Article Text

Abstract
Background Atezolizumab treatment improves survival, with manageable safety, in patients with previously treated advanced/metastatic non-small cell lung cancer. The global phase III/IV study TAIL (NCT03285763) was conducted to evaluate the safety and efficacy of atezolizumab monotherapy in a clinically diverse population of patients with previously treated non-small cell lung cancer, including those not eligible for pivotal trials.
Methods Patients with stage IIIB/IV non-small cell lung cancer whose disease progressed after 1–2 lines of chemotherapy were eligible for this open-label, single-arm, multicenter study, including those with severe renal impairment, an Eastern Cooperative Oncology Group performance status of 2, prior anti-programmed death 1 (PD-1) therapy, and autoimmune disease. Atezolizumab was administered intravenously (1200 mg every 3 weeks). Coprimary endpoints were treatment-related serious adverse events and immune-related adverse events.
Results 619 patients enrolled and 615 received atezolizumab. At data cutoff, the median follow-up was 12.6 months (95% CI 11.9 to 13.1). Treatment-related serious adverse events occurred in 7.8% and immune-related adverse events in 8.3% of all patients and as follows, respectively, in these subgroups: renal impairment (n=78), 11.5% and 12.8%; Eastern Cooperative Oncology Group performance status of 2 (n=61), 14.8% and 8.2%; prior anti–PD-1 therapy (n=39), 5.1% and 7.7%; and autoimmune disease (n=30), 6.7% and 10.0%. No new safety signals were reported. In the overall population, the median overall survival was 11.1 months (95% CI 8.9 to 12.9), the median progression-free survival was 2.7 months (95% CI 2.1 to 2.8) and the objective response rate was 11%.
Conclusions This study confirmed the benefit–risk profile of atezolizumab monotherapy in a clinically diverse population of patients with previously treated non-small cell lung cancer. These safety and efficacy outcomes may inform treatment decisions for patients generally excluded from checkpoint inhibitor trials.
- clinical trials
- phase IIII clinical trial
- immunotherapy
- lung neoplasms
- PD-L1 inhibitor
- checkpoint inhibitor
- metastatic
- subgroup analysis
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
- clinical trials
- phase IIII clinical trial
- immunotherapy
- lung neoplasms
- PD-L1 inhibitor
- checkpoint inhibitor
- metastatic
- subgroup analysis
Introduction
Anti–programmed death-ligand 1/programmed death 1 (anti–PD-L1/PD-1) monotherapy is the standard of care in patients with advanced or metastatic non-small cell lung cancer (NSCLC)1 2 after progression on platinum-based chemotherapy. Atezolizumab is a monoclonal antibody against programmed death-ligand 1 (PD-L1) that prevents its interaction with PD-1 and B7.1. Atezolizumab was approved for patients with locally advanced or metastatic NSCLC after platinum-based chemotherapy on the basis of the results of the phase III OAK study in patients with previously treated NSCLC,3 in which the median overall survival (OS) was 13.8 months with atezolizumab compared with 9.6 months with docetaxel.
Pivotal clinical trials for immunotherapies typically exclude patients with poor performance status, greater comorbidity, or concomitant autoimmune or chronic viral conditions.3–6 However, these patients typically account for 25%–40% of those with NSCLC,7–10 and more data in these subgroups are needed to guide immunotherapy treatment decisions.
TAIL (NCT03285763) is a global phase III/IV study to evaluate the safety and efficacy of atezolizumab in a diverse patient population with previously treated NSCLC. This study permitted enrollment of patients with poor prognostic risk factors who would have been excluded from pivotal studies such as OAK, about whom few or no prospective data are published. Safety and efficacy are described for the overall population as well as key subgroups.
Materials and methods
Study design and treatment
TAIL is a prospective, phase III/IV, open-label, single-arm, multicenter study conducted in patients with locally advanced or metastatic NSCLC with disease progression following standard chemotherapy. After a 28-day screening period, patients received 1200 mg intravenous atezolizumab on day 1 of each 21 (±5)-day cycle until radiographic disease progression per Response Evaluation Criteria in Solid Tumors V.1.1 (RECIST 1.1), unacceptable toxicity, or treatment withdrawal. Patients could continue atezolizumab after progression if they had evidence of clinical benefit in the opinion of the investigator (see online supplemental methods for criteria).
Supplemental material
Patients
Eligible patients had histologically or cytologically documented stage IIIB/IV NSCLC measurable per RECIST 1.1 that had progressed after one or two chemotherapy regimens (details in supplementary methods in online supplemental appendix A). Patients with any PD-L1 status (including those not tested) were eligible, as were those with high morbidity or risk, including those with treated or untreated asymptomatic central nervous system (CNS) metastases, autoimmune disease (AID), an Eastern Cooperative Oncology Group performance status (ECOG PS) of 2, active or chronic hepatitis B or hepatitis C (HBV/HCV) infection, severe renal impairment (estimated glomerular filtration rate (eGFR) ≥15 mL/min using the Chronic Kidney Disease Epidemiology Collaboration equation), and prior anti–PD-1 therapy.
