Article Text

Abstract
Background Low-density lipoprotein receptor-related protein 1b (encoded by LRP1B) is a putative tumor suppressor, and preliminary evidence suggests LRP1B-mutated cancers may have improved outcomes with immune checkpoint inhibitors (ICI).
Methods We conducted a multicenter, retrospective pan-cancer analysis of patients with LRP1B alterations treated with ICI at Duke University, Johns Hopkins University (JHU) and University of Michigan (UM). The primary objective was to assess the association between overall response rate (ORR) to ICI and pathogenic or likely pathogenic (P/LP) LRP1B alterations compared with LRP1B variants of unknown significance (VUS). Secondary outcomes were the associations with progression-free survival (PFS) and overall survival (OS) by LRP1B status.
Results We identified 101 patients (44 Duke, 35 JHU, 22 UM) with LRP1B alterations who were treated with ICI. The most common tumor types by alteration (P/LP vs VUS%) were lung (36% vs 49%), prostate (9% vs 7%), sarcoma (5% vs 7%), melanoma (9% vs 0%) and breast cancer (3% vs 7%). The ORR for patients with LRP1B P/LP versus VUS alterations was 54% and 13%, respectively (OR 7.5, 95% CI 2.9 to 22.3, p=0.0009). P/LP LRP1B alterations were associated with longer PFS (HR 0.42, 95% CI 0.26 to 0.68, p=0.0003) and OS (HR 0.62, 95% CI 0.39 to 1.01, p=0.053). These results remained consistent when excluding patients harboring microsatellite instability (MSI) and controlling for tumor mutational burden (TMB).
Conclusions This multicenter study shows significantly better outcomes with ICI therapy in patients harboring P/LP versus VUS LRP1B alterations, independently of TMB/MSI status. Further mechanistic and prospective validation studies are warranted.
- prostatic neoplasms
- lung neoplasms
- immunotherapy
- genetic markers
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Background
The encouraging success of the immune checkpoint inhibitors (ICI) has dramatically improved the care of patients in a growing number or cancer subtypes. These therapies have become first-line options for many cancers, including melanoma, non-small cell lung cancer (NSCLC) and renal cell carcinoma.1–3 However, not all patients respond to treatment with ICIs. The use of predictive biomarkers for response has been explored in many tumor types with varying degrees of success. Programmed-death ligand-1 (PD-L1) expression,1 microsatellite instability (MSI)/mismatch repair deficiency4 and tumor mutational burden (TMB)5 6 are the only FDA-approved predictive biomarkers for immunotherapy response. Other potential biomarker candidates include tumor infiltrating lymphocytes,7 immunophenotyping,8 inflamed gene expression profiling9 and the gut microbiome.10
One potential predictive biomarker candidate is the low-density lipoprotein receptor-related protein 1b, encoded by the LRP1B gene. LRP1B is a large gene located on chromosome 2q, containing >91 exons and spanning over 500 kilobases, and is a member of the LDL receptor family.11 The protein product of LRP1B is 4599 amino acids long. Liu et al demonstrated that 50% of NSCLC cell lines harbored alterations of the LRP1B gene (complete or partial homozygous deletions), and implicated LRP1B as a likely putative tumor suppressor.12 Subsequent investigation has shown that LRP1B may be a tumor suppressor in gastric cancer, where it is regulated by methylation.13 In addition, LRP1B is altered or inactivated in many other solid tumors and hematological malignancies.13–27 In a study of 3312 human cancer specimens, LRP1B was one of the top 10 most frequently deleted genes.28 Based on data from The Cancer Genome Atlas data available on the cBioPortal database, the frequency of somatic LRP1B alteration was 11.8% across all samples and >20% of cases of NSCLC, melanoma, esophageal, stomach, head and neck, uterine and bladder cancers.29 30
A number of studies have suggested a correlation between LRP1B alterations and improved outcomes with ICI.31–36 It remains to be determined if LRP1B alterations are simply a prognostic passenger biomarker of high TMB, or whether LRP1B may have true biologic relevance and can independently predict responses to ICI. Thus, we performed a multi-institutional retrospective study to describe the outcomes of patients across multiple tumor types harboring alterations in LRP1B who were treated with ICI, and to compare outcomes in patients with pathogenic/likely pathogenic (P/LP) and variant of unknown significance (VUS) LRP1B alterations.
Methods
Patients and eligibility
We performed a retrospective review of all patients with LRP1B alterations reported on tissue-based next-generation sequencing (NGS) panels who also received ICI at the Duke Cancer Center between July 2015 and October 2018, Johns Hopkins University (JHU) from May 2013 to September 2019 and University of Michigan (UM) from July 2013 to January 2020.
All patients with advanced or metastatic malignancies and a LRP1B alteration were eligible. Patients who received chemotherapy concurrently with ICI were excluded, and patients treated in the neoadjuvant/adjuvant setting were also excluded. Patients at Duke and JHU were identified as having LRP1B alterations from a genomic database, and ICI receipt was identified on chart review to generate the final patient list. At UM, patients were identified from an immunotherapy patient database, which was cross-referenced with patients who had tissue-based NGS.
