senescence and overcome the ACT unresponsiveness to solid tumors. We will continue our efforts to develop novel strategies combining ILT4 blockade with other immunotherapeutics in different tumor models.
In summary, we report that increased expression of ILT4 in various types of tumors, driving tumor growth, progression and poor clinical outcomes. Furthermore, tumor-derived ILT4 can induce senescence in effector T cells and promote T cell dysfunction for tumor immunity. In addition, we elucidate the molecular and metabolic mechanisms responsible for ILT4-induced T cell senescence, which involve initiation of DNA damage response, reprogramming of lipid metabolism and activation of MAPK ERK1/2 signaling in tumor cells. Using both breast cancer and melanoma tumor models, as well as the T cell adoptive transfer therapy model, we further establish the novel concept that ILT4 is a promising checkpoint target for tumor immunotherapy.
Acknowledgments
The authors would like to thank Joy Eslick and Sherri Koehm for FACS sorting and analyses.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors AG, YS and GP: Designed research, analyzed data, prepared figures and wrote the paper. AG, XL, WL, SW, FS, LH, YZ: Performed experiments. XL: Analyzed data and prepared partial figures. JW: Collected public datasets and analyzed the data. All authors approved the final version of the manuscript.
Funding This work was partially supported by grants from the Jinan Science and Technology Development Program (201907110, to AG), The Youth Training Program for High-level Project of Jinan Central Hospital (202007003, to AG), Academic Promotion Programme of Shandong First Medical University (2019QL025, to YS), Shandong Provincial Natural Science Foundation (ZR2019MHO42, to YS), Melanoma Research Alliance (to GP).
Competing interests None declared.
Patient consent for publication Not required.
Ethics approval Primary patient tissue samples were obtained from patients in Jinan Central Hospital with written informed consent, as approved by the local ethics committee. All the animal experiments have been approved by the Institutional Animal Care Committee in Saint Louis University (Protocol No. 2411).
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available in a public, open access repository. All data presented in this study are included in this published article and the supplementary information files. Datasets analyzed in the current study are available at Kaplan Meier-plotter and GEO DataSets (Accession: GSE50081 and GSE1456).
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.