Exclusion criteria included symptomatic CNS metastases, spinal cord compression, prior treatment with CD137 agonists or checkpoint inhibitor therapies (other than anti–PD-1 therapy), significant cardiovascular disease, and renal disorders requiring dialysis or transplant.
Endpoints and assessments
The primary endpoint was safety, measured by the coprimary endpoints: the incidence of treatment-related serious adverse events (SAEs) and treatment-related immune-related adverse events (irAEs; defined as adverse events (AEs) of special interest requiring corticosteroid treatment within 30 days of onset). Relationship to treatment was per investigator assessment. AEs were graded using National Cancer Institute Common Terminology Criteria for Adverse Events V.4.0. An independent data monitoring committee reviewed safety data every 6 months.
Secondary efficacy endpoints included OS, investigator-assessed progression-free survival (PFS), and objective response rate (ORR). Exploratory endpoints included safety and efficacy in key subgroups. In a prespecified subgroup analysis, patients who would have been included in the phase III OAK study3 defined the OAK-like population (see online supplemental methods).
PD-L1 immunohistochemistry (IHC) analysis was not mandatory, and results of testing performed locally (any validated assay; online supplemental table A1) or centrally (Ventana PD-L1 SP263 IHC assay) on formalin-fixed paraffin-embedded tumor tissue were collected. Local and central PD-L1 results were pooled and summarized as PD-L1 positive (tumor cell (TC) or tumor proportion score (TPS) ≥1%), negative (TC or TPS <1%), or unknown (no result).
Statistical analysis
There was no formal statistical hypothesis testing linked to the sample size calculation. The planned sample size was 600 patients, and the primary analysis was conducted approximately 6 months after the last patient was enrolled. Primary and secondary endpoints were assessed in all enrolled patients who received at least 1 dose of atezolizumab (safety population).
Incidence rates and 95% Clopper-Pearson CIs were used to summarize atezolizumab-related SAEs and irAEs. Time-to-event data were summarized using Kaplan-Meier methodology, and 95% CIs for the median overall survival and survival rates were calculated using Greenwood’s formula with SAS V.9.4.