Tissue-based NGS
Tissue-based NGS at Duke and JHU was performed on the Foundation Medicine platform. All Foundation Medicine panels used for this analysis were prior to the currently available CDX panel; at the time of this writing, LRP1B is not included on the FoundationOne CDX panel. Tissue-based NGS at UM was performed on an in-house NGS panel, Michigan Oncology Sequencing Center(MI-ONCOSEQ). LRP1B alterations were considered in three groups. P alterations were defined as any genomic alteration that would lead to a large deletion, truncation or loss of function (ie, nonsense mutation, homozygous loss, frameshift mutation, intragenic rearrangement, splice acceptor/donor mutation). Missense mutations were then further categorized as LP alterations if they were listed in the Catalogue of Somatic Mutations in Cancer (COSMIC) database with a Functional Analysis through Hidden Markov Models (FATHMM) score for likelihood of pathogenicity of >0.5.37 38 VUS were defined as missense changes not listed in COSMIC or having FATHMM scores of <0.5.
Patient variables
The variables collected included patient demographics (age, race, ethnicity), tumor type (lung, kidney, bladder and so on), treatment selection (pembrolizumab, nivolumab and so on), PD-L1 status and TMB. Patient outcomes included best radiographic response, progression-free survival (PFS) and overall survival (OS). On patients collected at Duke and JHU, best radiological response was characterized by RECIST V.1.1 criteria.39 At UM, best radiological response was determined using RECIST V.1.1 definitions in combination with clinical notes.
Statistical analysis
The primary outcome was the difference in overall response rates (ORR, complete or partial response) between the P/LP LRP1B alterations compared with the LRP1B VUS subgroup. Other secondary outcomes included PFS, defined as time from initiation of ICI to progression or death (progression was defined by radiographic criteria or clear clinical progression based on chart review of notes from the treating physician) and OS. Logistic regression was used to compare ORR between the P/LP and VUS subgroups using a prespecified two-sided alpha error of 0.05. The Cox proportional hazard model was used for time-to-event outcomes such as OS and PFS between the P/LP and VUS subgroups, using an alpha error of 0.05 for nominal significance testing between groups along with 95% CIs. Logistic regression and Cox proportional hazard models were used for post hoc subgroup analysis between different institutions as well as between tumor types. No formal sample size calculation was performed, as all cases across three institutions were included. With a final sample size of 101 patients (45 VUS and 56 P/LP alterations), and assuming an ORR of 25%, a post hoc calculation shows our study has 80% power to detect an ORR in the P/LP category of 54.2% or an OR of 3.55.
An additional analysis was performed to adjust for MSI status and TMB in which all patients with MSI-high (MSI-H) or MSI-unavailable status were excluded and then ORs for overall response and HR for PFS and OS were adjusted for TMB as a continuous variable as well as a binary variable (TMB >10 vs <10 mut/Mb). To compare the rate of TMB between genomic alteration groups, the Wilcoxon rank-sum test was used.
Results
Patients
A total of 101 patients were identified who harbored alterations in LRP1B and received therapy with ICI. Duke, JHU and UM identified 44, 35 and 22 patients, respectively (see Consolidated Standards of Reporting Trials diagram, figure 1A). Demographics are shown in table 1 according to LRP1B alterations. The P/LP are combined for comparison against the LRP1B VUS internal control group. The distribution of missense mutations for LP and VUS groups are shown in online supplemental figure 1. The most common ICI therapy was single-agent anti-PD-1 or anti-PD-L1 at 91% (61% of which was pembrolizumab and 32% nivolumab). Combination anti-PD-1 or anti-PD-L1 plus anticytotoxic T-lymphocyte-associated protein 4 (anti-CTLA-4) (primarily nivolumab and ipilimumab) was administered to 8% of subjects, which was over-represented in the P/LP group at 13% compared with 2% in the VUS group. Median time on therapy for all patients, P/LP cohort and VUS cohort was 4.1, 4.4 and 3.4 months, respectively. Median follow-up for all patients, P/LP cohort and VUS cohort was 10.3, 8.5 and 13.5 months, respectively.
Supplemental material

(A) Consolidated Standards of Reporting Trials diagram. (B) Distribution of malignancies identified for inclusion. ICI, immune checkpoint inhibitor; LRP1B, lipoprotein receptor-related protein 1b; NGS, next-generation sequencing; SCLC, small cell lung cancer; NSCLC, non-small cell lung cancer; SCC, squamous cell carcinoma.
