Article Text

Abstract
Background Limited therapeutic options are available for triple-negative breast cancer (TNBC), emphasizing an urgent need for more effective treatment approaches. The development of strategies by targeting tumor-associated macrophages (TAMs) to stimulate their ability of Programmed Cell Removal (PrCR) provides a promising new immunotherapy for TNBC treatment.
Methods CD47 is a critical self-protective “don’t eat me” signal on multiple human cancers against macrophage immunosurveillance. Using human and mouse TNBC preclinical models, we evaluated the efficacy of PrCR-based immunotherapy by blocking CD47. We performed high-throughput screens on FDA-approved anti-cancer small molecule compounds for agents potentiating PrCR and enhancing the efficacy of CD47-targeted therapy for TNBC treatment.
Results We showed that CD47 was widely expressed on TNBC cells and TAMs represented the most abundant immune cell population in TNBC tumors. Blockade of CD47 enabled PrCR of TNBC cells, but the efficacy was not satisfactory. Our high-throughput screens identified cabazitaxel in enhancing PrCR-based immunotherapy. A combination of CD47 blockade and cabazitaxel treatment yielded a highly effective treatment strategy, promoting PrCR of TNBC cells and inhibiting tumor development and metastasis in preclinical models. We demonstrated that cabazitaxel potentiated PrCR by activating macrophages, independent of its cytotoxicity toward cancer cells. When treated with cabazitaxel, the molecular and phenotypic signatures of macrophages were polarized toward M1 state, and the NF-kB signaling pathway became activated.
Conclusion The combination of CD47 blockade and macrophage activation by cabazitaxel synergizes to vastly enhance the elimination of TNBC cells. Our results show that targeting macrophages is a promising and effective strategy for TNBC treatment.
- macrophages
- triple-negative breast cancer
- phagocytosis
- immunotherapy
- tumor microenvironment
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Triple-negative breast cancer (TNBC) accounts for 15%–20% of all breast cancers and is a highly aggressive and the most difficult-to-treat subgroup of breast cancers,1 2 commonly associated with a poor patient prognosis and a median survival of approximately 18 months or less.3 TNBC is characterized by a lack of estrogen receptor (ER), progesterone receptor (PR) and human epidermal growth factor receptor 2 (HER2) expression, all of which are important therapeutic targets.1 2 TNBC is considered a single disease entity, with chemotherapy remaining the mainstay and standard treatment approach. However, further long-term clinical benefits have thus far been hindered by a poor prognosis and higher relapse rate caused by lingering cancer cells that were either not eradicated by chemotherapy, or had developed a resistance to it.
The understanding of the mechanisms by which the immune cells detect and attack cancer cells has enabled the development of effective immunotherapies against a range of cancers.4–10 A fraction of TNBC tumors upregulate programmed death ligand 1 (PD-L1) and are infiltrated with lymphocytes, providing the rationale for using immunotherapy for TNBC treatment.11 12 Clinical trials in advanced TNBC using atezolizumab, an antibody targeting PD-L1 that effectively blocks the interaction between PD-L1 and the immune checkpoint PD-1, resulted in significantly improved overall survival and progression-free survival.3 13 As a result, combinatory therapy using both atezolizumab and nab-paclitaxel has been recently approved by the Food and Drug Administration (FDA) in 2019 for the treatment of advanced TNBC.14 15 Atezolizumab currently remains the only FDA-approved immunotherapeutic agent for the treatment of TNBC. While substantial clinical benefits have been observed in patients receiving atezolizumab–nab-paclitaxel combination therapy, only a small fraction of patients showed a positive response,3 likely due to the high level of heterogeneity of TNBC tumors.13 16 17 Coupled with an overall poor patient prognosis, a limited number of therapeutic options for this aggressive disease has instilled urgency for developing novel efficacious therapies.