Results
Patient population
Between October 2017 and December 2018, 619 patients were enrolled at 112 sites across 24 countries (online supplemental appendix B). Four patients died before starting treatment; 615 patients who received atezolizumab monotherapy are described as the overall study population (figure 1). At the data cut-off (June 4, 2019), the median follow-up was 12.6 (95% CI 11.9 to 13.1) months. The OAK-like subgroup made up 66% of the overall study population (n=406; table 1); hence, approximately one-third of the TAIL population (34%) would have been ineligible for OAK for one or more reasons. Also included in the overall study population were 89 patients with CNS metastases (14.5%), 78 with renal impairment (12.7%, including two patients with severe renal impairment (eGFR 15–29 mL/min/1.73 m2)), 61 with ECOG PS 2 (9.9%), 39 who had received prior anti–PD-1 therapy (6.3%), 30 with baseline AID (4.9%), and 15 with active or chronic HBV/HCV (2.4%).
Supplemental material
Patient demographics and baseline disease characteristics
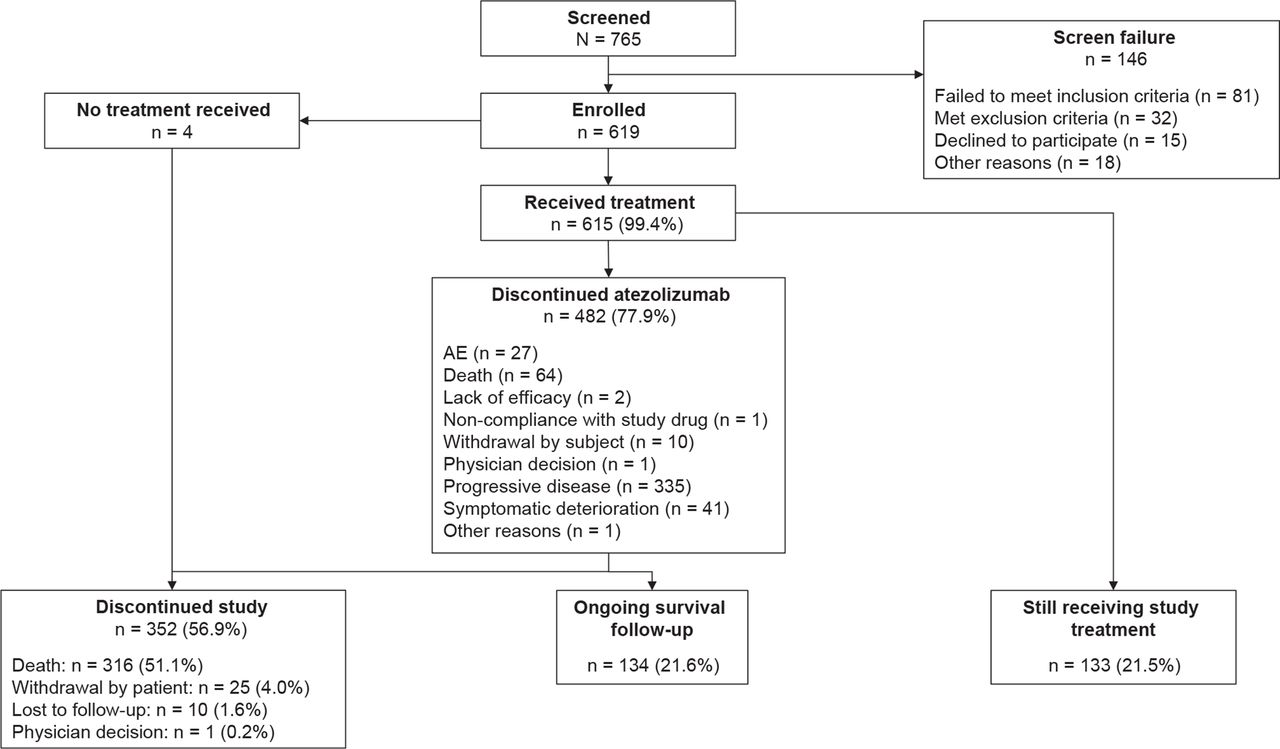
Trial profile and patient disposition. AE, adverse event.
At baseline, 35.3% (n=217) of the study population had received ≥2 prior lines of NSCLC therapy and 6.3% had received prior anti–PD-1 therapy, half of whom (20 of 39 patients) received ≥3 prior treatment lines (table 1). Patients with ECOG PS 2 had a higher incidence of metastases in bone (41.0%; n=25) and liver (29.5%; n=18) than those with ECOG PS 0 or 1. In the overall study population, 34.6% (n=213) were PD-L1 positive, 27.3% (n=168) were PD-L1 negative, and 38.1% (n=234) had not been tested. PD-L1 status distribution in the OAK-like subgroup was similar to that in the overall population. The PD-L1 22C3 IHC assay (Agilent) was the most frequently used (online supplemental table A1), and most patients were tested locally.
Safety
The median duration of treatment in the safety population at the cut-off date was 3.2 months (range 0–18.6; table 2) with a median of 5.0 cycles (range 1–27). Atezolizumab was discontinued in 482 patients (77.9%), most frequently due to progressive disease (54.1%) or death (10.3%; figure 1). Disease progression was the most common cause of 312 deaths (86.5%).