Demographics
Other notable differences between the P/LP and VUS groups include a higher proportion of MSI-H (9% vs 5%) and PD-L1 >1% (25% vs 18%) in the P/LP group. Notably, PD-L1 information was not available in a majority of patients (72%). The cancer type is shown in figure 1B for the entire cohort and each molecular subgroup. Lung cancer was the most common tumor type (41%, 40/41 NSCLC, 1/41 small cell lung cancer) followed by prostate cancer (9%), sarcoma (6%), breast cancer (5%), kidney cancer (5%) and melanoma (5%). Notable differences between the P/LP and VUS groups include imbalances in the rates of lung cancer (36% vs 49%), melanoma (9% vs 0%), esophagus (7% vs 0%), unknown primary (5% vs 0%), cutaneous or head and neck squamous cell carcinoma (7% vs 0%), and kidney (2% vs 9%) across LRP1B alteration groups.
Patient outcomes
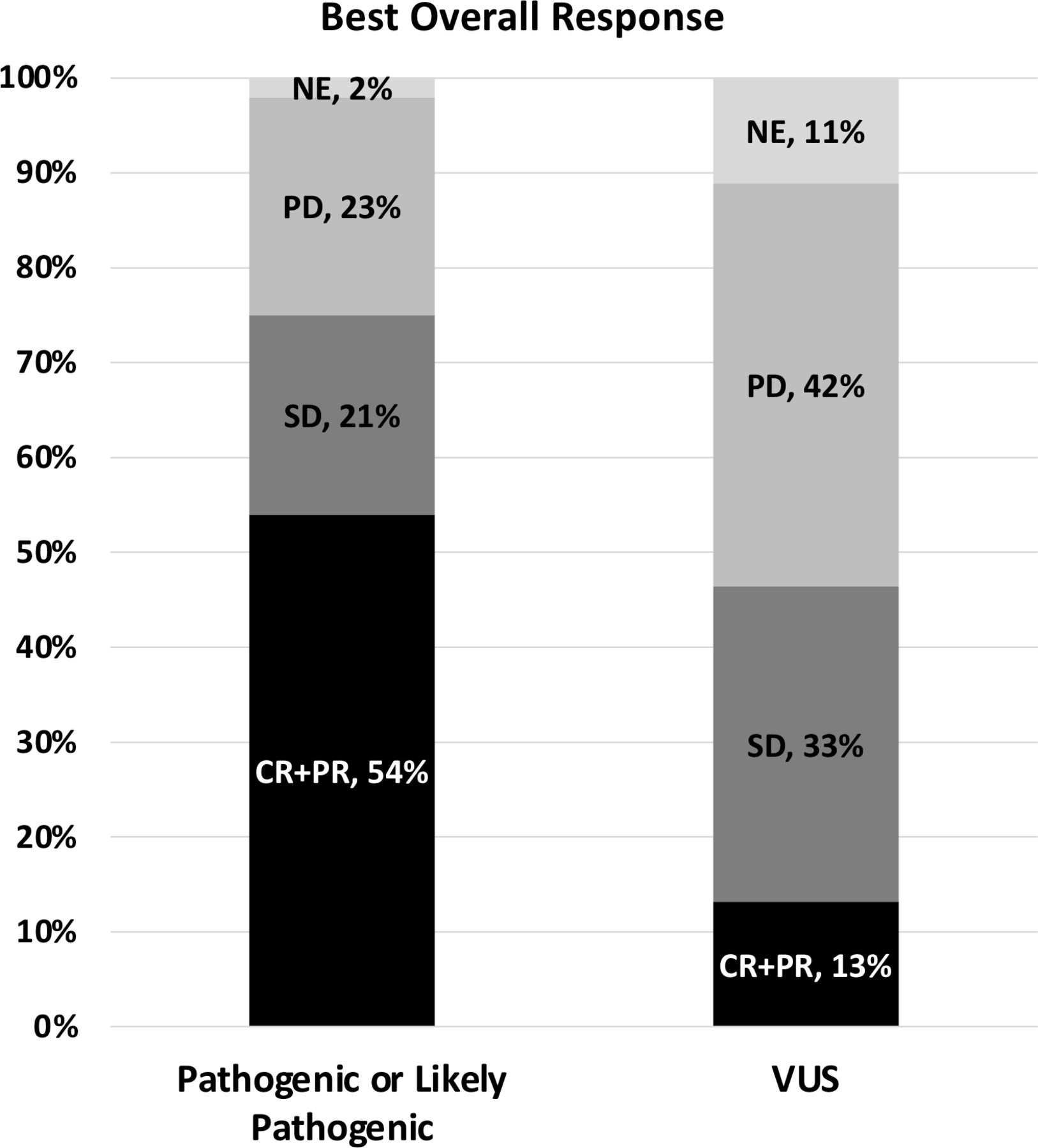
The primary outcome of radiographic response rates by molecular subtype are shown in figure 2 and table 2. The ORR (CR+PR) in the P/LP group was 30/56 (54%; 95% CI 40% to 67%) compared with 6/45 (13%; 95% CI 5% to 27%) in the VUS group. The OR by logistic regression for the ORR for P/LP versus VUS was significant at 7.5 (95% CI 2.9 to 22.3, p=0.0009).