During cancer development, circulating monocytes are constantly recruited to primary tumors and metastatic sites where they derive to macrophages.18 19 Tumor-associated macrophages (TAMs) represent the most abundant cell type in the tumor microenvironment, composing up to 50% of tumor mass in almost every type of solid cancer, including breast cancer.18–21 Targeting TAMs by activating them to attack malignant cells or inhibiting their function in supporting tumor progression, represents a new class of cancer treatment approaches. While directly depleting TAMs inhibited tumor growth in certain cancers, reprogramming TAMs to elicit their potent tumoricidal functions has become a highly attractive strategy bearing significant therapeutic potential, thus ushering in the next generation of cancer immunotherapy.22
Recent exciting breakthroughs in cancer immunology have begun to unveil the role of macrophages in directly recognizing and engulfing cancer cells, a process termed “Programmed Cell Removal” (PrCR).23–25 Cancer cells and macrophages communicate with each other, during which an imbalance of pro-phagocytic “eat me” pathways over anti-phagocytic “don’t eat me” pathways results in a downstream signaling cascade in macrophages.23–25 Subsequently, cytoskeleton rearrangement drives the engulfment of target cells by macrophages, and engulfed cells are digested in the lysosomes. PrCR is an efficient process of cancer immunosurveillance which can be initiated independent of the induction of cancer cell death.23–25 In tumors, malignant cells have developed self-protective mechanisms to facilitate their survival, such as the upregulation of “don’t eat me” signals, which transduce negative signaling to macrophages, therefore preventing PrCR. The blockade of such signals reinstates macrophage-mediated PrCR, leading to an effective elimination of cancer cells. To date, several potential “don’t eat me” signals have been identified, including CD24, CD47 and MHC I.26–29 Among these, CD47 has been demonstrated to be the most important “don’t eat me” signal. CD47 is ubiquitously expressed and particularly upregulated on a variety of human cancer cells, functioning via a cis-interaction with Signal Receptor Protein alpha (SIRPα) on macrophages to inhibit PrCR by effectively shutting down the machinery required to carry out PrCR.26 27 30 A blockade of CD47 has been shown to be effective in inducing PrCR, therefore eliciting potent anti-cancer effects in multiple preclinical cancer models with promising benefits in clinical trials when used in both monotherapy and combinatory therapy.26 27 30–37
Despite the encouraging progress, it remains to be explored whether the induction of PrCR can be used as a treatment approach for TNBC. CD47 was reported to be highly expressed on TNBC tumors and its expression was correlated with worse patient prognosis and outcomes.38 In this study, using TNBC preclinical models, we examined the efficacy and potential of treating TNBC with therapies aiming to induce PrCR. We demonstrated that although a blockade of CD47 was sufficient in inducing PrCR of TNBC cells, its efficacy was not satisfactory. We further identified that when used in combination with CD47 blockade, cabazitaxel, an FDA-approved chemotherapeutic agent,39 elicited strong anti-cancer effects on PD-L1+ and PD-L1− TNBC. This treatment regimen was thus termed “enhanced PrCR therapy”. More importantly, we discovered that cabazitaxel functions by directly promoting the ability of macrophages to perform PrCR independently of its cytotoxicity toward cancer cells. Furthermore, we identified that NF-kB signaling plays a key role in transcriptional landscape remodeling of these macrophages. Our discoveries uncovered a novel function and purpose for cabazitaxel in PrCR-based immunotherapy and highlighted the potential for PrCR induction to be a promising new direction for developing novel and more effective therapies to fight TNBC.
Materials and methods
Mice
BALB/c and RAG2−/− γc−/− BALB/c mice were bred in the Animal Resources Center at City of Hope Comprehensive Cancer Center. BALB/c mouse strain was purchased from the Jackson Laboratory. RAG2−/− γc−/− mouse strain was a generous gift from Dr. Irving L. Weissman at Stanford University. All animal procedures were in accordance with the guidelines and approved by the Administrative Panel on Laboratory Animal Care at City of Hope Comprehensive Cancer Center.
Cell culture
Human triple-negative breast cancer cell lines (MDA-MB-231, MDA-MB-453, MDA-MB-468), a mouse triple-negative breast cancer cell line (4T1), human non-Hodgkin’s lymphoma cell lines (Raji and Mac1) and a mouse monocyte/macrophage cell line (RAW264.7) were used in this study. MDA-MB-231, MDA-MB-453, MDA-MB-468, 4T1, Raji and RAW264.7 were purchased from ATCC. Mac1 was a generous gift from Dr. Marshall Kadin at Roger Williams Medical Center. Cryopreservation of large quantities of low passage (below 3) cells was performed and cells with passage number below 20 were used in this study. Cells were routinely cultured in high-glucose DMEM medium supplemented with 10% fetal bovine serum (MDA-MB-231, MDA-MB-453, MDA-MB-468 and RAW264.7) or RPMI-1640 medium supplemented with 10% fetal bovine serum (4T1, Raji and Mac1). All cell lines were maintained at 37°C in a humidified 5% CO2/95% air incubator. Mycoplasma examination was routinely performed every 2 months.
