Article Text

Abstract
Background Resident memory T lymphocytes (TRM) are located in tissues and play an important role in immunosurveillance against tumors. The presence of TRM prior to treatment or their induction is associated to the response to anti-Programmed cell death protein 1 (PD-1)/Programmed death-ligand 1 (PD-L1) immunotherapy and the efficacy of cancer vaccines. Previous work by our group and others has shown that the intranasal route of vaccination allows more efficient induction of these cells in head and neck and lung mucosa, resulting in better tumor protection. The mechanisms of in vivo migration of these cells remains largely unknown, apart from the fact that they express the chemokine receptor CXCR6.
Methods We used CXCR6-deficient mice and an intranasal tumor vaccination model targeting the Human Papillomavirus (HPV) E7 protein expressed by the TC-1 lung cancer epithelial cell line. The role of CXCR6 and its ligand, CXCL16, was analyzed using multiparametric cytometric techniques and Luminex assays.
Human biopsies obtained from patients with lung cancer were also included in this study.
Results We showed that CXCR6 was preferentially expressed by CD8+ TRM after vaccination in mice and also on intratumoral CD8+ TRM derived from human lung cancer. We also demonstrate that vaccination of Cxcr6-deficient mice induces a defect in the lung recruitment of antigen-specific CD8+ T cells, preferentially in the TRM subsets. In addition, we found that intranasal vaccination with a cancer vaccine is less effective in these Cxcr6-deficient mice compared with wild-type mice, and this loss of efficacy is associated with decreased recruitment of local antitumor CD8+ TRM. Interestingly, intranasal, but not intramuscular vaccination induced higher and more sustained concentrations of CXCL16, compared with other chemokines, in the bronchoalveolar lavage fluid and pulmonary parenchyma.
Conclusions This work demonstrates the in vivo role of CXCR6-CXCL16 axis in the migration of CD8+ resident memory T cells in lung mucosa after vaccination, resulting in the control of tumor growth. This work reinforces and explains why the intranasal route of vaccination is the most appropriate strategy for inducing these cells in the head and neck and pulmonary mucosa, which remains a major objective to overcome resistance to anti-PD-1/PD-L1, especially in cold tumors.
- adaptive immunity
- CD8-positive T-lymphocytes
- vaccination
- immunization
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Different subpopulations of memory CD8+ T cells have been characterized: (1) central memory CD8+ T cells, expressing CCR7 and CD62L lymph node-homing receptors, are mainly found in secondary lymphoid organs; (2) effector-memory CD8+ T cells, not expressing the receptors for migration to lymphoid organs, circulate in the periphery and in tissues and play an effector role (ie, release of cytotoxic granules and cytokine secretion); (3) CD8+ memory stem cells are poorly differentiated with a high capacity for self-renewal and proliferation and are able to reconstitute the whole spectrum of memory CD8+ T-cell populations; and (4) resident memory CD8+ T cells (TRM) expressing retention receptors such as CD103, CD49a and CD69, are found in non-lymphoid peripheral tissues and do not recirculate except for possible retrograde transport in lymph nodes.1 Recent studies have also reported that CD8+ TRM cells can give rise to circulating effector and memory T cells, but they remain predisposed to migrate back to their tissue of origin.2 These cells are among the first cells able to act in healthy or tumor tissues, and because of their local mobility, they play a major role in tissue immune surveillance.3 4 After their activation, these cells also allow the recruitment of circulating immune cells via the secretion of cytokines and chemokines in order to amplify the local immune response.5 These TRM cells appear to be more cytotoxic than other CD8+ T effector cells, as CD103 expression is correlated with the levels of granzymes.6
The role of TRM in antitumor immunity has recently been highlighted. For example, it has been shown in mice that these lymphocytes delay tumor progression.7 8 Our team has shown that TRM specifically induced by intranasal vaccination plays an essential role in controlling the growth of orthotopic murine head and neck or lung tumors.9 10 Other studies have identified TRM as a key mediator of cancer vaccines targeting mucosal tumors.11 12
In humans, TRM has been found in different types of cancer, such as melanoma, urothelial carcinoma, endometrial adenocarcinoma, and particularly lung cancer.9 13 14
Our team and other groups have shown that a larger CD103+CD8+ T-cell infiltrate was associated with better survival in lung cancer, even in multivariate analysis.6 9 13 In the present study, we wanted to analyze the mechanism by which intranasal vaccination promotes preferential intratumoral infiltration of TRM in head and neck cancer and lung tumors, compared with systemic vaccination. Previous results, based on phenotypical and transcriptomic analysis, from our team and other groups, have identified CXCR6 chemokine receptor as a core marker of TRM present in lung or head and neck tumors, but its function has not been investigated.6 9 15–18
CXCR6 binds a unique ligand, CXCL16, which can exist in transmembrane and soluble forms,19 the latter requiring cleavage from the membrane by a disintegrin and metalloproteinase domain-10, or a disintegrin and metalloproteinase domain-17.20 CXCL16 is produced by epithelial and immune cells and can serve as an alarmin to recruit cells to the site of inflammation.21 22
In contrast with TRM, CXCR6 is detected at very low levels on naive CD8+ T cells19 and is upregulated by priming by dendritic cells,23 or after T-cell receptor activation.24
CXCR6 promotes homing of lymphocytes to non-lymphoid tissues, such as the skin,25 the liver in hepatitis C,26 the synovium in rheumatoid arthritis27 and the brain in experimental autoimmune encephalomyelitis.28
Cxcr6 is also one of the 100 genes presenting the greatest difference in expression between TRM in the lung and Teff in the blood.29 The fact that interleukin-15 is required for the differentiation of TRM30 31 and upregulates the expression of CXCR632 reinforces the link between CXCR6 and TRM.
As the role of CXCR6 in the migration of tumor-specific TRM to mucosal cancers (head and neck and lung cancers) after intranasal vaccination has not yet been addressed, we chose in the present work to focus on this key question, as it might have direct consequences on patient response to cancer immunotherapy.
Materials and methods
Mice
Female C57BL/6J wild-type mice were purchased from Janvier Labs. CXCR6gfp/gfp mice, are homozygous CXCR6-deficient mice, in which the coding region of the chemokine receptor CXCR6 has been substituted with the coding region of the Enhanced Green Fluorescent Protein (eGFP). These mice were obtained from Jackson Laboratory (cat# JAX: 005693) and bred in our animal facility. Male CXCR6gfp/gfp mice were crossed with female C57BL/6J mice to generate CXCR6gfp/+ mice. B6.SJL-PtprcaPepcb/BoyJ (CD45.1) were purchased from Charles River. CD3 knockout (KO) mice were obtained from B Malissen’s laboratory (CIML, Marseille, France) and bred in our animal facility.
Mice were used in experiments at 8–10 weeks of age. All mice were housed in INSERM U970-PARCC animal facility under specific pathogen-free conditions.
Tumor cells
TC-1 cells expressing the Human Papillomavirus (HPV)16 E6–E7 proteins were obtained from the laboratory of T C Wu (Department of Pathology, School of Medicine, Johns Hopkins University, Baltimore, Maryland, USA). Cells were cultured in RPMI 1640 (Life Technologies) supplemented with 10% heat-inactivated fetal calf serum (FCS, GE Healthcare), 1 mM sodium pyruvate (Life Technologies), 1 mM non-essential amino acids (Life Technologies), 100 U/mL penicillin and 100 µg/mL streptomycin (Life Technologies), and 0.5 mM 2-β mercaptoethanol (Life Technologies), and incubated at 37°C in 5% CO2. They were regularly tested for mycoplasma contamination.