Stacked bar graph of best overall response among molecular subtypes. CR, complete response; NE, not evaluable; PD, progressive disease; PR, partial response; SD, stable disease; VUS, variants of unknown significance.
Best overall response and overall response rate among molecular subtypes
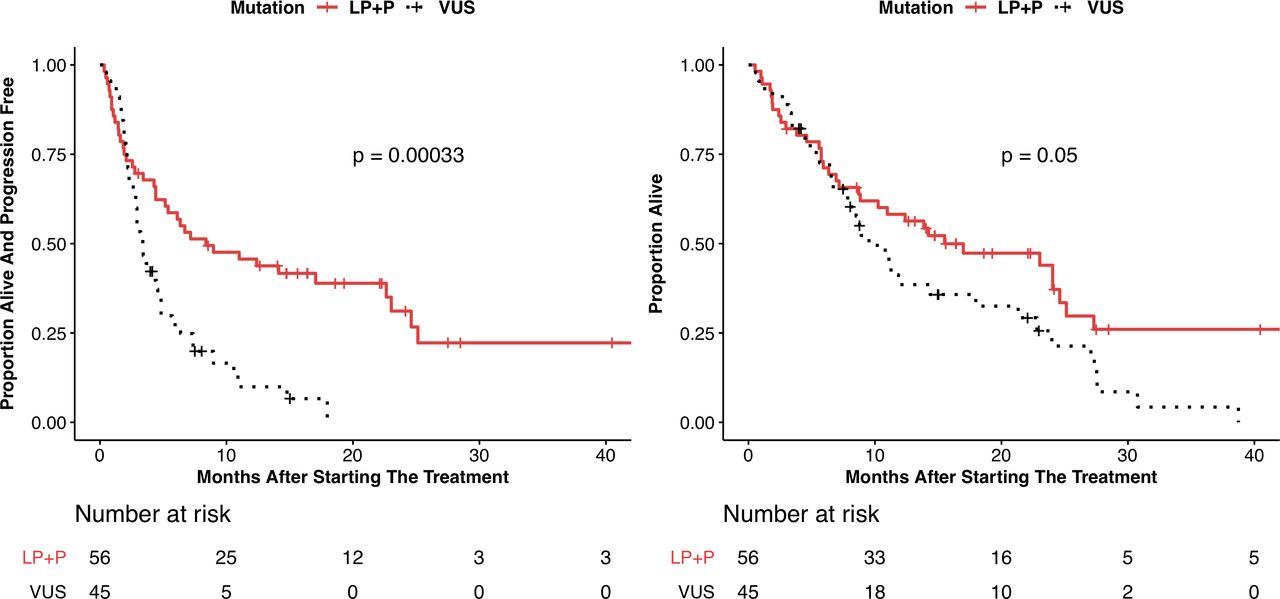
PFS and OS are shown in table 3, with Kaplan-Meier plots shown in figure 3 and online supplemental figure 4. The P/LP group showed a significantly improved PFS compared with the VUS group by Cox proportional hazard analysis (HR 0.42; 95% CI 0.26 to 0.68, p=0.0003) and median PFS of 8.4 months (95% CI 5.2 to 23) vs 3.4 months (95% CI 2.8 to 4.3), respectively. The P/LP group also showed an improvement in OS compared with the VUS group (HR 0.62; 95% CI 0.39 to 1.01, p=0.053) with median PFS of 15.5 months (95% CI 10.3 to 25.1) vs 9.5 months (95% CI 7.6 to 21.3), respectively. In total, there were 77 PFS events and 69 deaths in the entire cohort. The P/LP and VUS cohorts experienced 37 and 40 PFS events, respectively. The P/LP and VUS cohorts experienced 34 and 35 deaths, respectively.