Analysis of clinical datasets
Gene expression datasets collected by The Cancer Genome Atlas (TCGA) were obtained from UCSC Xena (https://xenabrowser.net/)40 in the form of log2 (normalized counts+1) values with the query “TCGA Breast Cancer (BRCA)”. From the dataset, the TNBC patient group was selected by the deficiency of ER, PR and HER2. Expression of CD47 and PD-L1 was analyzed and compared between non-TNBC and TNBC groups.
For analysis of immune composition in TNBC tumors, four independent studies were included into the analysis. Gene expression datasets of these studies (GSE25066; GSE58812; GSE76124; GSE21653)41–44 were obtained from Gene Expression Omnibus (GEO) (https://www.ncbi.nlm.nih.gov/geo/). The analytical tool CIBERSORT (https://cibersort.stanford.edu/)45 was used to estimate the relative proportion of the immune cell types.
To analyze the correlation of overall survival and the ratio of M1-like to M2-like TAMs in TNBC patients, TNBC samples from the TCGA dataset were deconvoluted by CIBERSORT.45 The samples were grouped by ratio of M1-like to M2-like TAMs and survival analysis was performed with overall survival information of each patient.
CRISPR/Cas9-mediated gene editing
The CRISPR/Cas9 system was used for suppressing gene expression in TNBC cells. Pairs of primers containing sequences of control sgRNAs or sgRNAs targeting human CD47, mouse CD47, mouse TLR2 or mouse TLR4 genes were designed and cloned into the all-in-one LentiCRISPR V2 vector.46 The LentiCRISPR V2 vector was transfected with the packing plasmids into 293 T cells to generate lentiviruses. Viruses were collected 48–72 hours after transfection and filtered through 0.45 µm filters to remove residual cells and cell debris. For the infection, target cells were incubated with lentiviruses for 48 hours in the presence of polybrene (8 µg/mL) and selected with puromycin (2 µg/mL) 24 hours after removal of the viruses.
The following sgRNA sequences were used:
Control 1 (Non-target): GAACGUAGAAAUUCCCAUUU46
Control 2 (LacZ): UUGGGAAGGGCGAUCGGUGC47
Human CD47: CUACUGAAGUAUACGUAAAG46
Mouse CD47: CCCUUGCAUCGUCCGUAAUG46
Mouse TLR2: CGCGGAUCGACUUUAGACUU46
Mouse TLR4: ACACGUCCAUCGGUUGAUCU46
Flow cytometry analysis
Anti-mouse CD47 (clone miap301; BioLegend), anti-human CD47 (clone B6H12; BD Biosciences), anti-mouse F4/80 (clone BM8; BioLegend), anti-mouse/human CD11b (clone M1/70; BioLegend), anti-mouse MHC-II (clone M5/114.15.2; BioLegend), anti-mouse CD206 (clone C068C2; BioLegend), anti-mouse Sirpα (clone P84; BioLegend) and anti-human CD206 (clone 15-2; BioLegend) were used for FACS analyses. Antibodies were phycoerythrin (PE), PE Cy7, APC, APC Cy7, Alexa Fluor 700, BV787, or BV605 conjugated, or fluorophore-conjugated secondary antibodies were used. Sytox blue was used to exclude dead cells. Annexin V (BD Biosciences) and 7-aminoactinomycin D (7-AAD; ThermoFisher) were used to evaluate cell viability of macrophages on cabazitaxel treatment. Flow cytometry was performed using the BD LSRFortessa cell analyzers.
Generation of macrophages
To generate mouse bone marrow–derived macrophages, BALB/c mice 6 to 12 weeks old were euthanized and femurs were flushed with a 25G needle and filtered through a 70 µm strainer. The filtrates were pelleted by centrifugation and red blood cells were lysed with ACK lysis buffer at room temperature for 2 min and washed with IMDM supplemented with 10% FBS. Subsequently, cells were suspended in IMDM supplemented with 10% FBS and 10 ng/µL murine MCSF. Macrophages from day 6 to day 8 were used for functional assays.
To generate human peripheral blood–derived macrophages, monocytes were enriched from human peripheral blood by magnetic-activated cell sorting using Whole Blood CD14 microbeads (Miltenyi). CD14+ monocytes were cultured in IMDM supplemented with 10% human serum. Macrophages from day 6 to day 8 were used for functional assays.
Phagocytosis assay
Macrophage-based phagocytosis was examined by flow cytometry-, luminescence- or microscopy-based assays.
