Article Text

Abstract
Background CAR-T cells immunotherapy is a breakthrough in the treatment of hematological malignancies such as acute lymphoblastic leukemia (ALL) and B-cell malignancies. However, CAR-T therapies face major hurdles such as the lack of tumor-specific antigen (TSA), and immunosuppressive tumor microenvironment sometimes caused by the tumorous expression of immune checkpoints (ICPs) such as HLA-G. Indeed, HLA-G is remarkable because it is both a potent ICP and a TSA. HLA-G tumor expression causes immune escape by impairing innate and adaptive immune responses and by inducing a suppressive microenvironment. Yet, to date, no immunotherapy targets it.
Methods We have developed two anti-HLA-G third-generation CARs based on new anti-HLA-G monoclonal antibodies.
Results Anti-HLA-G CAR-T cells were specific for immunosuppressive HLA-G isoforms. HLA-G-activated CAR-T cells polarized toward T helper 1, and became cytotoxic against HLA-G+ tumor cells. In vivo, anti-HLA-G CAR-T cells were able to control and eliminate HLA-G+ tumor cells. The interaction of tumor-HLA-G with interleukin (IL)T2-expressing T cells is known to result in effector T cell functional inhibition, but anti-HLA-G CAR-T cells were insensitive to this inhibition and still exerted their function even when expressing ILT2. Lastly, we show that anti-HLA-G CAR-T cells differentiated into long-term memory effector cells, and seemed not to lose function even after repeated stimulation by HLA-G-expressing tumor cells.
Conclusion We report for the first time that HLA-G, which is both a TSA and an ICP, constitutes a valid target for CAR-T cell therapy to specifically target and eliminate both tumor cells and HLA-G+ suppressive cells.
- immunotherapy
- adoptive
- tumor escape
- receptors
- chimeric antigen
- antigens
- tumor-associated
- carbohydrate
- biomarkers
- tumor
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
- immunotherapy
- adoptive
- tumor escape
- receptors
- chimeric antigen
- antigens
- tumor-associated
- carbohydrate
- biomarkers
- tumor
Introduction
CAR-T cell is a breakthrough immunotherapy that redirects T cell function against specific tumor antigens.1 2 Exceptional results were obtained in the treatment of B-cell malignancies, and recently approved by the Food and Drug Administration and the European Medicines Agency for acute lymphoblastic leukemia and diffuse large B-cell lymphoma treatments in adults.3 4 The main hurdles for the application to other tumors are the identification of proper tumor-specific antigens (TSA),5 and the existence of immune-suppressive tumor microenvironment (TME), particularly in solid tumors.6 7 Immune-suppressive TME is caused in part by tumor-driven immune-suppressive populations such as myeloid-derived suppressor cells, tumor-associated macrophages or neutrophils that secrete immune-suppressive cytokines,8 and by expression of immune checkpoints (ICPs). The negative impact of ICP on CAR-T cell therapy efficiency was demonstrated for programmed cell death protein 1 (PD-1)/programmed death-ligand 1 (PD-L1).9 10 Thus, in most CAR-T cell developments, the challenges are (i) to target a specific tumor antigen (to avoid ‘on-target off-tumor’ effect11 and (ii) to bypass inhibition by ICPs. The association of CAR-T cells with anti-PD-1 has shown benefits in clinical trials, by lifting PD-1-driven inhibition of CAR-T cells.12 However, this strategy increases the risk of a general immune system dysregulation, and also increases the already heavy burden on patients.13
HLA-G is an ICP molecule first shown to be expressed on fetal trophoblasts that invade the immune-aggressive maternal decidua.14–17 In adults, HLA-G expression is strictly restricted to few tissues,18–24 but in the context of cancer, it is often neo-expressed by tumor cells (for review, including per-cancer-type HLA-G expression data, see Loustau et al,25 Carosella et al26 and Lin and Yan27). The association of a physiological absence of expression and a common expression by tumor cells makes of HLA-G a particularly tumor-specific target.28 29 In this regard, HLA-G differs from other checkpoints that are physiologically expressed in adult tissues and therefore expressed by tumor cells ‘in addition to’ other tissues.
The primary function of HLA-G is to protect histologically incompatible fetal tissues against destruction by the maternal immune system. It does so by broadly inhibiting all actors of an immune response (B, T and natural killer (NK) cells, monocytes/dendritic cells, neutrophils) mainly through two inhibitory receptors: interleukin ILT2 and ILT4.26 HLA-G also induces a strong immune-suppressive microenvironment through the induction of suppressive NK cells,30 T cells31 and antigen-presenting cells.32 When expressed by tumor cells, HLA-G exerts the same functions and efficiently protects them against destruction. Consequently, HLA-G neo-expression in cancer was always associated with worse prognosis, poor clinical outcome of patients with cancer and with worse overall survival.25 Finally, the demonstration that HLA-G acts as an immune escape mechanism for tumors was formally made in animal models.33 34
Despite these characteristics, HLA-G was not targeted during the first wave of immune therapy development, probably because of its structural complexity and the lack of a murine homolog. Nevertheless, we reasoned that anti-HLA-G CAR-T cell innovative immune therapeutic approach would be highly relevant, HLA-G being a highly TSA, and an ICP, that is, a tumor-specific ICP. In this study, we generated several anti-HLA-G CARs using the scFv of high-affinity anti-HLA-G monoclonal antibodies. These were generated to specifically recognize the HLA-G isoforms interacting with the ILT2 and ILT4 receptors. We show that in vitro, anti-HLA-G CAR-T cells were specifically cytotoxic against HLA-G-expressing targets, insensitive to inhibition through the HLA-G:ILT2 pathway and that they acquire a T effector memory (TEM) phenotype on multiple antigenic stimulations by HLA-G. In vivo, anti-HLA-G CAR-T cells eradicated implanted HLA-G-expressing tumor cells. This is the first demonstration that anti-HLA-G CAR-T therapy may be a viable and flexible clinical approach to target many tumor types that may not express known TSA.
Materials and methods
Construction of CARs and T-cell culture transduction
Six anti-HLA-G CAR constructs were generated with the scFv of LFTT1 and 15E7 monoclonal antibodies (mAbs) developed and cloned into a third-generation lentiviral plasmid backbone under the regulation of a human EF1α promoter.35 CAR-T cells and activated non-transduced (NT) T cells were generated as previously decribed.35
Vector production
HIV-1-derived vector particles were produced by transient calcium phosphate co-transfection of HEK 293 T cells (American Type Culture Collection (ATCC)) with the vector plasmid pTRIP encoding the vector RNA, an envelope expression plasmid encoding the glycoprotein from VSV serotype Indiana, and the p8.74 encapsidation plasmid for the production of integrative lentiviral vector particles. Vector gene transfer capacity was determined by quantitative PCR after transduction of 293 T cells as previously described and was expressed as transduction unit/mL of vector.
Cell lines
In this study, we used the JEG-3 choriocarcinoma cell line that endogenously expresses HLA-G (ATCC), the HLA-class I-negative erythroblastoid cell line K562 (ATCC) and the 293 T cell line (ATCC) transfected with HLA-G5wt or mutated HLA-G5 isoforms. As shown in online supplemental figure 1, JEG-3 cells express only HLA-G1/β2m-associated isoform whereas K562-HLA-G1 cells express both HLA-G1/β2m-associated and HLA-G1/β2m-free isoforms.
Supplemental material
T cell isolation and activation
The peripheral blood mononuclear cells (PBMCs) from healthy donors were obtained by Ficoll isolation. T cells were sorted by column purification (Miltenyi), activated with CD3/CD28 microbeads (Miltenyi) and cultivated 48 hours at 37°C in a 5% CO2 incubator in RPMI 1640 (Gibco) supplemented with 10% fetal calf serum, 1% penicillin-streptomycin (Gibco), 50 μM beta-mercaptoethanol (Gibco), non-essential amino acid, 10 mM HEPES (Gibco), 1 mM sodium pyruvate (Gibco). Transduced cells were used 8 days after transduction.