Supplemental material
Progression-free survival (PFS) and overall survival (OS) between patients with pathogenic or likely pathogenic (P/LP) alterations compared with those with VUS
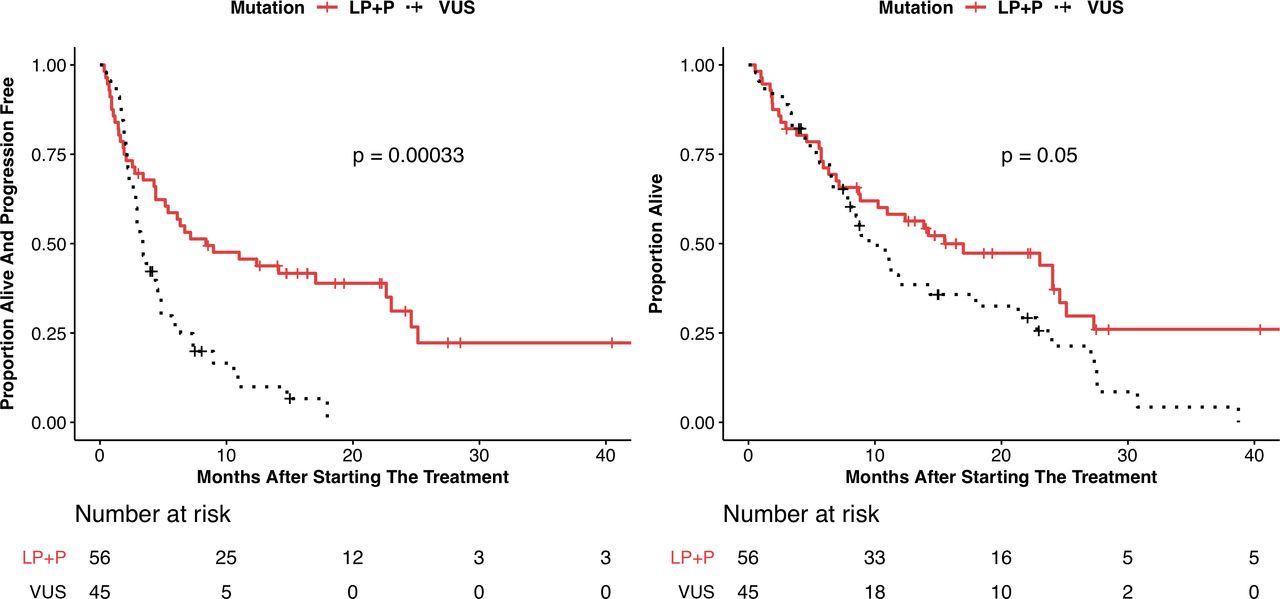
Kaplan-Meier curves showing (A) progression-free survival and (B) overall survival for subjects with pathogenic or likely pathogenic LRP1B alterations (P/LP) and variants of unknown significance (VUS). P values shown are determined from the log-rank test.
For a more comprehensive view of patient-level time-to-event data, a swimmer’s plot of all patients separated by molecular subtype (P/LP vs VUS) showing best radiographic response, treatment duration, progression and death is shown in online supplemental figure 2.
Supplemental material
Association with tumor mutational burden
Box-and-whisker plots for TMB are shown in online supplemental figure 3. Median TMB in the P/LP group was 10 mut/Mb vs 7.9 mut/Mb in the VUS group. By the Wilcoxon rank-sum test, the two groups were not different (p=0.32). The P/LP group showed more outliers with TMB >50 mut/Mb at 13 patients (23.2%) compared with the VUS group with three patients (6.7%). MSI-H was identified in 5/56 (8.9%) in P/LP group and 3/45 (6.7%) in the VUS group.
Supplemental material
Subgroup analysis
Shown in figure 4 are forest plots of ORs for overall response and HRs for OS and PFS across institutions and cancer subtypes (lung cancer and non-lung cancers). In the unadjusted analysis, ORs and HRs remained consistent in favor of the P/LP group across the three institutions. Overall response and PFS also were consistent in the unadjusted analysis across patients with lung cancer and those without lung cancer. The OS benefit in the P/LP versus VUS group appeared to be strongest and most significant in patients with lung cancer, HR 0.41 (95% CI 0.19 to 0.91), but not among those with non-lung cancers, HR 0.82 (95% CI 0.44 to 1.51).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Subgroup forest plots showing (A) ORs for overall radiographic response (complete or partial response) by logistic regression, (B) HRs for progression-free survival by Cox proportional hazard and (C) HRs for overall survival by Cox proportional hazard; 95% CIs are shown for OR and HR. When adjusting for tumor mutational burden, all patients with microsatellite instability-high (MSI-H) or MSI-unavailable status are excluded. TMB, tumor mutational burden.
Two adjusted analysis were performed to isolate the contribution of TMB and MSI status, as shown in figure 4. All patients who were MSI-H (n=8) or MSI-unavailable (n=13) or missing TMB data (n=8) were excluded, and then subsequently the three analysis of overall response, PFS and OS were adjusted by TMB in two fashions, as a continuous variable and as a binary variable using the cut-off of 10 mut/Mb on a subset of patients based on established cutoffs from published clinical trials in NSCLC.40 41 This analysis included 75 patients (3 patients were missing both TMB and MSI information). Our overall findings remained consistent across TMB as a continuous or binary variable. When controlling for TMB as a continuous variable, the OR for overall response was 6.75 (95% CI 2.32 to 23.00) and HRs for PFS and OS were 0.49 (95% CI 0.28 to 0.87) and 0.77 (95% CI 0.42 to 1.39), respectively. Similar to the unadjusted analysis for OS, the TMB-adjusted analysis for OS, but not overall response or PFS, showed that the adjusted OS benefit for the P/LP group over the VUS group appeared to driven by patients with lung cancer HR 0.34 (95% CI 0.12 to 0.94) as compared with those with non-lung cancers, 1.31 (95% CI 0.58 to 2.99).