Safety summary
The coprimary endpoint of treatment-related SAEs occurred in 7.8% of patients (95% CI 5.8 to 10.2) (figure 2A). Grade 3–4 treatment-related SAEs occurred in 3.7% and grade 5 treatment-related SAEs in 1.5%: pericarditis (in two patients), hepatotoxicity, pneumonitis, systemic inflammatory response, general physical health deterioration, pneumonia, respiratory failure and stress cardiomyopathy.
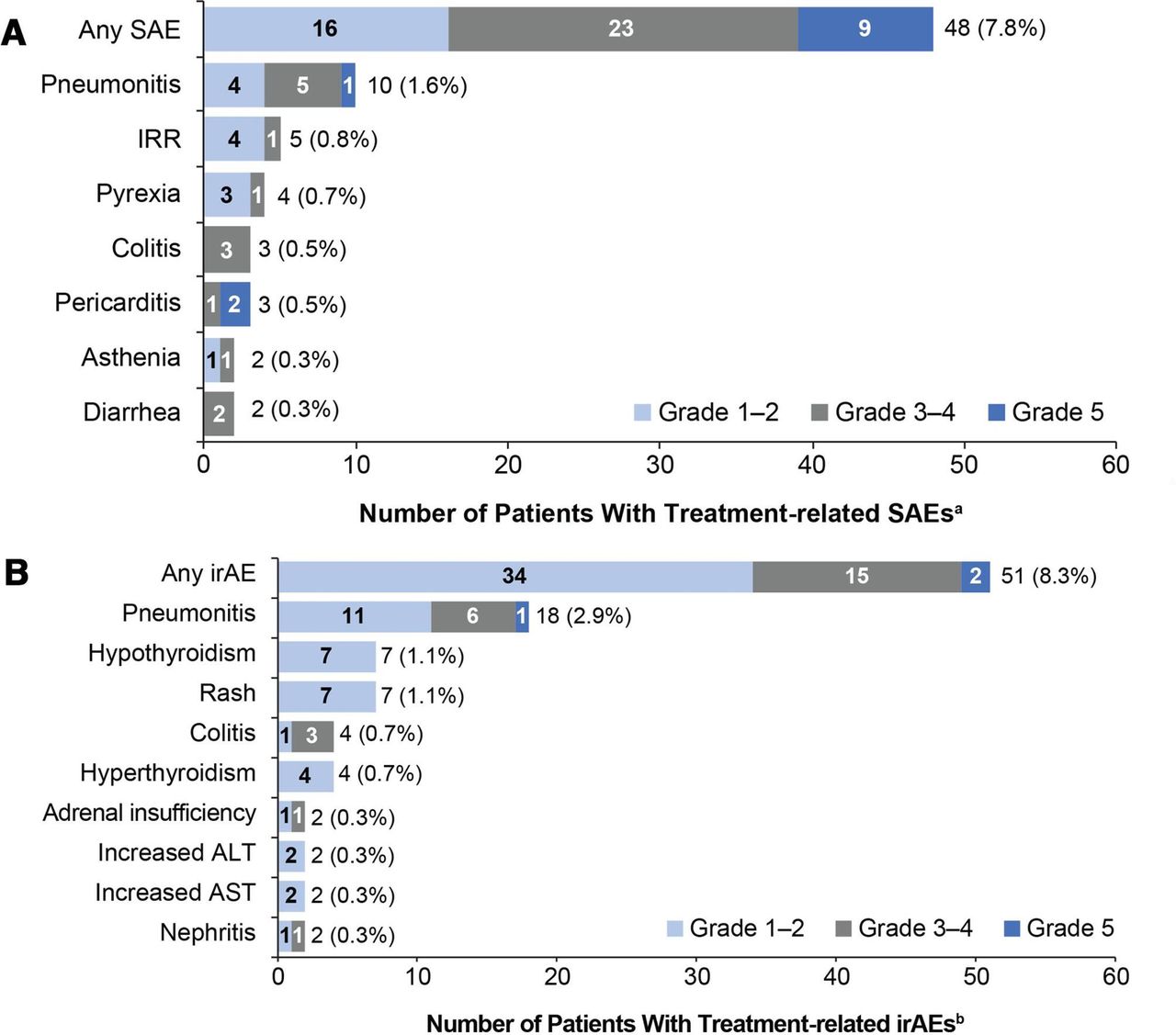
Primary endpoint: most common treatment-related SAEs and irAEs (in ≥2 patients) by CTCAE grade. (A) Treatment-related SAEs and (B) treatment-related irAEs. aAdverse events were defined as serious if they were fatal, were life threatening, required hospitalization, resulted in disability, or resulted in a congenital birth defect in an infant born to a mother exposed to study drug. birAEs were defined as any adverse event of special interest requiring corticosteroid treatment within 30 days of onset. ALT, alanine aminotransferase; AST, aspartate aminotransferase; CTCAE, Common Terminology Criteria for Adverse Events; irAEs, immune-related adverse events; IRR, infusion-related reaction; SAE, serious adverse event.
The second coprimary endpoint of treatment-related irAEs occurred in 8.3% of patients (95% CI 6.2 to 10.8), mostly at grade 1 or 3 severity (figure 2B). Grade 5 treatment-related irAEs occurred in 0.2% of patients (pneumonitis and hepatoxicity).
An overview of safety is shown in table 2. The most common grade 3–4 AEs were pneumonia (2.4%), anemia (2.0%), and worsening hyponatremia (1.8%) (online supplemental table A2). Grade 5 AEs occurred in 5.7% of patients; the most common were respiratory disorders (1.6%), infections (1.5%), general disorders and administration site conditions (1.1%) and cardiac disorders (0.8%). AEs that most frequently led to treatment discontinuation were pneumonitis (0.8%) and infusion-related reactions (0.5%). Treatment-related grade 3–4 events occurred in 8.8% of patients; the most common were pneumonitis (1.0%), fatigue and colitis (each 0.7%), and asthenia (0.5%). Treatment-related grade 5 events occurred in 1.5%; the only event to occur in more than one patient was pericarditis (0.3%).