Activation profile and degranulation assay
The day prior to the assay, 3×104 JEG-3 cells were labeled with 1 µM of CFSE (CellTrace, Thermo Fisher) and seeded in flat-bottom 96-well microplates. On the day of the assay, CAR-T cells were added at various E:T ratios to either the plated CFSE-labeled JEG-3 cells or 3×104 CFSE-labeled K562/K562-HLA-G1 cells. After 24 hours incubation, medium was collected and cells recovered, washed and labeled with antibodies against CD4 (clone RPA-T4, Biolegend), CD8 (clone RPA-T8, Biolegend), CD19 (HIB-19, Biolegend), CD25 (clone M-A251, BD Bioscience), CD69 (clone FN-50, BD Bioscience), PD-1 (EH12.2h7, Biolegend) and a viability dye (Invitrogen). For degranulation assays, co-cultures were set-up at an E:T ratio of 10:1. Anti-CD107a (clone eBioH4A3, Biolegend) was added at the start of the experiment, GolgiStop (BD Bioscience) was added after 1 hour. Five hours after the beginning of the assay, cells were collected and labeled with antibodies directed against CD4, CD8, CD19 and a viability dye. Acquisition was performed with a fluorescence-activated cell sorting (FACS) Attune (Thermo Fisher), and results were analyzed with FlowJo software.
Cytokine secretion profile
CAR-T cells 15E7CH2-CH3 (2×105) and LFTT1CH2-CH3 were seeded with 2×105 K562-HLA-G1 in a U-bottom 96-well plate and centrifuged at 100 g during 1 min. Cells were incubated 1 hour at 37°C in a 5% CO2 incubator then brefeldin A (5 µg/mL, BD Biosciences) was added. Cell were incubated 18 hours longer at 37°C in a 5% CO2 incubator. For FACS analysis, cells were labeled with anti-CD8 (clone RPA-T8, Biolegend), CD19 (HIB-19, Biolegend), interferon (IFN)γ (clone 4S.B3, Biolegend), tumor necrosis factor (TNF)⍺ (clone MAb11, Biolegend), IL-2 (MQ1-17H12, Biolegend) and a viability dye.
Phenotype variation after multiple stimulations
Co-cultures were set up with CAR-T cells and K562-HLA-G1 cells at an E:T ratio of 10:1 with no IL-2. Twenty-four hours after stimulation, 50 µL of medium were taken, stored for cytokine secretion analysis and replaced by complete RPMI. Seventy-two hours after stimulation, half of the medium was changed with complete RPMI supplemented with 50 U/mL of IL-2. Cells were maintained at 106/mL. Twelve days after stimulation, some cells were used for flow cytometry analysis, whereas the rest was used for a new round of stimulation. A total of three consecutive stimulations were performed. For flow cytometry analysis, cells were labeled with antibodies directed against CD4 (clone RPA-T4, Biolegend), CD8 (clone RPA-T8, Biolegend), CD19 (HIB-19, Biolegend), CD62L (clone DREG-56, BD Bioscience) and CD45RA (clone HI-100, Invitrogen). In these experiments, the phenotype of CAR-T cells and activated NT autologous controls was also established 24 hours prior to the assay. At the end of the experiment, all recovered medium was analyzed for IFNγ, TNFα and IL-2 secretion using a cytometric bead array (CBA) kit (BD Biosciences).
In vivo models
NOD/SCID/IL-2Rγc-deficient (NSG) mice (6–8 weeks of age) were purchased from Charles River Laboratories (L’Arbresle, France) and housed in filter-top cages with freely available food and sterile water (Plexx), at the UMR1098 Animal facility (agreement #C25-056-7). At day −1, a 2.5 Gy single-dose total body irradiation was applied. Twenty-four hours later, mice were inoculated intravenously with 106 luciferase-expressing K562-HLA-G1 cells. On day 3, 107 CAR-T cells were injected into the tail vein. Engraftment was monitored weekly by bioluminescence measurements: mice received 3 mg of luciferin (VivoGlo Luciferin, #P1043, Promega, Fitchburg, Wisconsin, USA) intraperitoneally within 10 min of imaging (IVIS Lumina Series III, Perkin Elmer, Waltham, Massachusetts, USA). Detailed methods are provided in online supplemental methods.
Results
Anti-HLA-G CAR-T cells targeting immunosuppressive HLA-G isoforms
HLA-G is a complex protein. First, its splicing pattern yields at least four membrane-bound isoforms that can be shed, and three soluble ones,36 and new splicing isoforms were recently described.37 In addition, HLA-G heavy chain may, or may not be associated with β2m, depending on the isoform and possibly on the environment. In sharp contrast with this diversity, little is known of HLA-G isoforms other than β2m-associated HLA-G1/HLA-G5 (α1-α2-α3 domains) and β2m-free HLA-G2/HLA-G6 (α1-α3 domains) for which immune-modulatory function has been well described in vitro.38 39 HLA-G1/β2m-associated isoform is mainly expressed in several tumor contexts, whereas HLA-G/β2m-free molecules were detected in melanoma.28
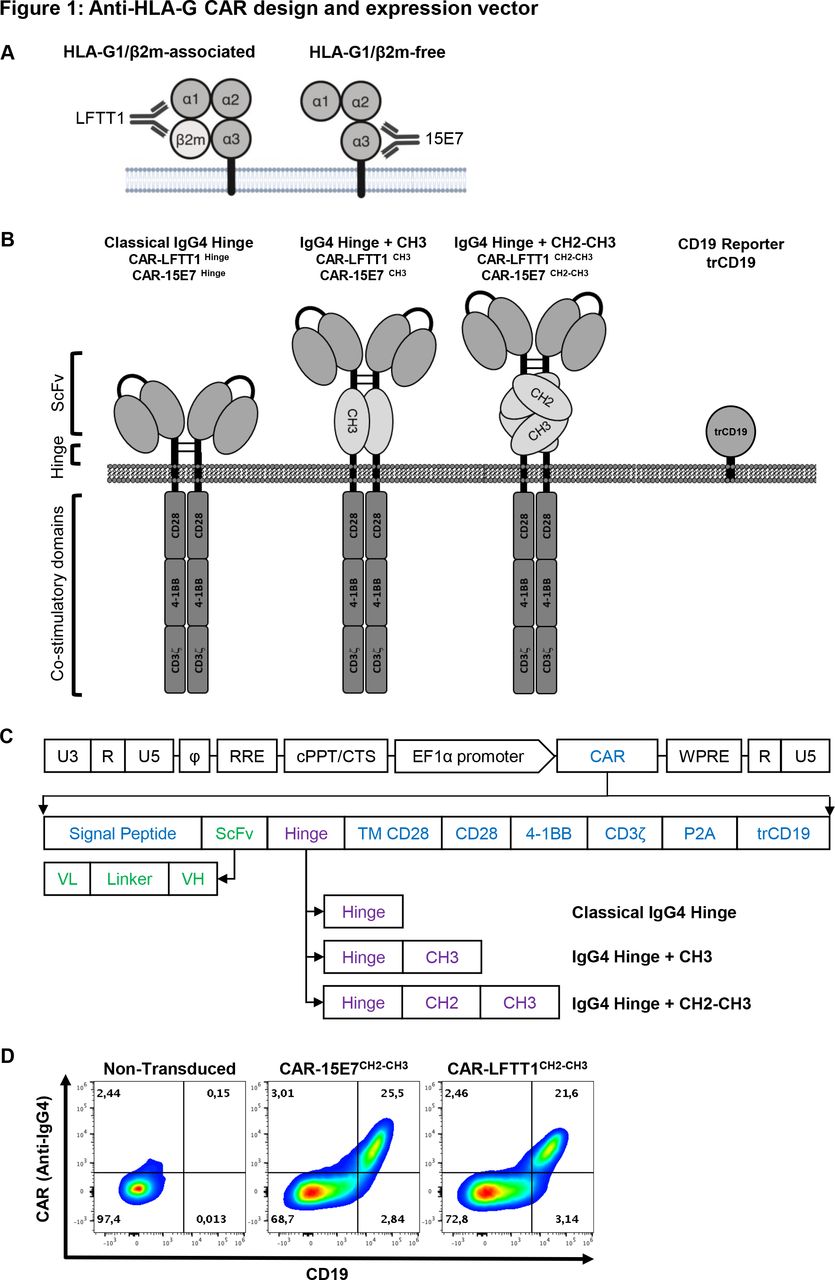
Our goal was to specifically target HLA-G1/β2m-associated and HLA-G1/β2m-free immunosuppressive isoforms whose inhibitory functions depend on the interaction with ILT2 and/or ILT4. For this purpose, we generated new antibodies: the LFTT1 monoclonal antibody is specific for the HLA-G-α1 domain of β2m-associated HLA-G1/HLA-G5 isoforms, whereas the 15E7 monoclonal antibody binds to β2m-free HLA-G isoforms and is specific for the unique F-D-Y amino acid loop in HLA-G-α3 domain (figure 1A). Information on these antibodies is provided in online supplemental figure 1.