An imbalance in the patients who received CTLA-4, primarily in combination with anti-PD-1 therapy inhibitors, was seen with a higher proportion of patients receiving anti-CTLA-4 therapy in the P/LP compared with the VUS group. To isolate the contribution of anti-CTLA-4 therapy, an analysis was performed excluding two patients in VUS group (one who received anti-CTLA-4 monotherapy and one who received combination anti-PD-1/anti-CTLA-4) and excluding seven patients in the P/LP group (all who received anti-PD-1/anti-CTLA-4). In this subset analysis, ORR continued to favor P/LP over VUS for the entire cohort 49% (95% CI 35 to 63) vs 14% (95% CI 4 to 24), respectively, for an OR of 5.9 (95% CI 2.2 to 17.9), and 53% (95% CI 30 to 75) vs 14% (95% CI 0 to 28), respectively, among patients with lung cancer for an OR of 7.0 (95% CI 1.4 to 25.9). This relationship was consistent in both the unadjusted analysis and when adjusting for TMB and excluding MSI-H (data not shown). Similarly, PFS continued to favor the LP/P group over VUS for all patients (HR 0.49, 95% CI 0.30 to 0.81) and patients with lung cancer (HR 0.47, 95% CI 0.22 to 1.0). OS was also numerically similar, but with a wider CI when excluding patients receiving anti-CTLA-4 in the entire cohort (HR 0.72, 95% CI 0.44 to 1.2) and for patients with lung cancer (HR 0.44, 95% CI 0.20 to 0.96). The HRs for OS and PFS and remained consistent when adjusting for TMB and excluding MSI-H (data not shown).
Extraordinary responder analysis
There were 24 out of 56 (42.9%) patients free from progression or death for 12 months after initiating ICI therapy and having a P/LP alteration in LRP1B. This compares to only 3 out of 45 (6.7%) patients with LRP1B VUS alterations who were free from progression or death after 12 months. Of those with pathogenic alterations in LRP1B, 10 patients had NSCLC with 4 having PD-L1 expression >1% (60%, 80%, 95% and >1%) and 5 having TMB>10 mut/Mb (12, 21.7, 31, 66.8 and 73 mut/Mb). Four patients with prostate cancer were identified (3 of 4 were MSI-H), and four patients with melanoma were identified (with TMBs of 8, 33, 62 and 107 mut/Mb). The other extraordinary responder cases included two with cutaneous SCC (TMB 87, 112 mut/Mb), one with esophageal cancer (TMB 116 mut/Mb), two with unknown primary (TMB 43, 92 mut/Mb) and one with clear-cell kidney cancer. Of the three exceptional responders in the VUS group, two had lung cancer (TMB 9 and PD-L1 95%; TMB 24.8 and PD-L1 unavailable) and one had colon cancer (microsatellite stable).
Discussion
We performed the largest multicenter retrospective chart review of patients with LRP1B alterations across multiple tumor type to our knowledge and found that patients with P/LP alterations in LRP1B have a higher ORR, improved PFS and improved OS compared with patients with LRP1B VUS when treated with immune checkpoint blockade. Our results are suggestive that P/LP LRP1B alterations may be a tumor-agnostic biomarker—one which may help predict more favorable outcomes with ICIs, particularly PD-1 pathway blockade. A strength of our study is that our findings were consistent across all three institutions and in adjusted analyses for TMB and excluding MSI-H disease. Larger prospective studies are needed for external validation, but these results suggest that LRP1B has clinical significance and should be included in targeted gene panels.
These results are supported by prior studies that have suggested that LRP1B alterations may be associated with improved outcomes with ICI. In 2016, Johnson et al found that alterations in LRP1B was associated with high TMB and significantly associated with response to ICI therapy in patients with metastatic melanoma, being present in 11/32 (34%) of responders compared with 1/33 (3%) in non-responders (p<0.001).31 A more recent study found that OS was better among patients with NSCLC and melanoma with LRP1B mutations compared with LRP1B wild-type tumors.32 They also found a significantly higher TMB rate among LRP1B-mutated compared with LRP1B wild-type tumors. Additionally, three case reports of patients having had unexpected responses to ICI also reported alterations in LRP1B in human papillomavirus-related small cell cancer of the head and neck, sebaceous carcinoma and renal cell carcinoma with rhabdoid features.33–35 Finally, a recent study from our group reporting outcomes among men with metastatic castration-resistant prostate cancer found that 3/4 (75%) patients with LRP1B alterations experienced durable prostate-specific antigen (PSA) declines of >50% as compared with 2/14 (14.2%) of patients with LRP1B wild-type cancer.36
A major unanswered question is the mechanism by which LRP1B inactivation might modulate response to ICI. Its function both in normal tissue and its role in cancer is poorly understood. Reconciling LRP1B’s role as a tumor suppressor14 with its immunomodulatory properties is difficult as one would not expect a tumor suppressor to affect immune evasion, or antigen presentation; however, LRP1B’s role as a tumor suppressor has not been definitively established. There is evidence that LRP1B may participate in extracellular ligand scavenging through its role as an endocytic receptor, thereby modulating the tumor microenvironment23 42 or could participate in cellular drug uptake.43 One previous study found that cell cycle and antigen processing pathways were significantly altered in tumors with LRP1B alterations, and patients with LRP1B mutations had higher T-cell inflamed gene expression scores.32 Finally, could LRP1B impact the immune cell infiltrate and therefore a functional LRP1B could diminish immune recognition of a malignant cell? Only further mechanistic studies in immunocompetent preclinical models of lung and other cancer subtypes treated with ICI therapy will address this functional relevance and mechanisms of immune recognition.