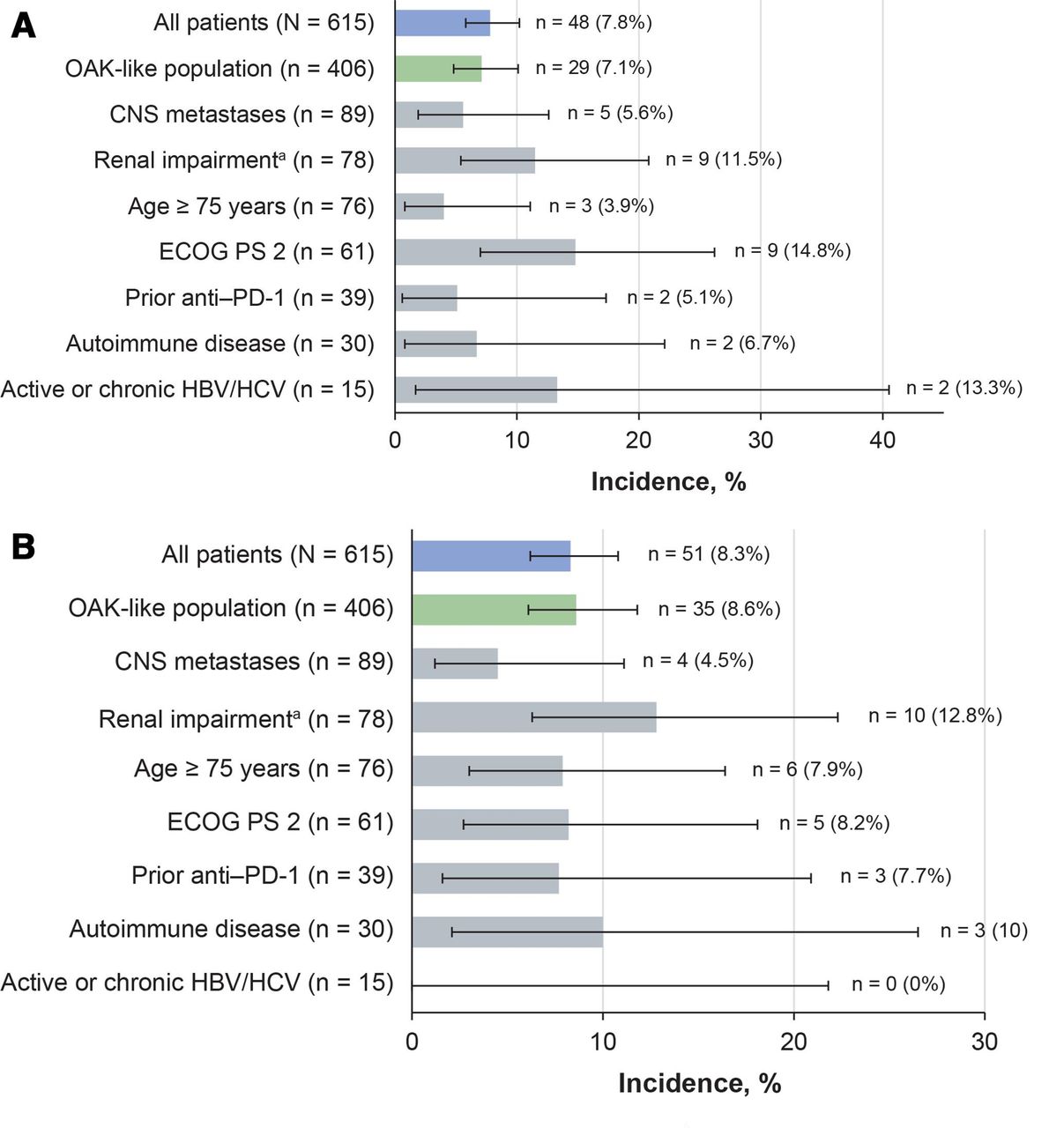
Incidences of treatment-related SAEs and irAEs in subgroups of special interest were generally comparable with those in the overall population (figure 3). In patients with ECOG PS 2, the incidence of treatment-related SAEs was higher than in the overall population, but treatment-related irAEs occurred at a rate similar to that in the overall population (figure 3). In this subgroup, the overall incidence of grade 3–4 AEs was higher than in the overall study population; however, fatigue was the only grade 3–4 AE to occur in more than two patients (8.2%). Other grade 3–4 events in this subgroup were mostly categorized as infection-related (9.8%) or respiratory-related disorders (8.2%). Grade 5 AEs in patients with ECOG PS 2 were stress cardiomyopathy, neurological deterioration, pneumonia, and pulmonary embolism, each in one patient (1.6%).
{kind=link}
{kind=link}
{kind=link}
Incidence of treatment-related SAEs and irAEs in key subgroups. (A) treatment-related SAEs and (B) treatment-related irAEs. The Clopper–Pearson method was used to calculate the 95% CI. aRenal impairment defined as estimated glomerular filtration rate <60 mL/min/1.73 m2. CNS, central nervous system; ECOG PS, Eastern Cooperative Oncology Group performance status; HBV/HCV, hepatitis B/C virus; irAE, immune-related adverse event; PD-1, programmed death-1; SAE, serious adverse event.