Anti-HLA-G CAR design and expression vector. (A) Schematic representation of anti-HLA-G monoclonal antibodies specificities: LFTT1 monoclonal antibody (mAb) is specific for HLA-G1/β2m-associated isoform and 15E7 mAb is specific for HLA-G1/β2m-free isoform. (B) Schematic representation of the third-generation anti-HLA-G CAR protein and lentiviral vector backbones used to transduce human CD3+ T cells. (C) Lentiviral constructs details; SP: signal peptide from mouse Igκ. scFv: single chain fragment from either LFTT1 (CAR-LFTT1) or 15E7 (CAR-15E7). Hinges: classical IgG4 hinge, IgG4 hinge+CH3 domain of human IgG4 and IgG4 hinge+CH2-CH3 domain of human IgG4. TM CD28: transmembrane domain of human CD28. CD28, 4-1BB, CD3ζ: endodomains of respectively CD28, 4-1BB and CD3ζ human proteins. P2A: cleavage site. trCD19: truncated CD19. U3, R, U5: HIV-1 LTR regions. Φ: encapsidation signal. RRE, Rev Response Element. EF1α, full length elongation factor-1α promoter. WPRE, Woodchuck Post-transcriptionnal Response element mutated for HBx ATG codon start. (D) Validation of transduction and co-expression of CAR using the CARCH2-CH3 construct proteins and trCD19 reporter for both CAR-LFTT1 and CAR-15E7 T cells from primary human T cells. Transduced T cells were labeled with CD19 and IgG4 antibodies and analyzed by flow cytometry.
Based on LFTT1 and 15E7 scFv, we generated two third-generation anti-HLA-G CAR sets: CAR-LFTT1 and CAR-15E7. The CAR-LFTT1 set targets the HLA-G-α1 domain, distal from the membrane, whereas the CAR-15E7 set targets HLA-G-alpha3, proximal to the membrane. Because accessibility to the epitopes by the CAR protein was an issue, each set comprises three CAR constructs, with different hinges. Hinges were: classical IgG4 (CAR-LFTT1 and CAR-15E7), IgG4+CH3 (CAR-LFTT1CH3 and CAR-15E7CH3) and IgG4+CH2-CH3 (CAR-LFTT1CH2-CH3 and CAR-15E7CH2-CH3) (figure 1B).40 CAR constructs also contained human-derived CD28 transmembrane domain, CD28 and 4-1BB co-stimulation domains to improve CAR-T cell cytotoxic function and persistence,41 and human CD3ζ chain. Anti-HLA-G CAR constructs were introduced in an HIV-1-derived lentiviral vector (figure 1C) co-expressing a truncated CD19 protein, reporter for CAR cell-surface expression on transduced cells (figure 1D).35
In all subsequent experiments, CAR expression was assessed using CD19 reporter (trCD19) expression levels as reported.35
Anti-HLA-G CAR-T are specific and efficient effector cells
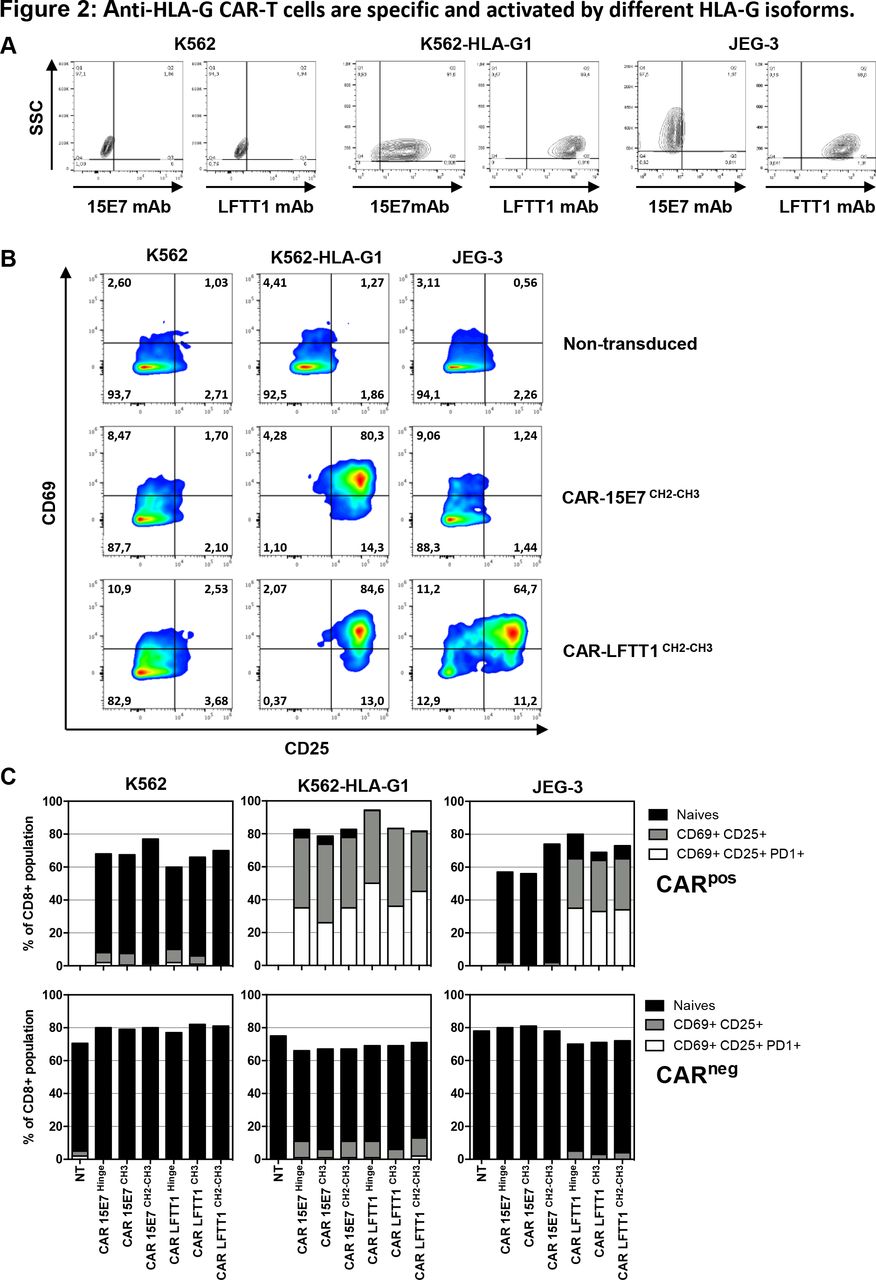
CAR-LFTT1 and CAR-15E7 function was expected to follow staining patterns of LFTT1 and 15E7 mAbs: control K562 cells are not stained by 15E7 or LFTT1 mAbs, whereas K562-HLA-G1 cells are stained by both, and JEG-3 cells are only stained by LFTT1 (figure 2A).