Conversely, there is evidence that LRP1B may be associated with a higher TMB given its large size as well as its location at the common fragile site, FRA2F.44 Our study did not note a statistical difference in TMB between the two LRP1B cohorts, although there were more outliers with TMB >50 mut/Mb in the P/LP group compared with the VUS group. A strength of our study is the comparison between outcomes of patients with pathogenic LRP1B alterations with those with LRP1B VUS to reduce confounding from TMB. Furthermore, our post hoc analysis excluding MSI-H cases and adjusting for TMB suggests that LRP1B may have independent predictive value beyond that which is mediated by TMB. Despite this, we do recognize that the lack of comparison to outcomes among patients with LRP1B wild-type malignancies is a significant shortcoming and limits its interpretation. Thus, prospective pan-cancer studies are needed comparing outcomes with ICI-treated patients with and without LRP1B alterations.
Our study included a diverse set of malignancies. Interestingly, we found that with regard to overall response and PFS, the improved outcomes in the P/LP group over the VUS group were consistent for both patients with lung cancer (the largest subset) and patients with non-lung cancers. However, the improved OS in the entire cohort appeared to be primarily driven by the patients with lung cancer. Given its high incidence as well as the frequency of LRP1B alterations, these findings require further investigation, specifically in lung cancer. Although ICI is part of the standard-of-care first-line regimen for all patients with NSCLC without a driver mutation (such as EGFR, ALK or ROS1), LRP1B alterations may identify a subpopulation of patients with lower PD-L1 expression who can be treated with ICI monotherapy. The size of our cohort limits our ability to make any inferences about specific tumor types other than lung cancer, but LRP1B potentially remains an intriguing tumor-agnostic biomarker candidate across multiple tumor types.
This study has several additional limitations. First, this is a small, retrospective study conducted entirely at academic institutions where many patients had been heavily pretreated prior to receiving immune checkpoint inhibition. Additionally, the only patients eligible were those who had tissue-based NGS testing. Additionally, given our limited understanding of the normal function of LRP1B, the definition of P/LP or VUS alterations may be problematic. We are particularly cautious about interpretations of this biomarker because this is not well defined. We took a prespecified systematic approach to our analysis to define the patient subgroups, but it is challenging to determine whether a detected genomic alteration is truly ‘pathogenic’. This definition will likely require further refinement over time with better understanding of LRP1B’s role in normal tissue and cancer. ICI therapy was not standardized across patients and the asymmetry between the distribution in tumor types across the LRP1B subgroups raises concerns over unmeasured confounders. Of note, there is a higher rate of anti-CTLA-4 therapy in the P/LP group compared with the VUS group, although the improvements in ORR, PFS and OS remained consistent even after excluding the nine patients who had received anti-CTLA-4 therapy from the analysis. As mentioned above, the choice of a control group is also a limitation. LRP1B VUS was chosen as a convenience control group, but no inferences can be made in comparison to patients with LRP1B wild-type malignancies. Wild-type malignancies were not used as a control group due to the challenges of appropriate controlling and matching leading to increased heterogeneity. Finally, although not a direct limitation on this analysis, LRP1B is no longer included in the current version of the FoundationOne CDX panel, which will limit its study and prospective validation moving forward. LRP1B is also not included on Guardant360, FoundationOneLiquid, or MSK-IMPACT panels, but is included on Caris Molecular Intelligence, Personal Genome Diagnostics and MI-ONCOSEQ panels.
In summary, in our multicenter, retrospective cohort study of patients with multiple tumor types, LRP1B P/LP alterations are associated with improved response rates, PFS and OS when treated with ICI compared with patients with LRP1B VUS alterations. Our findings were strongest in patients with NSCLC and were independent of high TMB and MSI status. Further mechanistic studies into the function of LRP1B are needed as well as prospective validation of this potential predictive clinical biomarker for ICI responses.
Acknowledgments
The authors would like to thank the patients and staff at the Duke Cancer Institute, Sidney Kimmel Comprehensive Cancer Center and Rogel Cancer Center.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Twitter @MattLabs831
Presented at These data were presented at the ASCO annual meeting 2020, Abstract # 3007.