Vaccine and adjuvant
The STxB-E7 vaccine was produced by the chemical coupling of the N-bromoacetylated E743–57 peptide to the sulfhydryl group of a recombinant nontoxic Shiga toxin B-subunit variant according to previously described procedures.33 After purification, endotoxin concentrations determined by the Limulus assay test (Lonza, Aubergenville, France) were <0.5 endotoxin unit/mg. Polyinosinic-polycytiylic acid-poly-l-lysine carboxymethylcellulose (Poly-ICLC) were obtained from Oncovir (Washington, USA).
Tumor challenge and vaccination protocol
Anesthetized mice were injected with 5×104 TC-1 tumor cells in the submucosal area of the tongue with a Hamilton Gastight syringe of 10 µL, or in the mucosal part of the cheek with a 26 G insulin syringe. Mice were daily monitored for survival analysis, and tumor size was measured every 2–3 days with a caliper (volume mm3=(length×width×width)/2).
Anesthetized mice were immunized two times per day (D)0 and at D14 by intranasal or intramuscular routes with STxB-E7 (20 µg) associated with poly-ICLC (10 µg) as adjuvant for the first immunization. Total volume injected was 25 µL for the intranasal route and 50 µL for the intramuscular route. In the prophylactic setting, tumors were grafted 7 days after the second immunization, whereas for the therapeutic setting, mice were vaccinated at D5 and 10 after tumor graft for tongue tumor and at D7 and D14 for cheek tumor.
The mean survival time was calculated using the Kaplan–Meier method and statistical analysis was performed using a log-rank test. Analysis of differences in tumor volume were performed with two-way analysis of variance (ANOVA) and Bonferroni or Tukey post hoc test.
Isolation of lymphocytes from bronchoalveolar lumen fluid, lung parenchyma, tumors and spleen
Intravascular staining was performed to discriminate between tissue-localized and blood-borne cells as described by Anderson et al.34 Briefly, 5 µg of anti-CD8a APC-efluo780 (clone 53-6-7, ebioscience/Thermofisher) was injected intravenously 3 min prior to bronchoalveolar lavage (BAL) and tissue harvest.
Bronchoalveolar lavage (BAL) was obtained by flushing the lungs with phosphate-buffered saline(PBS)-EDTA 0.5 mM via a cannula inserted in the trachea (5 washes×1 mL).
Lungs were perfused with PBS-EDTA 0.5 mM and digested in RPMI-1640 medium supplemented with 1 mg/mL collagenase type IV (Life Technologies/Thermofisher) and 30 µg/mL DNase I (Roche). Lungs were dissociated using the GentleMACS (Miltenyi Biotec, France) lung programs 1 and 2, with gentle shaking at 37°C for 30 min in between both steps. Then, the obtained single‐cell suspensions were filtered through a 70 μm strainer washed with PBS–FBS 2%, suspended in 40% Percoll solution and layered over 75% Percoll solution (Sigma-Aldrich), and centrifuged at 600×g for 20 min at room temperature (RT). Interface cells were collected and washed. Tumors were harvested, minced and placed into GentleMACS C-tube with PBS-FCS 2%, dissociated mechanically with GentleMACS dissociator (Miltenyi) according to the manufactor’s standard protocol, then filtered on a 70 μm strainer.
Spleens were dissected and pressed through a 40 µM cell strainer, red blood cells were lysed with osmotic lysis buffer.
Cheek tumors were harvested and mechanically dissociated by using the GentleMACS (Miltenyi, Bergisch Gladbach, Germany) (program m-imp-tumor-01–01), then filtered on a 70 µm strainer.
Flow cytometry
After FcR blocking with CD16/32 Ab (clone 93, ebioscience/ Life Technologies), cells were first incubated for 30 min at RT with PE-conjugated DbE749–57 (R9F) tetramer (Immudex). Then, cells were washed and stained 20 min at 4°C with the following Abs : anti-mouse CD8b AF700 or BUV495 (clone YTS156), CD3 PercpCy5.5 (clone 145 2C11, ebioscience/Life Technologies), CD103 Pacific Blue (clone 2E7, Biolegend), and CD49a Alexa 647 (clone Ha31/8, BD Biosciences). For chemokine receptor analysis, cells were stained 30 min at RT with anti-mouse CCR5 PE-CY7 (clone HM-CCR5), CCR6 BV785 (clone 29–2 L17), CXCR3 BV650 (clone CXCR3-173), CXCR6 BV711 (clone SA051D1) (all Abs from Biolegend). Then staining was performed as described previously. All the cells were labeled using the live/dead cell aqua blue viability (Life Technologies).
Non-circulating CD8+ T cells were defined as CD3+CD8anegCD8b+. Acquisitions were performed on BD Fortessa X20 (Becton Dickinson), and data were analyzed on live singulet cells with FlowJo Software (BD).
Chemokines multiplex assays and ELISA
Supernatants from the first BAL’s washes were collected. Serum samples were prepared from blood collected from retro-orbital sinus. Proteins were extracted from perfused lungs and from spleen by using Bioplex lysis buffer (Bio-Rad), and total proteins were determined by bicinchoninic acid (BCA) assay (Pierce) according to the manufacturer’s protocol.
Chemokines was measured by bead-based multiplex immunoassay: CXCL16, MIP1a/CCL3,MIP1b/CCL4,RANTES/CCL5,IP10/CXCL10, CCL20 (R&D Systems Biotechne), according to manufacturer protocol and were analyzed on Bio-Plex 200 (Bio-Rad). The analyte concentration was calculated using a standard curve (5 Parameter logistic (PL) regression), with Bio-Plex manager software. When indicated, CXCL16 was measured by ELISA (R&D Systems Biotechne) according to manufacturer protocol.
Transcriptomic analysis
C57BL/6 mice were immunized with STxB-E7 (20 µg)+α-GalCer (2 µg) by intranasal or intramuscular route and then immunized a second time with STxB-E7 (20 µg) 14 days later (prime-boost). Seven days after the second immunization, the BAL or spleen of the mice were recovered, and the CD44hitetramer+CD8+T cells were sorted by BD FACSAria II cell sorter. Then, RNA of the cells was extracted using the QIAGEN RNeasy Plus Micro Kit and then genotyped by DNA chip (Affymetrix) by the genomics and transcriptomics platform of the Cochin Institute.
Data were normalized using Robust Multiarray Averaging (RMA) algorithm in Bioconductor with the custom CDF vs 18 (Dai M et al Nucleic Acids Res 2005). Statistical analysis was carried out with the use of Partek GS. All the data have been deposited in NCBI’s Gene Expression Omnibus44 and are accessible through GEO Series accession number GSE77366 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?token=ghsrcweibruzvsj&acc=GSE77366).
First, variations in gene expression were analyzed using unsupervised hierarchical clustering and PCA to assess data from technical bias and outlier samples. To find differentially expressed genes, we applied one-way ANOVA for each gene and made pairwise Tukey’s post hoc tests between groups. Then, we used p values and fold changes to filter and select differentially expressed genes. Differentially expressed genes (DEG) enrichment analysis was carried out using Ingenuity (Ingenuity Systems, USA; www.ingenuity.com).
Data availability
All data were deposited in NCBI’s Gene Expression Omnibus and are accessible through GEO Series accession number GSE77366 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?token=ghsrcweibruzvsj&acc=GSE77366).