Anti-HLA-G CAR-T cells are specific and activated by different HLA-G isoforms. (A) Anti-HLA-G monoclonal antibodies specificity against HLA-G–/+ cell lines. Representative dot plot of K562, K562-HLA-G1 and JEG-3 tumor cell lines labeling with the anti-HLA-G 15E7 and LFTT1 monoclonal antibodies. Upregulation of activation-associated markers CD69, CD25 and programmed cell death protein 1 (PD-1) were monitored on activated non-transduced T cells, CAR-15E7 and CAR-LFTT1 T cells following a 6-hour incubation with K562, K562-HLA-G1 or JEG-3 cell lines. (B) Representative dot plot of the upregulation of CD69 and CD25 on activated non-transduced T cells and CAR-T cells sets. (C) Upregulation of activation-associated markers CD69, CD25 and PD-1 was determined on CAR expressing T cells (CD8+/trCD19+) in comparison to CAR negative T cells (CD8+/trCD19-) following incubation with K562, K562-HLA-G1 or JEG-3 cell lines (n=3, NT, non-transduced).
All CAR-T cells were tested against HLA-G-expressing K562-HLA-G1 and JEG-3 targets, using K562 cells as HLA-G-negative controls, and autologous activated T cells as non-transduced controls. At the end of the experiments, we investigated the upregulation of activation-associated markers (CD25, CD69, PD-1), degranulation (CD107a upregulation) and tumor cell lysis.
Upregulation of CD25 vs CD69 is shown in figure 2B for a representative experiment and the classical hinge: on stimulation by HLA-G-positive cells, anti-HLA-G CAR-T cells expressed both CD25 and CD69 markers at their surface, following the specificity pattern of LFTT1 and 15E7 mAbs. As shown in figure 2C for three individual experiments, upregulation of CD25, CD69 and PD-1 occurred in 30%–50% of CAR-expressing CD8+ T cells (trCD19+ fraction) on incubation with cells expressing the HLA-G structures targeted by LFTT1 or 15E7 mAbs. All CAR-T cells behaved similarly, regardless of hinge. No upregulation of CD25, CD69 or PD-1 was observed in control conditions.
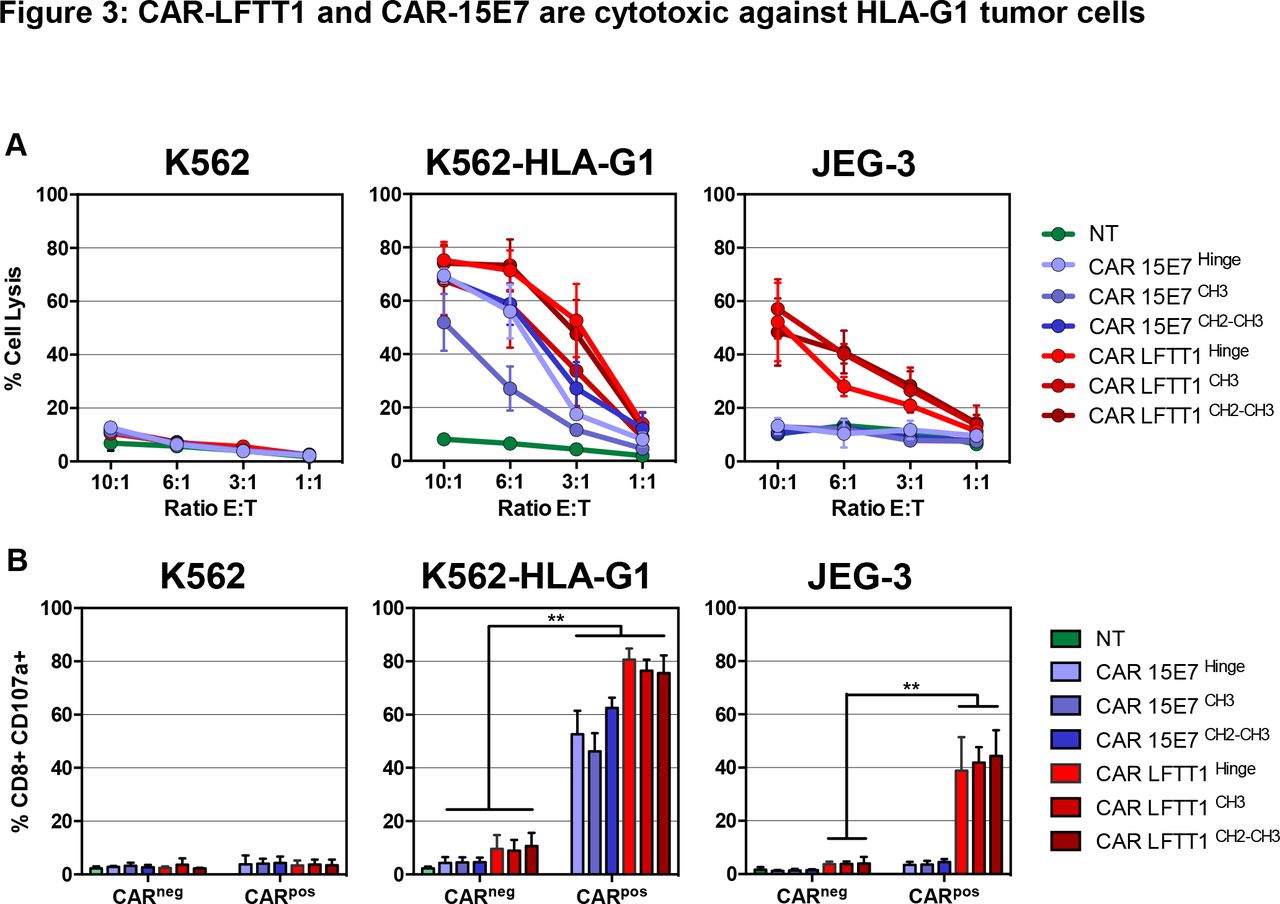
In cytotoxicity assays (figure 3A), while no lysis was detected in control conditions, CAR-LFTT1-set T cells were cytotoxic against both K562-HLA-G1 and JEG-3 cells that both express β2m-associated isoforms, whereas CAR-15E7-set T cells were cytotoxic only against K562-HLA-G1 cells, the only cells that express β2m-free isoforms. Independently on the HLA-G isoform targeted, CAR-T cells with classical hinge (CAR-LFTT1 and CAR-15E7) and with CH2-CH3 hinge (CAR-LFTT1CH2-CH3 and CAR-15E7CH2-CH3) showed similar cytotoxicity, whereas that of CAR-T cells with the CH3 hinge (CAR-LFTT1CH3 and CAR-15E7CH3) was lower in the six independent experiments. CD107a upregulation was investigated to gain insight in the proportion of CAR-T cells actually performing a cytotoxic function after being activated by targets. As shown in figure 3B, CAR-LFTT1 T cells increased CD107a expression when stimulated by HLA-G1/β2m-associated expressing cells (75%±2.7% against K562-HLA-G1 cells, 44%±4% against JEG-3 cells), whereas CAR-15E7 T cells displayed CD107a only when stimulated by HLA-G1/β2m-free K562-HLA-G1 cells (62%±1.5%). No impact of hinge was observed.