Contributors LCB: writing—original draft, investigation, formal analysis. MDT, RS, EBS, CK, MKL, RTG, DM: investigation. JZ: conceptualization, investigation. SG, YW: formal analysis. TZ, MH, DG: writing—review and editing. AA, EA: supervision, writing—review and editing. AJA: conceptualization, supervision, writing—review and editing.
Funding This research received no external funding. AJA is supported by the following federal funding: NIH P30 CA014236, NIH R01 1R01CA233585-01. EA is supported by NIH grant P30 CA006973 and DOD grant W81XWH-16-PCRP-CCRSA.
Competing interests JZ: consulting: NGM Biopharmaceuticals, UroToday; Travel Expenses: UroToday. RTG: speakers bureau: Bayer Pharma AG; Consulting Fees: Bayer Pharma AG, Invivo Corp, Bard. TZ: research funding: AstraZeneca, Janssen, OmniSeq, PGDx, Pfizer, Merrimack, AbbVie/Stemcentrx, Novartis, Merck, Mirati and Regeneron; Advisory/consultant role: Genentech Roche, Exelixis, Bayer, AstraZeneca, Pfizer, Sanofi-Aventis, Janssen, Foundation Medicine, Amgen, Bristol-Myers Squibb, Merck, Pharmacyclics and Seattle Genetics; speakers bureau: Genentech Roche, Exelixis, Sanofi-Aventis, Genomic Health; Stocks/Employment: Capio Biosciences (spouse), Archimmune Therapeutics (spouse). MH: consultant: AstraZeneca, Bayer, Bristol-Myers Squibb, Exelixis, Genentech, Janssen, Pfizer; speakers bureau: Exelixis and Genentech; Research Funding (to institution): Acerta, Bristol-Myers Squibb, Clovis, Exelixis, Genentech, Merck, Pfizer, Seattle Genetics. DG: Acerta Pharmaceuticals—Research, American Association for Cancer Research—Sr Editor, Astellas—Consultant, Research, Advisory Board, AstraZeneca—Consultant, Advisory Board, Axess Oncology—Independent Contractor, Bayer H/C Pharmaceuticals—Research, Consultant, Speaker, Honorarium, Travel accommodations, SC, Bristol-Myers Squibb—Consultant, Research, Steering Committee, Calithera—Research, Capio Biosciences—Scientific Advisory Board, EMD Serono—Honorarium, Exelixis, Inc—Research, Consultant, Speaker, Honorarium, Travel accommodations, Flatiron—Consultant, Ipsen—Honorarium, Janssen Pharmaceuticals—Research, Consultant, Independent Data Monitoring Committee (IDMC), Leidos Biomedical Research—Consultant, Merck Sharp & Dohme—Consultant, Michael J Hennessey Associates – Honorarium, Consultant, Millennium Medical Publishing, Clinical Advances in Hematology & Oncology—Co-Editor-in-Chief, Modra Pharmaceuticals—Advisory Board, Myovant Sciences, Inc—Consultant, Nektar Therapeutics—Steering Committee, Novartis—Research, Physician Education Resource LLC—Consultant, Pfizer—Research, Consultant, Steering Committee, Honorarium, Sanofi—Research, Consultant, Speaker, Honorarium, Travel accommodations, UroGPO—Honorarium, UroToday—Honorarium, Travel accommodations, Vizuri Health Sciences, LLC—Consultant, NCI—Steering Committee. AJA: consultant and/or advisor for AstraZeneca; Bristol-Myers Squibb; Merck & Co; Pfizer, EMD Serono. Paid DSMB member: Eisai. Grant/Research Support from AstraZeneca; Bristol-Myers Squibb and Merck & Co. Other financial or material support from Arcus Biosciences; Astellas Pharma US; Celgene Corporation; Clovis Oncology; Pfizer; Prometheus Biosciences and Seattle Genetics in the form of research support to institution. EA: grants and personal fees from Janssen, personal fees from Astellas, grants and personal fees from Sanofi, grants and personal fees from Dendreon, personal fees from Pfizer, personal fees from Invitae, grants and personal fees from AstraZeneca, grants and personal fees from Clovis, grants and personal fees from Merck, grants from Johnson & Johnson, grants from Genentech, grants from Novartis, grants from Bristol Myers-Squibb; and a patent (PCT/US2015/046806; US20170275673A1) on an AR-V7 biomarker technology that is licensed to Qiagen. AJA: consulting fees: AstraZeneca, Merck, Dendreon, Janssen, Clovis, Bayer and Medivation/Astellas; speaking fees: Bayer and Dendreon, Research funding to Duke: Janssen, Medivation/Astellas, Sanofi-Aventis, Active Biotech, Bayer, Dendreon, Merck, AstraZeneca, Genentech/Roche, Bristol-Myers Squibb, Constellation, Novartis and Pfizer.
Patient consent for publication Not required.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available on reasonable request.