Human lung cancer
Lung cancer biopsies were obtained from non-treated patients with non-small-cell lung carcinoma who underwent a lobectomy at the thoracic surgery department of European Georges Pompidou Hospital. Briefly, biopsies were digested for 45 min at 37°C with DNAse I (30 IU/mL, Roche) and collagenase type IV (1 mg/mL, Life Technologies/Thermofisher), then filtered through a 70 µm strainer washed with PBS–FCS 2%. Flow cytometry analysis of tumor-infiltrating lymphocytes was performed. : Receptors for the Fc region (FcRs) were first blocked with Human TruStain FcX (Biolegend) 5 min at RT, then stained with anti-human CD3 APC-CY7 (clone HIT-3a, Biolegend), CD8 BUV395 (clone RPA-T8, BD Biosciences), CD103 Percpcy5.5 (clone Ber-ACT8, Biolegend), CD49a PE (clone TS2-7, Biolegend), PD1 BV650 (clone EH12.2H7, Biolegend), CXCR6 biotin (clone K041E5, Biolegend) and steptavidin BV711 (Biolegend). All cells were labeled using the live/dead cell aqua blue viability (Life Technologies). Acquisitions were performed on BD Fortessa X20 (Becton Dickinson), and data were analyzed on live singulet cells with FlowJo Software (BD).
Statistical analysis
Data are presented as mean±SEM. Statistical comparisons were done with Prism V.8 GraphPad Software (San Diego, California, USA). Analysis of difference between two groups was performed with Mann-Whitney t-test; analysis of difference between more than two groups was performed with a one-way ANOVA and Holm-Sidak or Tukey post hoc. Mice survival was estimated using the Kaplan-Meier method and log-rank test. P values lower than 0.05 (*) were considered significant.
Results
CXCR6 expression is upregulated on CD8+ T cells with a TRM phenotype induced by ntranasal vaccination
Our previous study showed that in contrast to the spleen-derived E739–47-specific CD8+ T cells induced by intramuscular immunization, the BAL’s E749-57-specific CD8+ T cells induced by intranasal immunization express the core gene defining TRM (Itgae encoding CD103, Itga1 encoding CD49a and Itgb1, which associates with Itga1 to form the VLA-1 integrin).9 Here, we have completed this analysis by a transcriptomic analysis, showing that E749–57-specific CD8+ T cells with induced by intranasal immunization in the BAL and displaying a TRM phenotype preferentially express CXCR6 compared with those induced in the spleen after intramuscular immunization (fold change 6.2, p=0.00035) (figure 1, right). As we have previously reported, intramuscular vaccination did not induce TRM in the BAL and we could not check for their expression of CXCR6 after this route of immunization.9

Cxcr6 upregulation on CD8+ T cells with a TRM phenotype induced by intranasal vaccination revealed by heat MAP analysis. Mice were vaccinated via intranasal or intramuscular routes (n=4 mice per group) with STxB-E7 and the adjuvant α-GalCer (prime D0) and boosted at D14 with STxB-E7. BAL from in vaccinated mice and spleens from intramuscularly vaccinated mice were collected at day 21. H2-Db E749-57 tetramer+ CD44hi CD8+ T cells were sorted and the RNA was extracted. Whole gene expression microarray analysis was performed according to the procedure described in the methods section. Left: total gene analysis already reported in Nizard et al9 and not detailed here. Rright: focus on the expression of CXCR6 by TRM cells. Top: BAL CD8+ T cells (lanes 1–3) showed a typical TRM gene expression profile (CD103+ and CD49a+), which was not observed in splenic CD8+ T cells (lanes 4–6) (red square means overexpressed, while green square means underexpressed). Bottom: table of the transcriptomic characteristics of the Itga1, Itgb1, Itgae and Cxcr6 RNAs, with their p value and ‘fold change’. The fold change represents the difference in quantity between the genes expressed by CD8+ T cells from BAL after intranasal immunization and from the spleen after intramuscular immunization. These extractions were repeated at least three times. BAL, bronchoalveolar lavage.
CXCR6 is preferentially expressed on intranasal vaccine-induced E7-specific CD8+ T cells displaying resident memory T cell (TRM) phenotype
To investigate the potential role of CXCR6 in the establishment of lung-homing CD8+ T-cell populations, we used heterozygous Cxcr6 gfp/+ mice, in which one allele of the Cxcr6 gene has been replaced by the coding region of gfp.35 We also examined the expression of CXCR6 on lung resident CD8+ T cells at the protein levels using a recently available anti-CXCR6 antibody. In the first series of experiments, Cxcr6gfp/+ mice were first grafted with TC-1 cells in the tongue, then intranasally immunized with STxB-E7 at D5 and D10, and then tumor, BAL, lung and spleen were harvested at D15 (figure 2A). We then analyzed CXCR6 expression on E749-57-specific CD8+ T cells with a TRM phenotype, defined as CD8+ T cells expressing CD103 and/ or CD49a as previously reported,9 or with an effector phenotype without the expression of these markers. CXCR6 was analyzed by flow cytometry based on GFP expression or with the use of anti-CXCR6 mAb (figure 2B).9 In tumor, lung parenchyma and BAL, we found a statistically significant increase in membrane CXCR6 expression on E7-specific CD8+ T cells with a TRM phenotype, when compared with cells with an effector phenotype (figure 2C).

Cxcr6 (membrane expression) is preferentially expressed on CD8+ resident memory T cells in mice and humans. (A) Cxcr6gfp/+ mice were grafted with TC-1 cells (5×104) in the tongue at D0, then vaccinated via the in route with STxB-E7 and poly-ICLC at D5 and D10 and sacrificed at D15. (B) Gating strategy for TRM (in blue: Boolean gate CD103+ and/or CD49a+) and Teff (in orange: CD103−CD49a−). Representative flow plots showing GFP expression versus surface expression of CXCR6 on TRM and Teff. (C) Percentage of CXCR6 membrane expression in tumor, lung parenchyma (CD8a IV−) and BAL. Mean±SEM, n=4/group. Results are representative of two experiments. (D–F) Fresh biopsies from a patient with lung cancer (n=4) were dissociated and digested, and flow cytometry analysis of tumor-infiltrating lymphocytes was then performed. (D) Representative flow plot of CD103 and CD49a expression and gating strategy on live CD8. (E) Representative histograms of CXCR6 expression (left), percentage (middle) and MFI (right) of CXCR6 with TRM phenotype (Boolean gate CD103+ and/or CD49a+) and CD103−CD49a−CD8+ T cells. (F) Representative histograms (left) and percentage of PD1 (right) and Tim-3 (left) among CXCR6+ and CXCR6− TRM. Mean±SEM paired t-test. Poly-ICLC, polyinosinic-polycytiylic acid-poly-l-lysine carboxymethylcellulose. GFP, Green Fluorescent Protein. *P<0.05, **P<0.01. BAL, bronchoalveolar lavage.
Surprisingly no difference in the expression of GFP could be detected between the TRM and the effector T cells in the various organs (online supplemental figure S1). Differences in the expression of GFP and membrane CXCR6 had already been observed with these mice.36 We considered that the membrane expression of CXCR6 was the most relevant cellular localization for the function of a chemokine receptor.
Supplemental material
Next, we validated this preferential expression of CXCR6 on human TRM derived from lung tumors. Remarkably, 58% of intratumoral TRM expressed CXCR6, while it is detected on only 20% of intratumoral effector CD8+ T cells (figure 2D,E). CXCR6 Mean fluorescence intensity (MFI) expression was also higher on TRM than on effector cells (figure 2E). Phenotype characterization of these CXCR6+ TRM showed that they express PD-1 more strongly than CXCR6 negative TRM (figure 2F). These results show that both mouse and human tumor TRM cells preferentially expressed CXCR6 compared with effector T cells.