CAR-LFTT1 and CAR-15E7 are cytotoxic against HLA-G1 tumor cells. (A) K562, K562-HLA-G1 or JEG-3 tumor target cells were exposed to activated non-transduced T (NT) cells, CAR-15E7 or CAR-LFTT1 T cells sets at different E:T ratio. Target cells were labeled with CFSE and % of tumor cell lysis represent the percentage of CFSE+ cells labeled with live/dead at 24 hours posteffector exposure (n=6). Significance was determined using a Mann-Whitney U test of unpaired t-test (*p<0,05; **p<0002) (n=6). (B) CD107a degranulation of CAR-LFTT1, CAR-15E7 sets or activated NT human T cells was monitored on CD8+/trCD19+ T cells following 6 hours exposure to K562, K562-HLA-G1 and JEG-3 cells at a E:T ratio of 10:1 (n=6).
Together, these results indicate that anti-HLA-G CAR-LFTT1 and CAR-15E7 T cells are specifically activated by, and cytotoxic against cells expressing the HLA-G isoforms that the LFTT1 and 15E7 mAbs recognize. All three hinges allowed cytotoxic function, even though CAR-T cells based on CH3 hinge were less efficient than CAR-T cells using the classical and CH2-CH3 hinges.
Anti-HLA-G CAR-T cells differentiate into long-term effector memory cells on repeated stimulations
Anti-HLA-G CAR-T cells demonstrated both specificity and efficiency after a single stimulation by HLA-G1-expressing target cells. Next, we studied their differentiation after repeated stimulations. For this, anti-HLA-G CAR-T cells were repeatedly stimulated every 12 days with K562-HLA-G1 cells (figure 4A). Expression of CD62L and CD45RA differentiation markers on CAR-T cells was monitored by flow cytometry prior to, and 24 hours after stimulation (figure 4B). CAR-T IFNγ, TNFα and IL-2 secretion was monitored at days 13, 26 and 39. After Stim-1, CAR-15E7CH3 could not control K562-HLA-G1 cells proliferation.

Anti-HLA-G CAR-T cells differentiated into long-term memory effector cells after repeated stimulation with HLA-G+ tumor cells. (A) Schematic representation of anti-HLA-G CAR-T cells repeated stimulation with K562-HLA-G1 tumor cells. Transduced CAR-LFTT1, CAR-15E7 sets or activated non-transduced T cells were repeatedly stimulated every 12 days with K562-HLA-G1 cells at a 10:1 E:T ratio. Expression of differentiation markers was monitored prior to and 24 hours after the stimulation and cytokines secretion levels were monitored at days 13, 26 and 39. (B) Representative flow cytometry analysis of the expression of CD62L and CD45RA differentiation markers on activated non-transduced, CAR-15E7 and CAR-LFTT1 T cells prior to and 24 hours after stimulations rounds. (C) Day prior stimulation and after each repeated stimulation with K562-HLA-G1 cells, differentiation of T cells was determined on the basis of their CD62L/CD45RA cell-surface expression. CAR-T cells were gated on CD8 and trCD19 reporter co-expression. (D) Co-culture medium was recovered the day after each stimulation with K562-HLA-G1 cells and analyzed for interferon (IFN)γ, interleukin (IL)-2 and tumor necrosis factor (TNF)α concentration (n=3). Columns represent means±SEM. For each panel, after first stimulation round, activated non-transduced T cells were removed from the experiment due to absence of cytotoxicity against K562-HLA-G1 cells.
Prior to stimulation with HLA-G1-expressing cells (Stim-0), activated non-transduced and anti-HLA-G CAR-T cells contained: CD62L+CD45RA- central memory (TCM) cells (>50%), CD62L-CD45RA- T effector memory (TEM) cells (<30%), few remaining CD62L+CD45RA+ naïve T cells (10%) and barely detectable (<1%) effector memory RA T cells (TEMRA). From the first to third stimulation, both CAR-LFTT1 and CAR-15E7 presented the same differentiation profile: a predominant TEM population (>60%), a contraction of the TCM population (<30%), an almost extinction of the CD25-CD69- population and the induction of a small population of TEMRA cells (figure 4B,C).
CAR-LFTT1 displayed more TCM than the CAR-LFTT1CH2-CH3, which differentiated in TEM after repeated stimulations. All CAR-LFTT1 sets secreted IFNγ, TNFα and IL-2 cytokines (figure 4D). For both CAR-LFTT1 and CAR-LFTT1CH2-CH3, IFNγ secretion was strongly induced after Stim-1 and then remained stable, IL-2 secretion strongly increased between Stim-1 and Stim-2, and TNFα secretion was increased between Stim-1 and Stim-2. CH2-CH3 hinge secreted lower levels of these cytokines compared with classical hinge. Even though CAR-LFTT1CH3 secreted IFNγ after Stim-1, this secretion dramatically decreased along repeated stimulations. Also, IL-2 secretion for the CH3 hinge was weaker than for the classical or CH2-CH3 hinges, and TNFα secretion was barely detectable. For CAR-15E7 sets, IFNγ secretion also increased between Stim-1 and Stim-2 for all hinges, but its level was weaker than that of the CAR-LFTT1 sets. IL-2 and TNFα secretions were almost not detected for CAR-15E7 sets.
Anti-HLA-G CAR-T cells function is not inhibited by HLA-G:ILT2 interaction
Targeting HLA-G with CAR-T cells makes sense only if anti-HLA-G CAR-T cells are not inhibited by HLA-G itself. HLA-G inhibits T cells through the ILT2 receptor. Thus, unresponsiveness to HLA-G would occur if (i) anti-HLA-G CAR-T cells did not express ILT2, or if (ii) anti-HLA-G CAR-T cells were insensitive to HLA-G inhibition, or both.
Ten per cent to 20% of circulating CD8+ T cells express ILT2 in young healthy individuals, and this proportion increases with age. In patients with bladder cancer, CD8+ILT2+ T cells were shown to represent up to 80% of total CD8+ T cells.42 T cells expressing ILT2 are often antigen-experienced, and were shown to upregulate ILT2 on contact with HLA-G-expressing tumor cells.43 Thus, we compared ILT2 cell-surface expression on CAR-T cells before and after repeated stimulation with K562-HLA-G1. In these experiments, PD-1 expression levels were used as a marker of expanded and reactive T cells.44–47 After activation, ILT2 was expressed by 20% of non-transduced CD8+ T cells, and only by 10% of CAR-T cells (figure 5A). After three rounds of stimulation, no ILT2 upregulation was observed on CARpos T cells, whereas a slight increase was observed on CARnegCD8+ T cells (25% ILT2+CARneg T cells). By contrast, PD-1 expression was equivalent on both CARneg and CARpos CD8+ T cells prior to stimulation (8% PD-1+CARneg/pos), and after three stimulations, PD-1 expression was significantly upregulated in the CARpos population only (15% for CAR-15E7 and 22% for CAR-LFTT1) (figure 5B). As expected, ILT2+CD8+ T cells had a more differentiated phenotype before the first stimulation compared with their negative counterpart. Indeed, prior to stimulation, ILT2+CAR-LFTT1 and CAR-15E7 T cells contained 44% and 41% TEM cells, respectively, whereas their ILT2-negative counterparts contained only 23% and 25% TEM cells, respectively. However, after three stimulations, most CARpos-T cells had differentiated into TEM cells and these differences were no longer observable (figure 5C).