In patients with renal impairment, moderately higher incidences of treatment-related SAEs, irAEs (figure 3), and grade 3–4 AEs (table 2) were observed than in the overall study population. The most common grade 3–4 AEs were pulmonary embolism (5.1%), and fatigue, asthenia, and pneumonia (each 3.8%). Six patients with renal impairment (7.7%) had grade 3–4 renal events (including acute kidney injury and renal failure, each reported in one patient (1.3%)), but none led to treatment discontinuation. Grade 5 AEs in the renal impairment subgroup were two sudden deaths (2.6%), and pneumonia and pulmonary sepsis (each 1.3%).
The rates of treatment-related SAEs and irAEs in patients with AID were similar to those in the overall population (figure 3). In this subgroup, AEs with moderately increased incidences were generally respiratory or GI disorders of any grade; decreased appetite (26.7%), nausea (26.7%), fatigue (23.3%), and dyspnea (23.3%) were the most common. Nine patients (30%) experienced grade 3–4 events, none of which occurred in more than one patient. Pneumonitis occurred as a treatment-related grade 5 event in one patient (3.3%) and led to treatment discontinuation in two patients (6.7%).
In the 15 patients enrolled with HBV or HCV infection, treatment-related SAEs occurred at higher rates than in the overall population, but there were no treatment-related irAEs (figure 3A). Grade 3–4 AEs had higher incidence rates than the overall population (table 2), but none occurred in more than one patient, nor did they lead to treatment discontinuation. Safety results in patients with CNS metastases and those aged 75 years or older were similar to those in the overall population even though differences in median treatment durations were seen between these groups (figure 3, table 2). Most AEs in the elderly patients occurred at grade 1–2 severity, with most grade 3–5 events categorized as general/site administration (10.5%), infectious (9.2%), respiratory (6.6%), or metabolic disorders (5.3%).
Efficacy
In the overall study population, median OS was 11.1 months (95% CI 8.9 to 12.9), with a 12-month OS rate of 47.8% (table 3). The median PFS in the overall population was 2.7 months (95% CI 2.1 to 2.8), and the ORR was 11.1% (95% CI 8.7 to 13.8), which included a 0.5% complete response rate and a 10.6% partial response rate (table 3). Efficacy outcomes were similar in patients with squamous and non-squamous histology. The OAK-like subgroup showed a median OS of 13.7 months (95% CI 11.6 to 15.5) and 12-month survival rate of 54.3%.
Efficacy in all patients and selected subgroups
Patients in the subgroups of renal impairment, age ≥75 years, AID, and chronic/active HBV/HCV had efficacy outcomes similar to those in the overall and OAK-like populations (table 3). Patients in the ECOG PS 2, prior anti–PD-1 therapy, and CNS metastasis subgroups generally experienced poorer outcomes.
The median OS for PD-L1-positive patients was 12.6 months (95% CI 8.7 to 15.5) in the overall population and 15.5 months (95% CI 11.7 to NE) in the OAK-like population (online supplemental table A3). In PD-L1-negative patients, the median OS was 8.7 (95% CI 6.5 to 11.7) and 11.7 months (95% CI 8.0 to 13.7) in the overall and OAK-like populations, respectively. In untested patients, median OS was 12.5 months (95% CI 8.6 to 15.0) and 13.8 months (95% CI 9.0 to NE), respectively.
Discussion
In TAIL, atezolizumab monotherapy was evaluated in a diverse population of patients with previously treated advanced or metastatic NSCLC who more closely resemble a ‘real-world’ patient population than those typically enrolled in a pivotal phase III clinical study. The primary endpoint findings confirmed the safety profile of atezolizumab monotherapy in previous NSCLC studies.3 11 12 One-third of the patients enrolled in TAIL would have been excluded from the pivotal phase III OAK trial, principally due to their baseline comorbidities (eg, severe renal impairment and AID), laboratory parameters, ECOG PS 2, untreated CNS metastases, or prior treatment with anti–PD-1. Safety findings in these patient subgroups were as expected, with no new safety findings for atezolizumab. Despite the inclusion of these subgroups in TAIL, the median OS in the overall study population was 11.1 months at the clinical cut-off date, with a 12-month OS rate of 47.8%. The results of our exploratory analyses in the OAK-like subgroup corroborated the OS and ORR observed in OAK,3 as well as the histology findings and pooled PD-L1 IHC results. These findings are generally consistent with those for other checkpoint inhibitors in real-world settings and in key subgroups similar to those examined here.10 13 14
The ECOG PS 2 subgroup had higher incidences of grade 3–4 AEs, SAEs, and treatment-related SAEs than the overall population (reflecting the higher comorbidity at enrollment and increased hospitalizations), but rates of irAEs were consistent with those in the overall population and comparable with those reported with other checkpoint inhibitors.15 Efficacy outcomes in the ECOG PS 2 subgroup reflected in part their poorer prognosis. Nevertheless, these findings were in line with other published data in patients with ECOG PS 2 with nivolumab or pembrolizumab in the same setting and add evidence to support the safe use of checkpoint inhibitors in this difficult-to-treat population, although with lower efficacy.15–17 It remains to be demonstrated whether this seemingly lower efficacy of immunotherapy in patients with ECOG PS 2 is related to their inability to mount an effective immune response, or to a short-course outcome per se, which does not leave adequate time for the patient to benefit from anti–PD-1/PD-L1 treatment.