CXCR6 deficiency impairs induction of antigen-specific CD8+ T cells and TRM in the airway after intranasal vaccination
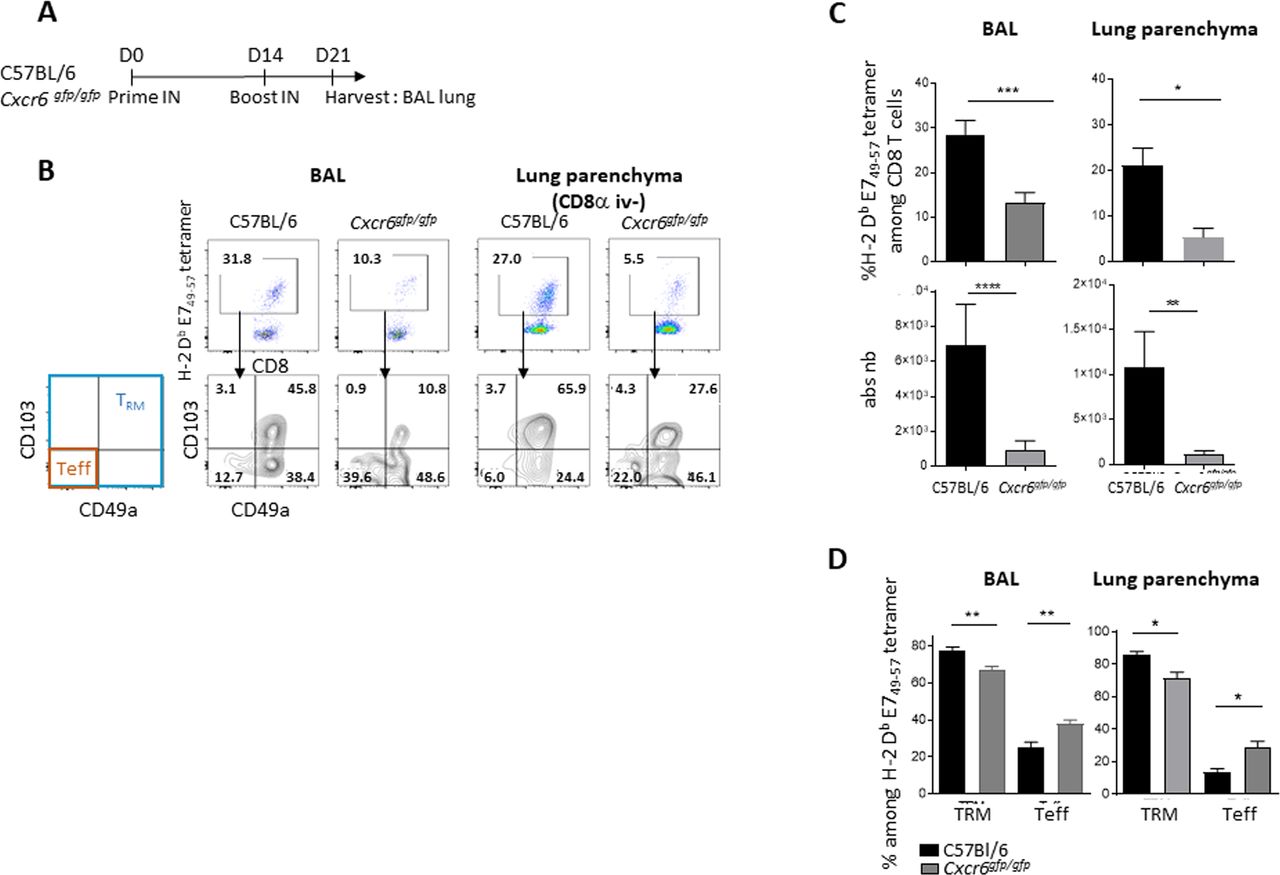
We then investigated the role of CXCR6 in the induction of E749–57-specific CD8+ T and TRM cells in BAL and spleen using mice lacking this chemokine receptor (Cxcr6gfp/gfp) (figure 3A). We observed a statistically significant reduction in the total number of E749–57-specific CD8+ T and TRM cells in BAL and lung parenchyma of Cxcr6-deficient mice, when compared with C57BL/6 wild-type mice (figure 3B,C). This decrease is more pronounced on E7 specific T cells with a TRM phenotype than with an effector phenotype (figure 3D). The latter population is even increased in Cxcr6-deficient mice compared with wild-type mice (figure 3D). After vaccination, we did not find any difference in the E749-57 tetramer levels in the spleen between wild-type and Cxcr6gfg/gfpmice (online supplemental figure S2A,B). Notably, CXCR6 membrane expression on E749-57 specific CD8+ T cells was not detected in the spleen of Cxcr6gfp/+ mice (online supplemental figure S2C).
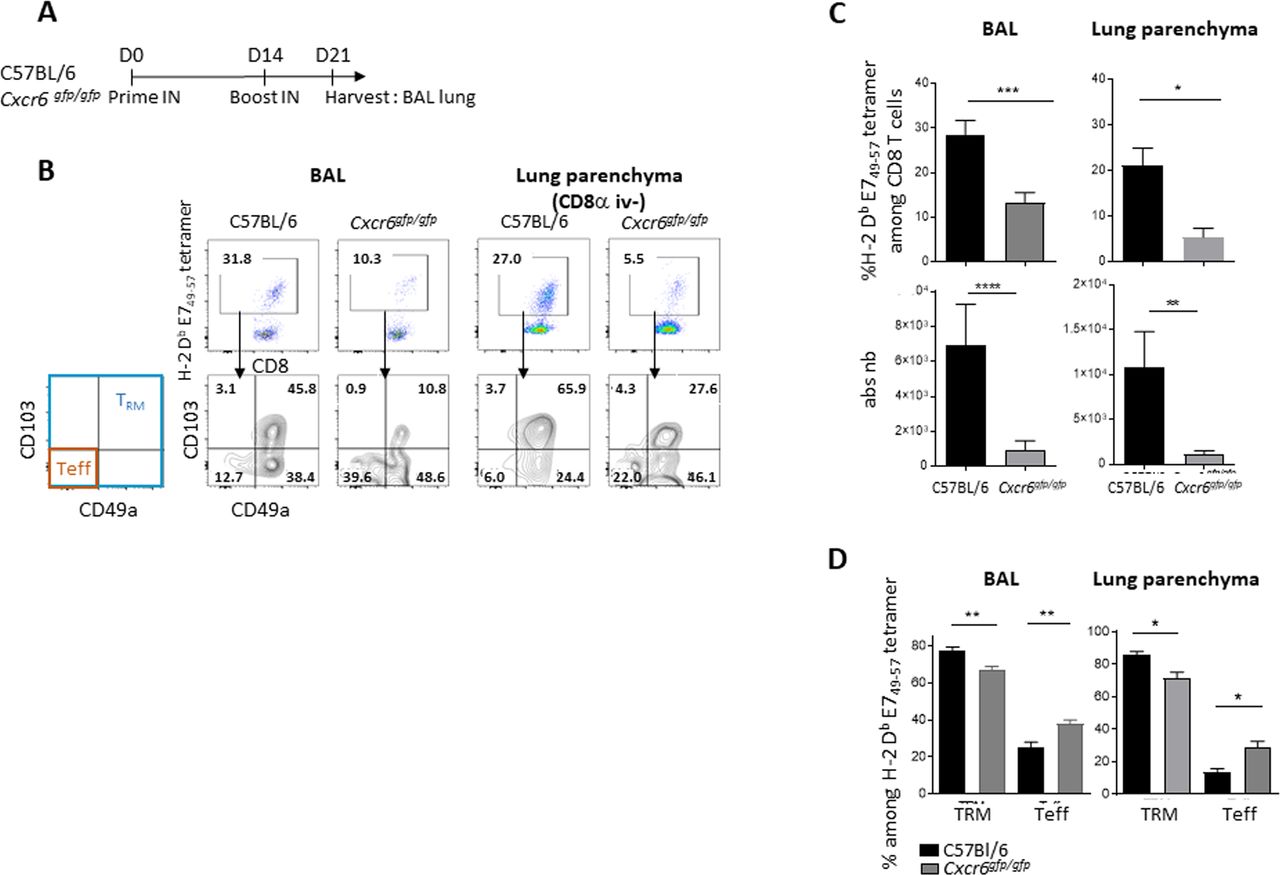
Induction of specific CD8+ T cells and TRM in the lung and airway is impaired in Cxcr6-deficient mice (A) C57BL/6 or Cxcr6gfg/gfp mice were vaccinated with STxB-E7 via the in route at days 0 and 14, then sacrificed at day 21. (B) Representative flow plots of H-2 Db E749-57 tetramer gated on CD8+ T cells (top), and TRM (Boolean gate CD103+ and/or CD49a+) and effector T cells (teff) (CD103−CD49a−) in BAL and lung parenchyma gated on H-2 Db E749-57 tetramer (bottom). (C) Percentage and absolute number of H-2 Db E749-57 tetramer in BAL and lung parenchyma. (D) Percentage of TRM and teff in BAL (n=21–22 mice/group) and lung parenchyma (n=5–7 mice/group). these experiments were repeated three times. Mean±SEM Mann-Whitney t-test. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001. BAL, bronchoalveolar lavage.