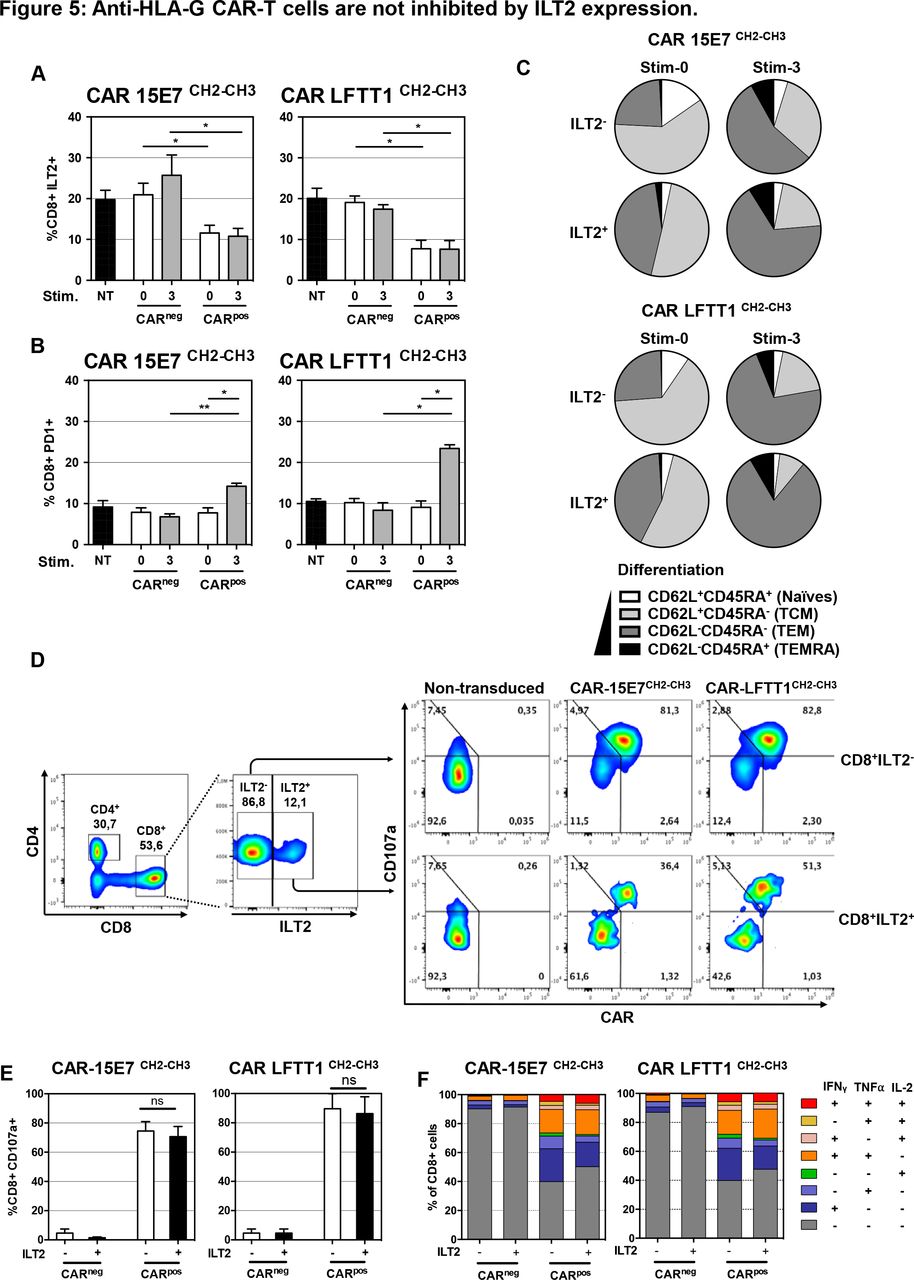
Anti-HLA-G CAR-T cells are not inhibited by ILT2 expression. Following transduction of T cells, CAR-T cells displaying or not the CAR construct at their surface (respectively CARpos and CARneg T cells) were analyzed for their ILT2 and programmed cell death protein 1 (PD-1) cell-surface expression after repeated stimulation with K562-HLA-G1 cells at a 10:1 E:T ratio. CH2-CH3 hinge was used to determine the CAR cell-surface expression CAR-LFTT1 or CAR-15E7 T cells. (A) ILT2 expression levels on CARneg/pos cells prior and after repeated stimulations with K562-HLA-G1 were compared with activated non-tranduced (NT) T cells. (B) PD-1 expression levels on CARneg/pos cells prior and after repeated stimulations with K562-HLA-G1 were compared with activated NT T cells. Significance was determined using a Friedmann test (*p<0,05; **p<0002) (n=6). (C) Differentiation of CAR-15E7 and CAR-LFTT1 T cells expressing or not ILT2 was determined on the basis of their CD62L/CD45RA expression on CARpos T cells before and after the repeated stimulations with K562-HLA-G1 cells. (D) 15E7CH2-CH3 or LFTT1CH2-CH3 CAR-T cells were generated from ILT2+ and ILT2- sorted human CD8+ T cells before being co-cultured with K562-HLA-G1 cells. (E) CD107a degranulation of ILT2–/+ CARneg/pos T cells was monitored by labeling of CD107a at the surface of CARneg/pos CD8+/trCD19+ T cells after 6 hours exposure to K562-HLA-G1 target cells. Columns represent means±SEM. Significance was determined using a Mann-Whitney U test (ns, non significative) (n=3). (F) ILT2 expression impact on anti-HLA-G CARneg/pos T cells. T helper 1 cytokines expression profile was evaluated after 18 hours co-incubation with K562-HLA-G1 cells. Cytokines (interferon (IFN)γ, tumor necrosis factor (TNF)α, IL-2) secretion by CARneg/pos CD8+ cells was determined by FACS and proportion of each combination are represented in cumulative histograms.
In order to determine if anti-HLA-G ILT2+CAR-T cells were capable of lysing HLA-G-positive target cells despite ILT2 cell-surface expression, we isolated and transduced ILT2+ and ILT2- CD8+ T cells. Transduced cells were then phenotyped and used in cytotoxicity assay against K562-HLA-G1 target cells. As shown in figure 5D, at the time of the assay, ILT2-CD8+ T cells had been efficiently transduced by both LFTT1 and 15E7 CAR constructs (82% and 81%, respectively), whereas transduction of ILT2+CD8+ T cells had been less efficient (51% and 36% of ILT2+CD8+ T cells, respectively). At the end of the cytotoxicity assay, the proportion of CD107a+ cells in ILT2-positive and ILT2-negative CARposCD8+ T cells was identical, for both LFTT1 and 15E7 CAR constructs, indicating that anti-HLA-G CAR-T cells expressing ILT2 had not been inhibited by the HLA-G/ILT2 interaction in terms of cytotoxicity (figure 5E) and T helper 1 cytokines secretion profile (figure 5F).