Whether retreatment of NSCLC with checkpoint inhibitors is safe and effective has not yet been demonstrated. Patients enrolled in TAIL who had received prior anti–PD-1 therapy may have discontinued it for any reason, including toxicity or progression. More than half these patients had received ≥3 lines of prior NSCLC therapy (compared with 6.5% of the overall population), suggesting that they had checkpoint inhibitor-resistant disease with a poorer prognosis. Nevertheless, one patient in this subgroup had a long-lasting partial response, consistent with previous reports that individuals may respond to retreatment with checkpoint inhibitors.18–20
Patients with AID are typically excluded from randomized controlled trials to avoid potential exacerbation or reactivation of their underlying immune conditions. Prospective studies of checkpoint inhibitors that include patients with AID are therefore scarce. Although relatively few patients with AID were enrolled in TAIL, their safety and efficacy outcomes were similar to those in the overall population and their moderate increases in AE tended to be respiratory or GI disorders and representative of the underlying disease. The slightly higher rate of irAEs in TAIL (13.3%) was consistent with other findings in patients with NSCLC and underlying AID who were treated with checkpoint inhibitors. In a retrospective cohort study of cancers treated with anti–PD-1 agents, 65.5% of the patients had NSCLC, of whom 11.3% had pre-existing AID.21 As in TAIL, the incidence of irAEs was higher in patients with AID than without (66% vs 40%); of the 11 patients with AID who had irAEs, 13% required systemic corticosteroid treatment. In the only other prospective cohort data, a higher incidence of irAEs was again observed in patients with AID versus those without (44% vs 29%), with six patients (13.3%) requiring systemic corticosteroid treatment.22 In TAIL, the irAEs observed in patients with AID were consistent with the known safety profile of atezolizumab, and reactivation of underlying diseases was not observed. Data from TAIL’s AID subgroup may help inform treatment decisions for patients with advanced NSCLC and AID and suggest that their inclusion in future clinical trials of anti–PD-L1 therapies is warranted.23
As in the AID subgroup, efficacy outcomes in the renal impairment subgroup were not impacted and safety remained manageable. Despite the higher incidence of grade 3–4 AEs in this subgroup (primarily renal events), only 6.4% of patients discontinued study treatment due to any AE. Rare cases of acute kidney injury and renal failure have been reported with cancer immunotherapy treatment, with an incidence of approximately 2%,24–26 and both events were reported with similar incidences in renally impaired patients in TAIL. In patients aged 75 years or older, despite a longer median treatment duration, the safety and efficacy outcomes were consistent with those of the overall TAIL population and with findings from other studies of checkpoint inhibitors in elderly patients.27–29 The pooling of locally and centrally tested PD-L1 data permitted the analysis of efficacy outcomes by PD-L1 status irrespective of PD-L1 IHC assay used. Although three-quarters of these PD-L1 data were generated from local assays, the efficacy outcomes in the overall population were improved in patients who were PD-L1 positive. Efficacy outcomes in the OAK-like subgroup were consistent with those in OAK for PD-L1-positive (TAIL median OS, 15.5; OAK median OS, 15.7 months) and PD-L1-negative patients (TAIL median OS, 11.7; OAK median OS, 12.6 months).3 Although analytical concordance among PD-L1 IHC assays differs, retrospective analyses of OAK30 and other NSCLC studies using various assays (Ventana SP142, SP263 or 22C3 pharmDx)31 have shown similar clinical outcomes among PD-L1 subgroups with atezolizumab monotherapy.30
The strength of TAIL’s design is its broad patient population, but the study nonetheless demonstrated safety results consistent with those of OAK,3 in which the patients had more favorable prognostic factors. Although there was no direct comparator in TAIL, efficacy outcomes for the OAK-like cohort were consistent with those in OAK3 and support the beneficial effect of atezolizumab seen across PD-L1 subgroups. In general, too few patients were enrolled from understudied NSCLC populations to permit meaningful conclusions about atezolizumab treatment in these subgroups. Nevertheless, TAIL does provide valuable information on the safety and efficacy of atezolizumab in a broader population of patients with NSCLC than is generally studied, including those with a poorer prognosis or comorbidities that could be exacerbated by treatment with immunotherapy.
Conclusions
This study confirmed that the benefit–risk profile of atezolizumab monotherapy for previously treated NSCLC was acceptable in diverse patient subgroups, including those with an ECOG PS of 2, renal impairment, and AID. These findings may help inform treatment decisions for patients generally excluded from NSCLC pivotal trials and could encourage broadening of study populations enrolled in future studies.