As Cxcr6 deficiency is not restricted to T cells only, we performed a transfer experiment, in CD3-deficient mice, with T cells derived from wild-type mice (CD45.1) or Cxcr6-deficient mice (CD45.2). We show that mice which had received the Cxcr6-deficient T cells had a decrease in the absolute number of anti-E749-57 CD8+ T cells in the BAL compared with mice which had received T cells derived from wild-type mice. A similar trend exists in the pulmonary parenchyma. This result confirms the intrinsic role of Cxcr6 deficiency in T cells on the decrease of the local TRM after vaccination by the intranasal route (online supplemental figure S3).
CXCR6 deficiency partially reverses cancer vaccine control of tumor growth in orthotopic head and neck tumor models
To assess the consequence of Cxcr6 deficiency on the recruitment of CD8+ T cells in the tumor microenvironment and on the control of tumor growth, we set up two orthotopic models using TC-1 tumor cells that were grafted in the cheek or in the tongue.
C57BL/6 wild-type or Cxcr6 gfp/gfp mice were challenged with TC-1 cells in the submucosal lining of the cheek (intracheek) then vaccinated or not with STxB-E7 by intranasal route at D7 and D14, and the tumor volume was monitored (figure 4A). Vaccinated C57BL/6 wild-type mice showed significant inhibition of tumor growth based on the measurement of tumor weight or tumor size in comparison to non-vaccinated mice (figure 4B–D). In contrast, we observed a partial loss of tumor control by the vaccine in Cxcr6 gfp/gfp mice (figure 4B–D).
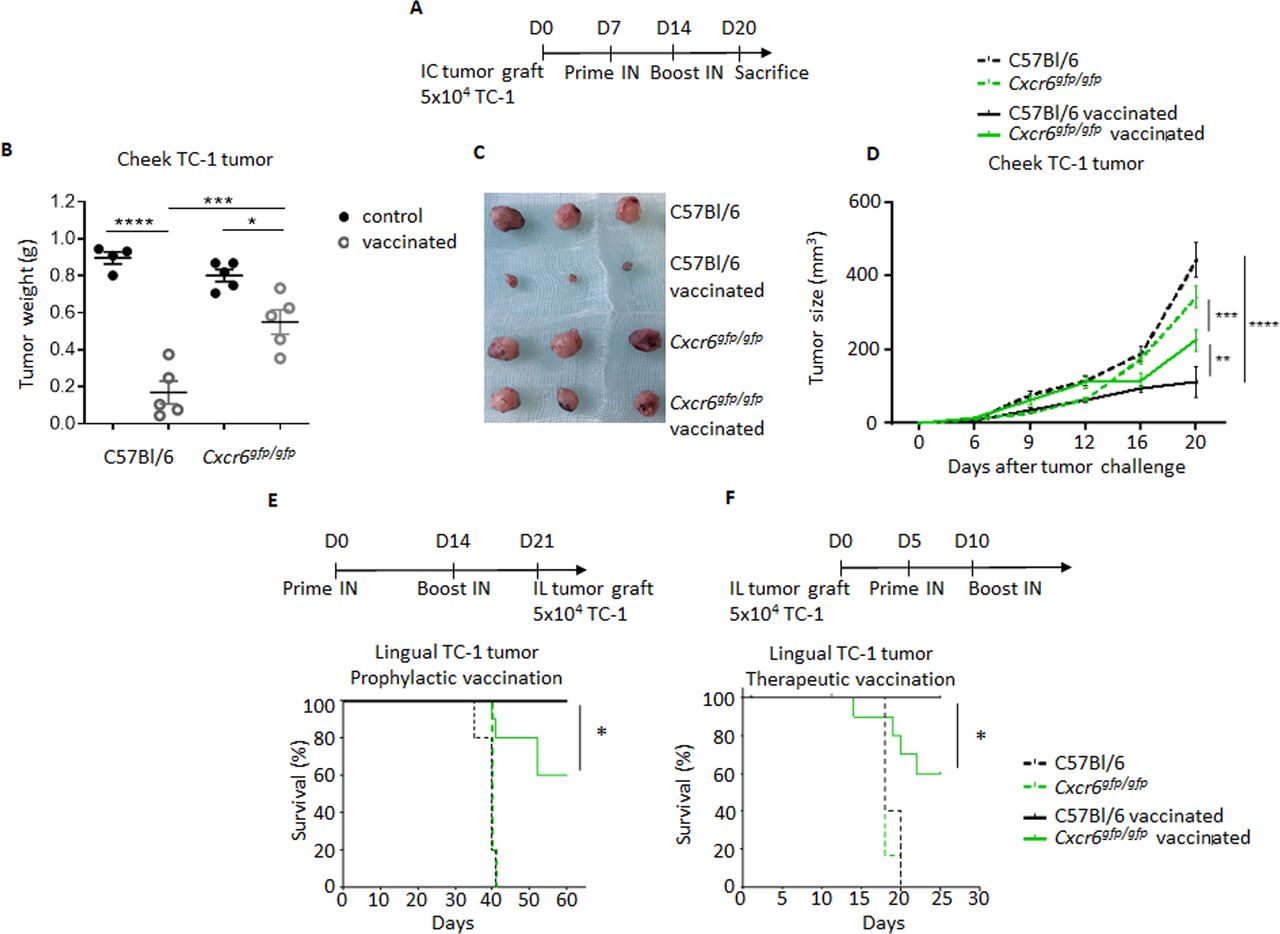
Cxcr6 deficiency impairs tumor control in orthotopic tumors. (A) C57BL/6 or Cxcr6gfg/gfp mice were grafted in the submucosal lining of the cheek (IC) with TC-1 tumor cells, then vaccinated or not with STxB-E7 via the in route at days 7 and 14, and sacrificed at day 20. (B–D) Tumor weight (B,C) and tumor size (D) were measured at day 20. (D,E) In a second orthotopic model, TC-1 was grafted in the sublingual mucosa (IL); mice were then vaccinated following a prophylactic (E) or therapeutic protocol (F), and survival was monitored. All data are representative from two independent experiments. Five mice/group (B–D) and 10 mice/group (E,F). Mean±SEM analysis of within-group differences was performed with a two-way analysis of variance and post hoc Tukey test (B–D), and survival was compared between groups with a Kaplan-Meier curve (log-rank test) (E,F). *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001. IC, intracheek; IL, intralingual.