Anti-HLA-G CAR-T cells in vivo functionality
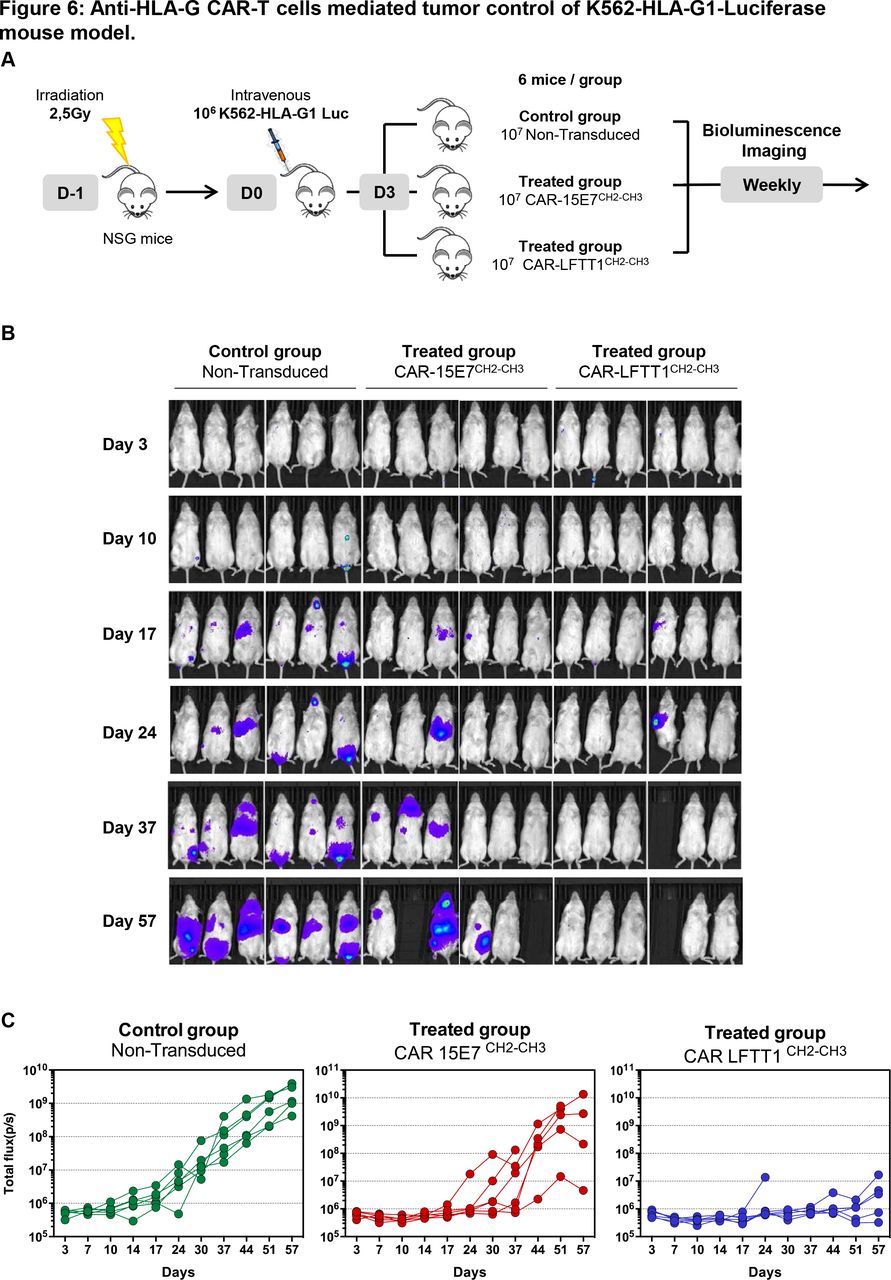
We sought to prove that our CAR strategy was suitable for in vivo application. At day 0, immune-deficient NSG mice were injected intravenously with 106 K562-HLA-G1 cells expressing luciferase Firefly reporter protein. At day 3, mice were injected with either 107 CAR-LFTT1, CAR-15E7 or control T cells (figure 6A). Tumor progression was monitored by bioluminescence every week. The results for each individual mouse can be seen in figure 6B,C, and results for six mice per group are shown in online supplemental figure 2. Already at day 17, K562-HLA-G1 tumors were detectable in all mice of the control group, but only in one mouse from CAR-15E7 and CAR-LFTT1 groups. At day 37, three out of six mice of the CAR-15E7 group, and five out of six in the CAR-LFTT1 group were still tumor-free. At day 57, only one mouse in the CAR-15E7 group, but still five out of six in the CAR-LFTT1 group were tumor-free. This demonstrated that CAR-15E7 T cells had delayed tumor growth by about 2 weeks, whereas CAR-LFTT1 T cells had efficiently eradicated K562-HLA-G1 cells in vivo.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Anti-HLA-G CAR-T cells mediated tumor control of K562-HLA-G1-luciferase mouse model. (A) Schema of experimental procedure: NOD/SCID/IL-2Rγc-deficient (NSG) mice were irradiated the day before being intravenously injected with 106 K562-HLA-G1-luciferase cells (K562-HLA-G1-Luc). Mice received activated non-transduced T cells or CAR-T (CAR-15E7CH2-CH3 or CAR-LFTT1CH2-CH3) cells on day 3 and were monitored by bioluminescence imaging over time. (B) Representative bioluminescence intensity of mice over time. (C) Total flux of each mouse in the three groups at different points.
Discussion
Despite exceptional promises in the treatment of cancer, adoptive T cell therapies are still impaired by (i) the lack of TSA, especially some that would be common to multiple/most tumor types5 and (ii) tumor expression of ICPs inhibiting antitumor immune responses, even those of CAR-T cells. Anti-ICP CAR-T cell therapy would take advantage of already implemented antibody-based immune therapies (eg, PD-1/PD-L1), but the lack of tumor-specific expression of these molecules raises safety concerns because of ‘on target, off tumor’ effects.48 49 This might not apply to HLA-G. Indeed, HLA-G is special among ICPs because notwithstanding its broad inhibitory function, its physiological expression is mainly fetal. Consequently, HLA-G is absent in most adult tissues while most tumor types neo-express it to various degrees.25 In patients with cancer, this makes of HLA-G a de facto TSA, and so effectively a tumor-specific ICP. For monomorphic ICP molecules, the difference between ‘TSA’ and ‘tumor-specific ICP’ might not exist, but for HLA-G, it is a key issue because it is a complex molecule with several isoforms, not all of them associated with β2m, not all of them inhibitory, and not all of them functionally described.
Thus, in the design of anti-HLA-G CAR constructs, we aimed at targeting the known ICP function of HLA-G by restricting our targets to isoforms known to be inhibitory through interaction with ILT2 (β2m-associated HLA-G1/HLA-G5) or ILT4 (β2m-free HLA-G1/HLA-G5, and HLA-G2/HLA-G6). Thus, new antibodies were generated for the paratope design of CARs. LFTT1 recognizes the α1 domain of β2m-associated HLA-G isoforms, as do other antibodies such as MEM-G/9 or 87G. For the generation of 15E7, because the structures of β2m-free isoforms may vary, we targeted a unique motif of the ILT4-interacting sequence (HPVFDYEATL) located in the HLA-G-α3 domain.50 By targeting the α3 domain, even newly described isoforms not containing an α1 domain37 may be targeted.
Both anti-HLA-G CAR-T cell sets specifically recognized, got activated by, and lysed their cognate HLA-G isoforms expressed by targeted tumor cells. Although CAR-LFTT1 T cells were more efficient than CAR-15E7 T cells in our experimental models, CAR-15E7 T cells interest resides in targeting tumor cells expressing HLA-G2 and/or HLA-G new isoforms, devoid of β2m and/or α1 domain but engaging ILT2/4 via their α3 domain. Also, the development of bispecific CAR-T cells, combining their respective scFv specificities, would allow recognition of the full repertoire of immunosuppressive HLA-G isoforms, and avoid a possible escape from CAR-LFTT1 T cells through downregulation of the β2m by tumor cells.51–53
CAR-T cell-based immunotherapies rely on persistence of their function over time after administration. We demonstrate that both CAR-LFTT1 and CAR-15E7 T cells differentiated mainly in TCM cells, which have been associated with greater in vivo antitumor activity and persistence.54 Concerning the cytokine secretion profile, CAR-LFTT1 T cells showed a significant increase of IL-2 secretion between first and second stimulation likely related to a stronger activation following antigen stimulation and higher proliferation, resulting in an enrichment of the most responsive T cells. Rossi et al determined that a polyfunctional phenotype of CAR-T cells, with increased IL-2, IFNγ and TNFα secretion, is associated with better clinical outcome in patients treated with anti-CD19 CAR-T cells in non-Hodkgin’s lymphoma.55 Therefore, for CAR-LFTT1, the construct based on a classical hinge that demonstrated a stronger secretion of IFNγ, IL-2 and TNFα cytokines levels in comparison to CH3 and CH2-CH3 hinges might be the best choice for clinical use. Regarding CAR-15E7 T cells, increasing the flexibility or the access to the membrane-proximal HLA-G-α3 domain with extended hinges did not improve the functionality. CAR-15E7 T cells activation after K562-HLA-G1 stimulation appeared less efficient than CAR-LFTT1 T cells. However, it is important to note that the K562-HLA-G1 cells used as model to test both CARs, express more β2m-associated HLA-G1 than β2m-free isoforms, favoring CAR-LFTT1 T cells cytotoxicity. CAR-15E7 T cells efficiency should be determined in a more controlled β2m-free system, to model the commonly observed context of tumors in which β2m is downmodulated. Given these, anti-HLA-G CAR-T cells clinical trial phase I will be based on the classical hinge with the LFTT1 paratope.