Acknowledgments
We would like to thank the patients and their families, the investigators, and the clinical study sites. This study is sponsored by F. Hoffmann-La Roche, Ltd. Medical writing support was provided by Preshita Gadkari, PhD, and Samantha Santangelo, PhD, of Health Interactions and funded by F. Hoffmann-La Roche, Ltd.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors Conceptualization: AA, JT, TN-D, KT, AC, SA, HP, PP-M, and BR-V. Data curation: AC, SA, BR-V, and KS. Formal analysis: TN-D, AC, HP, and PP-M. Funding acquisition: PP-M. Investigation: AA, DR-A, TN-D, KT, SA, PP-M, BR-V, HJMS, JY, KS, and JA-A. Methodology: TN-D, KT, AC, HP, and PP-M. Project administration: SA and PP-M. Resources: DR-A, KT, SA, BR-V, JY, KS, and JA-A. Software: AC. Supervision: AA, JT, and PP-M. Validation: AC, SA, PP-M, and KS. Visualization: AC, SA, HP, BR-V, and JA-A. Writing–original draft preparation: PP-M. Writing–review and editing: all authors. All authors read and approved the final manuscript.
Funding This work was supported by F. Hoffmann-La Roche/Genentech, which sponsored the study, provided the study drugs, had a role in the decision to submit the paper for publication, and collaborated with the academic authors on the study design, data collection, analysis, and interpretation. All manuscript drafts were prepared by the authors with editorial assistance funded by the sponsor. All authors approved the submission and vouched for data accuracy and completeness.
Competing interests Support of the parent study and funding of editorial support were provided by F. Hoffmann-La Roche/Genentech. AA is a consultant to Merck Sharpe & Dohme, AstraZeneca, Bristol Myers Squibb, and F. Hoffmann-La Roche and has received research funding from Bristol Myers Squibb, F. Hoffmann-La Roche, and Celgene and honoraria from Eli Lilly and Pfizer. SA is a consultant to and has received research funding, honoraria, and travel expenses from F. Hoffmann-La Roche, Bristol Myers Squibb, and Celgene, is a consultant to Merck Sharpe & Dohme, and has received research funding, travel expenses, and honoraria from Novartis, Eli Lilly, and AstraZeneca. BRV is a consultant to Merck Sharpe & Dohme and Eli Lilly and has received honoraria and travel expenses from F. Hoffmann-La Roche, Eli Lilly, Bristol Myers Squibb and Merck Sharpe & Dohme. DR-A is an advisor to and has received speaker honoraria from Bristol Myers Squibb, Boehringer Ingelheim, Eli Lilly, Merck Sharpe & Dohme, F. Hoffmann-La Roche, Pfizer, and AstraZeneca. JA-A is a consultant to, on a speaker bureau for, and has received travel expenses from Merck Sharpe & Dohme, AstraZeneca, Pfizer, Boehringer Ingelheim, Eli Lilly, Novartis, F. Hoffmann-La Roche, and Bristol Myers Squibb. HJMS is a consultant to Merck Sharpe & Dohme, AstraZeneca, and Bristol Myers Squibb and is secretary of the oncology section of NVALT (Dutch lung physician’s organization) and president of the Dutch Lung Cancer Audit. JY has nothing to disclose. KS is on a speaker bureau for Merck Sharpe & Dohme, has received research funding from F. Hoffmann-La Roche and Novartis, has received research funding and travel expenses from Bristol Myers Squibb, and has received travel expenses from Genesis. KT, JT, and AC are employees of F. Hoffmann-La Roche. HP and PP-M are employees of Genentech. TN-D is a consultant to Amgen, Bayer, AstraZeneca, Bristol Myers Squibb, Boehringer Ingelheim, Eli Lilly, Merck Sharpe & Dohme, Novartis, Otsuka, Pfizer, Roche, and Takeda, and is on a speaker bureau for AstraZeneca, Merck Sharpe & Dohme, F. Hoffmann-La Roche, and Takeda.
Patient consent for publication Not required.
Ethics approval The study was conducted in full accordance with the E6 guidelines for Good Clinical Practice and the Declaration of Helsinki. The protocol was approved by each institution’s institutional review board or ethics committee, and informed consent was obtained from all patients prior to their inclusion in the study.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement All data relevant to the study are included in the article or uploaded as supplementary information. Qualified researchers may request access to individual patient level data through the clinical study data request platform (https://vivli.org/). Further details on Roche’s criteria for eligible studies are available here (https://vivli.org/members/ourmembers/). For further details on Roche’s Global Policy on the Sharing of Clinical Information and how to request access to related clinical study documents, see here (https://www.roche.com/research_and_development/who_we_are_how_we_work/clinical_trials/our_commitment_to_data_sharing.htm).
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.