To confirm these results in a second orthotopic tumor model, C57BL/6 wild-type mice and Cxcr6gfp/gfp mice were engrafted in the sublingual mucosa with TC-1 cells and were vaccinated prophylactically (before the graft) (figure 4E) or therapeutically (after the graft) (figure 4F) with STxB-E7 by intranasal route.
In both settings, all vaccinated C57BL/6 wild-type mice were alive at D60 for the prophylactic model (figure 4E) and at D25 for the therapeutic model (figure 4F). In Cxcr6gfp/gfp, only 60% of vaccinated mice were alive in both models at the same time points. Survival curves indicate that vaccinated Cxcr6gfp/gfp mice showed decreased survival compared with vaccinated C57BL/6 wild-type mice. These results indicate that impairment of CXCR6 expression has a strong negative impact on antitumor immunity.
We then addressed whether this partial loss of tumor growth control after vaccination is associated with a reduction of intratumoral infiltration by CD8+ T cells. Tumor-infiltrating lymphocytes were analyzed by flow cytometry in C57BL/6 wild-type or Cxcr6ggfp/gfp mice engrafted with TC-1 tumor cells in the submucosal lining of the cheek, and vaccinated or not with STxB-E7 at D7 and D14. A significant decrease in the absolute numbers of intratumoral CD8+ T cells, total H-2Db-E749-57 CD8+ T cells, and H-2Db-E749-57 TRM cells was observed in Cxcr6gfp/gfp mice, when compared with C57BL/6 wild-type mice (figure 5A,B). To reinforce the role of this impaired migration on CD8+ T cells in Cxcr6-deficient mice on antitumor immunity, we showed that these specific CD8+ T cells did not exhibit defaults in cytotoxicity mechanisms (online supplemental figure S4).
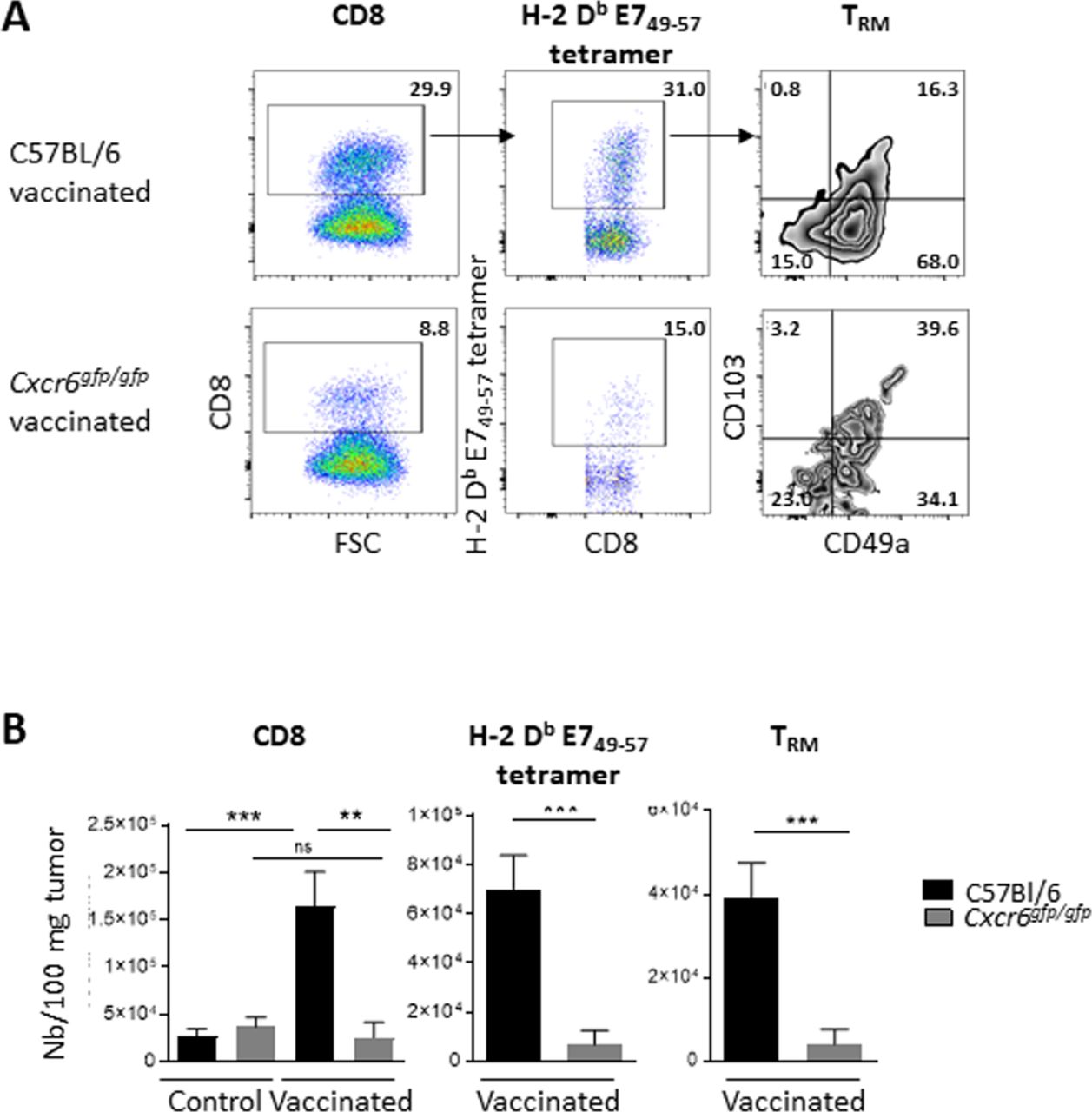
Cxcr6 deficiency impairs CD8+ T-cell infiltration in cheek tumor after in vaccination analysis of CD8 and H-2 Db E749-57 specific cells infiltrating a TC-1 cheek tumor in C57BL/6 and Cxcr6gfg/gfp mice, vaccinated or not with STxB-E7 via the intranasal route (D7 and D14) at day 20 after tumor graft. (A) Representative flow plots and (B) total number of CD8 T cells, H-2 Db E749-57 tetramer and TRM (Boolean gate CD103+ and/or CD49a+). Data are representative of two independent experiments. Five mice/group. Mean±SEM analysis of difference within groups was performed with a one-way analysis of variance and post hoc Tukey test, and between two groups with Mann-Whitney t-test. **P<0.01, ***P<0.001.