Macrophages were harvested by TrypLE and scrapers, and equally divided into FACS tubes with 1×105 cells per tube. Target cancer cells were either GFP-expressing or labeled with cell-permeant calcein AM. For calcein labeling, cells were incubated with 200 nM calcein AM in PBS at 37°C for 20 min and washed twice with DMEM medium supplemented with 10% FBS. Cancer cells were added and mixed with macrophages with the addition of antibodies (anti-CD47, BD Biosciences; anti-Sirpα, BioLegend) or IgG control (BioLegend), and incubated at 37°C for 1–2 hours. Anti-CD47 clone B6H12 was used for most experiments and clone CC2C6 was used for the experiment in online supplemental figure 3E. Macrophages were stained with anti-F4/80 antibody conjugated with PE-Cy7. The phagocytosis index was quantified as the percentage of macrophages that phagocytosed cancer cells during the incubation, namely the ratio of GFP+PE-Cy7+ cells to PE-Cy7+ cells. For pre-treatment experiments, cancer cells or macrophages were pre-treated with various concentrations of cabazitaxel for 24 hours followed by phagocytosis assays. Phagocytosis index was normalized to the maximal response in the experiments. In indicated experiments, phagocytosis of cancer cells was also examined with fluorescence microscopy.
Supplemental material
For examining the long-term effects of phagocytosis, the phagocytic clearance of cancer cells mediated by macrophages was quantified by detection of the luminescence signals from surviving cancer cells after coculture with macrophages. Specifically, luciferase equipped cancer cells were co-cultured with macrophages for 24 hours in the presence or absence of antibodies and/or cabazitaxel. Wells containing cancer cells alone (without macrophage addition) were used as a control for calculation (phagocytosis rate=0%). Luciferin was added and the luminescence signals were read with Cytation 3 which were used to quantify the remaining live cancer cells. The phagocytosis rate was calculated as the ratio of signals in the treatment group to the signal of wells containing cancer cells alone. Phagocytosis index was normalized to the maximal response in the experiments.
Mouse models
MDA-MB-231 cells expressing a luciferase-eGFP fusion protein were used for the human TNBC tumor model in RAG2−/− γc−/− mice. Cells were suspended in high glucose DMEM medium containing 25% Corning Matrigel Matrix (v/v) and injected to the mammary fat pad of RAG2−/− γc−/− mice with 5×105 cells per mouse. Nine days after the engraftment, the mice were treated with vehicle (Control), CD47-blocking antibody (clone B6H12; BioXCell), cabazitaxel (Cayman Chemical), or both, once every week. CD47-blocking antibodies were administered intravenously (4 mg/kg body weight), and cabazitaxel was administered intratumorally (40 µg/kg body weight).
Raji cells expressing a luciferase-eGFP fusion protein were used for the human NHL tumor model in RAG2−/− γc−/− mice. Cells were suspended in high glucose DMEM medium containing 25% Corning Matrigel Matrix (v/v) and injected subcutaneously on the back of RAG2−/− γc−/− mice with 5×105 cells per mouse. Nine days after the engraftment, the mice were treated with vehicle (Control) or CD47-blocking antibody (clone B6H12; BioXCell) once every week. CD47-blocking antibodies were administered intravenously (4 mg/kg body weight).
CtrlKD and CD47KD 4T1 cells were used for the mouse TNBC tumor model in immune competent mice. Cells were suspended in RPMI-1640 medium containing 25% Corning Matrigel Matrix (v/v) and injected to the mammary fat pad of BALB/c mice with 5×104 cells per mouse. Nine days after the engraftment, the mice were treated with vehicle or cabazitaxel (80 µg/kg body weight) intratumorally, twice every week.
CtrlKD and CD47KD 4T1 cells expressing a luciferase-eGFP fusion protein were used for the mouse metastatic TNBC model. Cells were suspended in RPMI-1640 medium and injected intravenously to BALB/c mice with 5×105 cells per mouse. Three days after the engraftment, when lung colonization by 4T1 cells was confirmed by bioluminescence imaging, the mice were treated with vehicle or cabazitaxel (2 mg/kg body weight) intravenously, twice every week for up to 3 weeks, as indicated in Results section. For cabazitaxel treatment, the formulations of vehicle are 5% ethanol+5% polysorbate-80+90% of glucose in sterile water (v/v).
Tumor cell engraftment and growth in vivo were monitored by bioluminescent imaging. D-Luciferin potassium salt (Biosynth Carbosynth) was dissolved in PBS to a final concentration of 16.6 mg/mL. Mice were injected intraperitoneally with luciferin solution at a dose of 0.139 g luciferin/kg body weight. Bioluminescent imaging was performed at indicated time points. Images were captured and analyzed with a Lago X (Spectral Instruments Imaging) and bioluminescence signals were analyzed with Aura Image software.