A major hurdle for CAR therapeutic application is T cell inhibition by ICP. ILT2 is expressed by some CD8+ T cells, and upregulated following cytotoxic T cell activation.43 ILT2 exerts its inhibitory function through four immunoreceptor tyrosine-based inhibitory (ITIM) motifs,56 while CAR cytoplasmic signaling pathway relies on tyrosine-based activating motifs (ITAM) motifs of CD3ζ.40 Therefore, it was possible that HLA-G+ targets could inhibit CAR-T cytotoxic function through ILT2 engagement. This is most relevant on two fronts: (i) ILT2 expression is upregulated on stimulation and therefore anti-HLA-G CAR T cells could also upregulate it, becoming sensitive to inhibition by HLA-G and (ii) ILT2+CD8+ T cells might be present in the PBMC used for transduction since this population increases with age, representing sometimes >80% of CD8 T cells, and since patients with cancer are often elderly. Here, we demonstrated that ILT2 expression might not be a problem, eventually. First, during generation of CAR-T cells, CD8+ILT2+ population showed a lower transduction efficacy (50%) compared with ILT2- T cells (83%), explained by the fact that ILT2 expression is associated with a downregulation of CD28,57 resulting in a suboptimal activation by CD3/CD28 beads and a lower transduction efficiency. Second, after chronic stimulation, ILT2 was not upregulated on resting CAR-T cells, unlike PD-1. Third, the presence of ILT2 did not affect CAR-T cell cytotoxicity, meaning that HLA-G:ILT2 signaling did not inhibit CAR activation signaling. This could be explained by a higher expression level of CAR at the membrane of T cells compared with ILT2 due to EF1α, a strong promoter in human T cells,35 or by the fact that the affinity of the original antibodies for HLA-G (Kd <5 nM) is 100 times higher than that of ILT2 (Kd <50 µM) (online supplemental figure 1).58
In tumors, soluble HLA-G molecules (secreted or shed) may be present. Thus, it can be argued that such cell-free HLA-G molecules could decrease anti-HLA-G CAR-T cell functions by interacting with their ILT2. As shown in this manuscript, HLA-G:ILT2 interaction was not sufficient to affect CAR-T cell function. Alternatively, cell-free HLA-G could bind to the CAR construct and block its interaction with membrane-bound HLA-G from tumor cells. The cell lines used here, JEG-3 and K562-HLA-G1, secrete soluble HLA-G and shed membrane HLA-G isoforms,59 60 but anti-HLA-G CAR-T cells lysed those targets anyway in vitro and in vivo. This indicates that in patients, soluble HLA-G released by tumors is unlikely to act as a decoy and significantly block anti-HLA-G CAR-T cell functions.
HLA-G is also involved in the generation of a tumor tolerogenic microenvironment through regulatory cells: its expression is correlated with accumulation of MDSC,61 increased CD4+CD25+FoxP3+ population62 and secretion of IL-10 and TGF-β that are responsible for the TME maintenance.17 Furthermore, HLA-G is expressed by tolerogenic immune cells such as CD4+FoxP3-HLA-G+ Treg,63 suppressive NK cells,30 DC-10 cells,64 infiltrating monocytes65 and tumor-associated macrophages (TAM),66 67 all involved in the TME. We did not formally demonstrate in the present manuscript that anti-HLA-G CAR-T cells were capable of killing real ex vivo tolerogenic HLA-G-expressing cells such as DC-10 cells. However, in order to demonstrate the possibility that they do, we tested in vitro anti-HLA-G CAR-T cells on LCL-HLA-G1 cells whose immune regulatory functions have been previously demonstrated,68 69 and on the myeloid cell line KG1 transduced with HLA-G1 before and after differentiation in monocyte-derived DC.70 In these three cases, anti-HLA-G CAR-T cells lysed HLA-G-expressing cells, but not their HLA-G-negative controls (online supplemental figure 3). This demonstrates that the capability of anti-HLA-G CAR-T cells to lyse HLA-G-expressing regulatory cells is a real possibility. In this context, anti-HLA-G CAR-T cells could target HLA-G-expressing tumor cells and HLA-G-positive suppressive cells, potentially allowing the treatment of tumors for which immunosuppressive TME could represent a major issue.
Rossig et al have recently developed a CAR-NK therapy against GD2 in Ewing sarcoma,71 but in vivo experiments showed that CARs failed to eliminate GD2-expressing EwS xenografts. Histopathology analysis revealed upregulation of HLA-G in tumor autopsies.72 73 Supporting the relevance of this finding, co-incubation of NK cells with EwS cells induced upregulation of ILT2.72 It is therefore possible that in this instance, the HLA-G:ILT2 interaction inhibited CAR-T cells, causing treatment failure. This is an example where the combinatory therapy with anti-HLA-G CAR-T cells may cancel the immune-resistance generated by the TME. Strikingly, tumors can escape from the immune response if only 10% of tumor cells express HLA-G.74 Thus, patients could benefit from anti-HLA-G CAR-T cell therapy even in tumors with low/heterogeneous HLA-G expression strengthening the case for targeting HLA-G in new combinatory immunotherapies.
Altogether, we report here the first anti-HLA-G CAR-T cells targeting HLA-G which is both a TSA and an ICP. Since HLA-G is expressed on both tumors and suppressive immune cells in the TME, anti-HLA-G CAR-T cells would participate in the elimination of both, improving infiltration of anti-HLA-G CAR-T cells and TILs in the tumor.
Acknowledgments
The authors would like to thank Dr Patrick England (Institut Pasteur, Paris) for the SPR analysis of 15E7 mAb. The authors would also like to thank the animal technicians for their expertise in animal care and management. The authors would like to thank Dr. Claire Germain for cytometry analysis and the animal technicians for their expertise in animal care and management.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors FA, EB-R, JLM, MLe, JMC and PS performed the experiments; FA, EB-R, MLe and ME analyzed data; FA and ME performed statistical analysis; JLM, JC, PS, FG, OA, PL-D, MLo and JC provided guidance and expertise in their respective areas of study. FA, JLM, EB-R, MLo and JC wrote the manuscript; MLo and JC supervised research. All authors provided input, edited and approved the final version of the manuscript.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Patient consent for publication Not required.
Ethics approval Peripheral blood cell samples of healthy donors were collected at the French Blood Center (EFS BFC, Besançon, France) after obtaining written informed consent. Blood samples collection was approved by the French Ministry of Higher Education and Research (agreement number #AC-2015-2408, May 22, 2015). All experimental studies complied with European legislation and were approved by local (Animal Ethics Committee of Besançon (Comité d'Ethique Bisontin en Experimentation Animale)) and national (French Ministry of Higher Education and Research (Ministère de l'Education Nationale, de l'Enseignement Supérieur et de la Recherche)) authorities for the care and use of animals.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available on reasonable request. Data could be available on request to julien.caumartin@invectys.com.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.