CXCL16 does not act as a local adjuvant after intranasal vaccination likely due to endogenous production of the chemokine
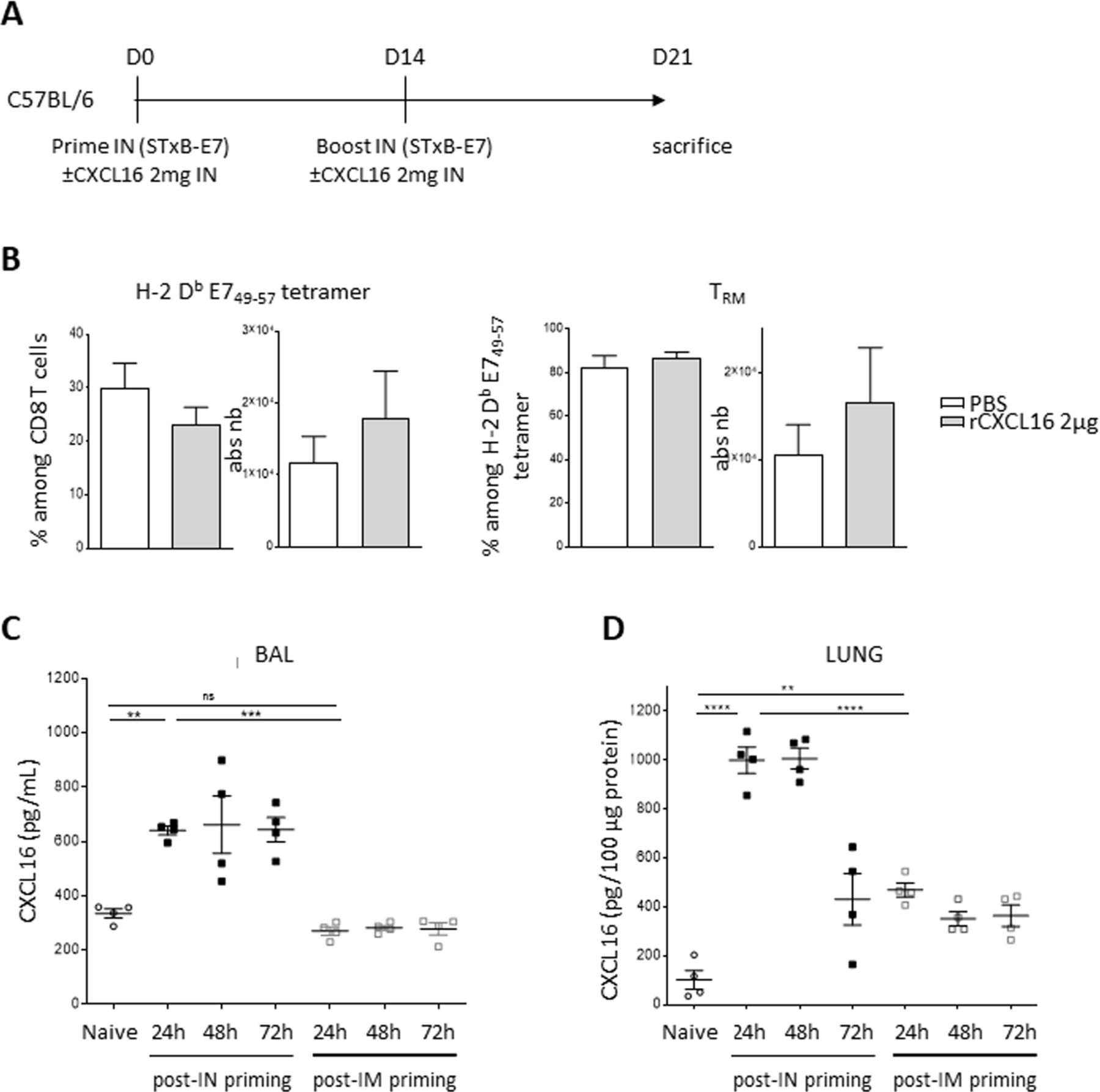
One of the potential applications of analyzing the role of these chemokine receptors and their ligands is to use them as adjuvants for local homing of CD8+ T cells to the lung. For this purpose, we vaccinated wild-type mice with STxB-E7 in the presence or absence of recombinant CXCL16 during priming and boosting and analyzed the recruitment of specific CD8+ T cells in BAL or lung parenchyma. The addition of recombinant CXCL16 did not show a significant increase in total specific CD8+ T cells nor of those with a TRM phenotype in the BAL (figure 6A) or lung (data not shown). Similar results were observed when recombinant CXCL16 was administered only at prime or boost.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Recombinant CXCL16 did not amplify recruitment of TRM likely due to endogenous CXCL16 induced after vaccination. (A) C57BL/6 mice were vaccinated with STxB-E7 via the in route at days 0 and 14 in the presence or absence of recombinant CXCL16 (2 mg), then sacrificed at day 21. (B) Percentage and absolute number of specific H-2 Db E749-57 tetramer within CD8 T cells (left) and TRM within tetramer (right) in the BAL are shown. n=8 mice/group. (C,D) C57BL/6 mice were vaccinated with STxB-E7 (20 µg)+poly ICLC (10 µg) via the intranasal or intramuscular route. CXCL16 was measured by ELISA at the indicated times after vaccination in BAL (C) and lysate from the lung (D). n=4 mice/group. The experiments were repeated twice. **P<0.01, ***P<0.001, ****P<0.0001. BAL, bronchoalveolar lavage; ns, not significant.
To better understand this observation, we measured the endogenous induction of CXCL16 before and after intranasal vaccination in the BAL and pulmonary parenchyma. We observed an induction of CXCL16 by a factor of 4–5 in the BAL and pulmonary parenchyma with a peak at 48 hours after immunization. Chemokine levels then decline but remain above baseline concentrations for at least 72 hours after vaccination in the lung parenchyma (figure 6C). As other chemokine receptors (CCR5, CXCR3, and CCR6) and their ligands have been implicated in the migration of CD8+ T cells into the lung,29 37 38 we also measured the induction of these chemokines. In the pulmonary parenchyma, we found an increase in chemokines binding CCR5 (CCL3, CCL4, and CCL5), CCR6 (CCL20), CXCR3 (CXCL10) as early as 24 hours after intranasal vaccination of wild-type mice (online supplemental figure S5). However, in contrast to CXCL16, this induction is transient, and the concentrations of these chemokines return to basal levels within 48–72 hours (online supplemental figure S5). Similar results were found in BAL (data not shown).
Cheek and lung epithelial cells may constitute one source of this production of CXCL16 (online supplemental figure S6). Interestingly, intramuscular immunization with the same vaccine also induces an increase in CXCL16 only in lung parenchyma, but at much lower levels (figure 6C,D) than those observed after intranasal immunization.This high endogenous CXCL16 induction after intranasal vaccination reinforces the physiological role of the CXCL16-CXCR6 axis in the migration of CD8+ T cells to the lung. It may also explain why the intranasal route is more efficient to recruit CD8+ T cells in the lung than the intramuscular route of administration.
Discussion
In this work, we confirmed, at the protein level, that CXCR6 was predominantly expressed on lung TRM as compared with effector CD8+ T cells, whereas memory CD8+ T cells of the spleen did not express this receptor after vaccination.
In addition, we demonstrate for the first time that vaccination of Cxcr6-deficient mice causes a defect in recruitment of antigen-specific CD8+ T cells in the lung, especially the TRM subsets. In contrast, this decrease in antigen-specific CD8+ T cells was not observed in CD8+ T cells derived from the spleen of Cxcr6-deficient mice. A decrease of total CD8+ T cells in the lung in CXCR6-deficient mice has also been reported in infectious models, while the proportion of these cells remained stable in the spleen and bone marrow.24
In order to evaluate the consequences of this CD8+ T-cell migration defect in a tumor context, we showed that Cxcr6-deficient mice bearing an orthotopic tumor in the head and neck regions are less well protected by an HPV vaccine, after both prophylactic and therapeutic vaccination. The present study is therefore the first to report the direct role of CXCR6 in antitumor immunity.
We have not formally demonstrated that the deficiency of Cxcr6 in CD8+ T cells directly explains partial loss of cancer vaccine efficacy, as only an inducible Cxcr6 KO specific to CD8+ T cells, not yet available, would allow specific deletion of these cells. However, the transfer of T cells derived from Cxcr6-deficient or wild-type mice confirms the intrinsic role of Cxcr6 deficiency on T cells in the reduction of TRM in the lung after vaccination by the intranasal route (online supplemental figure S3).
In addition, we have previously shown that this selected TC-1 epithelial tumor model, expressing the HPV E6–E7 protein, is dependent on CD8+ T cells, because in the absence of CD8+ T cells and especially CD8+ resident memory T cells, vaccine efficacy is abolished.9 10 The present study also demonstrated that the microenvironment of tumors derived from Cxcr6-deficient mice and vaccinated with a therapeutic vaccine directed against HPV E7 protein is less intensively infiltrated by total, antigen-specific CD8+ T cells and CD8+ resident memory T cells (figure 5A,B).
CXCR6 does not appear to be involved in the functional activity of CD8+ T cells, because memory CD8+ T cells either expressing or not CXCR6 produce similar levels of cytokines and exhibit equivalent levels of cytotoxicity markers (online supplemental figure S3).39
This role of CXCR6 associated with a protective phenotype has also been observed in an infectious model, as when CXCR6-positive and CXCR6-negative CD8+ T cells were isolated and sorted from the lung of mice previously immunized with a vaccine against Mycobacterium and injected into naive mice, and challenged with Mycobacterium tuberculosis, mice transferred with CXCR6+CD8+ T cells were better protected than mice receiving CXCR6 negative CD8+ T cells.40
The absence of CXCR6 does not totally reverse the effect of the cancer vaccine on controlling tumor growth. Other chemokine receptors have been involved in CD8+ T-cell migration into the lung, as it has been shown that CXCR3 and CCR5 are also important for promoting CD8+ T-cell homing into the lung.37 38 41 T cells present in BAL fluid of sarcoidosis patients coexpressed CXCR6 and CXCR3.32 Transcriptomic analyses showed that TRM expressed CXCR6, CCR5, and CXCR3.29 In our model, in contrast to the sustained induction of CXCL16, we showed a transient increase of CCL3, CCL4, CCL5 and CCL20 chemokines, the respective ligands of these receptors after intranasal vaccination, which could participate in the early recruitment of T cells in the tumor (online supplemental figure S5).
Earlier work by our team has shown that intranasal vaccination induced higher concentrations of antigen-specific CD8+ T cells in the lung or head and neck parenchyma than systemic vaccination.9 10 The present work demonstrates that these intrapulmonary CD8+ T cells express CXCR6, as already observed in lung derived CD8+ T cells from intranasal immunized mice with recombinant virus.40 42 The mechanisms explaining this preferential recruitment of CD8+ T cells in the lung and head and neck tumor microenvironment are poorly elucidated. We have shown that lung dendritic cells, rather than spleen derived dendritic cells, can induce an increase of CD49a on CD8+ T cells, which may explain the phenotype of the induced TRM cells but not the migration step.10 In this work, we report that intranasal vaccination induces a much greater increase in the chemokine CXCL16 in the BAL fluid and lung parenchyma, compared with intramuscular vaccination. This local increase in CXCL16 may explain the preferential recruitment of CXCR6-expressing CD8+ T cells into the lung and the impact of loss of expression of this receptor on CD8+ T-cell migration into the lung. However, we cannot exclude a role of the CXCL16–CXCR6 axis in the persistence of CD8+ T cells in the lung, as already suggested by another study.21 It has also recently been shown that the CXCR6–CXCL16 axis plays a role in the seeding of airway TRM from lung interstitium.36 43 While this work and our study show a role for CXCR6 in the recruitment of CD8+ T cells in the lung, other groups did not find an impact of CXCR6 in the pulmonary recruitment of CD4+ T cells after infection.44
CXCL16 can be produced by bronchial epithelial cells, dendritic cells, T cells and alveolar macrophages.15 23 The origin of pulmonary CXCL16 induction after intranasal vaccination remains to be determined. The Demaria group reported that irradiation induced the production of CXCL16 by murine and human tumor lines. Interestingly, in CXCR6-deficient mice, irradiation no longer resulted in recruitment of CD8+ T cells.45
Nevertheless, the CXCL16–CXCR6 axis is not specific to head and neck or pulmonary tissue because CXCR6 expression has also been demonstrated on iNKT cells in the liver.19 46 Cxcr6-deficient mice have a reduced number of TRM in the skin.25 One limitation of CAR T cells is their inefficient homing to solid tumors. The transduction of CXCR6 in CAR T cells could improve their migration into CXCL16 producing solid tumors, such as lung cancers and other tumors (pancreas, head and neck, stomach, ovarian and renal cancers).
Based on our results, CXCL16 could be considered to be an attractive mucosal adjuvant. Presumably because of the high endogenous CXCL16 concentrations, we could not show a role for CXCL16 when combined with an intranasal vaccine (figure 6). Similarly, a prime-pull strategy with a vaccine administered by the intramuscular route and nasal injection of CXCL16 did not significantly improve CD8+ T-cell recruitment (results not shown). Unlike other mucosal sites, local antigen is required for induction of TRM cells in the lungs, which could explain this result.47
Induction of TRM in the pulmonary mucosa is an important objective both in oncology to optimize anti-PD-1 therapy in patients with no pre-existing CD8+ T-cell response and in infectious diseases (tuberculosis, influenza, and coronavirus-associated diseases), where these cells have a demonstrated role in the control of infection.48–51 This work more clearly explains the unique potency of the intranasal route of vaccination to induce these memory T cells in the head and neck and pulmonary mucosa and may participate in improving current cancer vaccine strategies.
Supplemental material
Acknowledgments
We thank the staff of the European Georges Pompidou Hospital tumor banks (D Geromin and M Largeau) for providing sample materials and the PARCC Histology (C Lesaffre) and cytometry (C Knosp) platform as well as animal facilities (C Suldac and N Perez)
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
SK, CB and TT contributed equally.
Contributors IG-F, AM, ED, MA, and LP performed experimental work and analyzed and interpreted the data. SK, CB, and TT performed experimental work, analyzed and interpreted the data, and designed the figures. EF, LG, FL-B, CB, DD, and NB contributed to sample collection and analyzed and interpreted the data. NG, CT, ED, SK, and FM-C analyzed and interpreted the data. LJ and RG designed the study and revised the manuscript. TE designed the study, supervised the project, and wrote the manuscript. All authors were involved in the final approval of the manuscript.
Funding This work has been funded by Fondation ARC, INCA PLBio, Labex Immuno-Oncology, Site intégré de recherche en cancérologie (SIRIC CARPEM, SIRIC Curie), Cancéropole d’Ile de France, Carnot Curie Cancer, FONCER. SK is supported by the European Research Council (grant 756017, ARMOR-T).
Competing interests None declared.
Patient consent for publication Not required.
Ethics approval Experimental protocols in mice were approved by Paris Descartes University ethical committee (CEEA 34) in accordance with European guidelines (EC2010/63). The human study was approved by the local ethics committee (CPP Tours. CNRIPH n°18.11.21.67518. 05.03.2019). Informed consent was obtained from all subjects.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available in a public, open access repository. The data are available upon request from ET (eric.tartouraphp.fr).
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.