Tumor-associated macrophage analysis
BALB/c mice were engrafted with 4T1 cells by injection to the mammary fat pad and treated with vehicle or cabazitaxel. The resultant tumors were collected, minced into pieces with diameters less than 1 mm, and dissociated in DMEM with Liberase TM enzymes (ThermoFisher) and DNase I (ThermoFisher) at 37°C for 1 hour to achieve a single-cell suspension. Cells were pelleted by centrifugation and red blood cells were lysed with ACK lysis buffer, washed twice with FACS buffer (PBS with 2% FBS) and filtered through a 70 µm cell strainer. Samples were treated with FcR blocker (Miltenyi) before being stained with the indicated antibodies, and subjected to flow cytometry analysis. Tumor-associated macrophages were gated as CD45+, CD11b+, F4/80+ and Zombie Violet−. The expression of MHC-II and CD206 on TAMs was examined.
Macrophage depletion
BALB/c mice were injected with 200 µL of control (PBS) liposomes or clodronate liposomes (Liposoma) intravenously 1 day before the transplantation of 4T1 cells, and 100 µL of the control liposomes or clodronate liposomes were injected every 4 days thereafter throughout the duration of the experiment. The mice were engrafted with CtrlKD or CD47KD 4T1 cells by injection to the mammary fat pad and treated with vehicle or cabazitaxel. To verify the efficacy of macrophage depletion, the resultant tumors were collected, dissociated to single-cell suspension, and subjected to flow cytometry analysis. Tumor-associated macrophages were gated as CD11b+, F4/80+.
Bone marrow–derived macrophage (BMDM) polarization
BMDMs or RAW264.7 cells were seeded in 12-well plates at a density of 1.5×106 cells per well and stimulated for 48 hours with medium for M0-like, LPS (50 ng/mL) and IFNγ (10 ng/mL) for M1-like, or IL4 (10 ng/mL) for M2-like polarization.
NF-kB reporter assay
The GFP-based NF-kB reporter was a generous gift from Dr. Susan Carpenter at the University of California, Santa Cruz. In this system, 5x NF-kB-binding motifs was upstream of the minimal CMV promoter for driving GFP expression.48 We generated a luminescence-based NF-kB reporter by cloning the firefly luciferase gene into this vector to replace the GFP gene. RAW 264.7 cells were infected with lentiviruses expressing the luminescence-based NF-kB reporter to generate the reporter line. CtrlKD, TLR2KD or TLR4KD RAW264.7 NF-kB reporter lines were cultured in 96-well plates overnight, and then stimulated with LPS (10 ng/mL) or cabazitaxel (20, 10, 5, 2.5 µM) for another 8 hours. Unstimulated cells were used as the negative control. Luciferin was added to the cells and luminescence signals were measured using a luminometer (Cytation3 imaging reader, BioTek).
RNA sequencing
BMDMs from BALB/c mice after 7 days of differentiation were treated with DMSO (control) or 10 µM cabazitaxel for 24 hours, after which total RNA was extracted with an RNA extraction kit (Qiagen) and used for library preparation. BMDMs were generated from three individual mice for triplicate samples. The samples were submitted to Novogene for RNA sequencing. The libraries were sequenced on the Illumina NovaSeq 6000 platform. Clean reads were generated after removing reads containing adapter or poly-N and reads of low quality. Next, mapped reads were obtained through aligning clean reads to the reference genome mm10 using STAR V.2.5.3a. The number of mapped clean reads was then counted and normalized into fragments per kilobase of transcript per million reads (FPKM). FPKM value of 17,891 genes obtained through the RNA-seq were used as the original raw data and analyzed by GSA algorithm to find differentially expressed genes, presented by the volcano plot. The genes located out of the borders of the lines (fold change FC >2 and p value<0.05) were considered to be differentially expressed genes (DEGs). In this data set, 452 DEG genes were upregulated and 491 DEG genes were downregulated after treated by cabazitaxel. Gene Set Enrichment Analysis was performed using the GSEA software and the Molecular Signatures Database.49 50
Multidimensional scaling (MDS) analysis
Microarray raw data GSE69607 and RNA sequencing raw count data GSE103958 were downloaded from NCBI GEO separately as reference 1 and reference 2 gene expression of M0, M1 and M2 macrophages.51 52 For reference 1, the expression of 20,556 genes was extracted and annotated by Affymetrix Mouse Genome 430 V.2.0 using R package affy (V.1.62.0), and gcrma (V.2.56.0) for background noise removal and log2 transformation. The microarray gene expression was scaled to have zero mean and unit variance, and quantile normalized before performing MDS. For reference 2, the expression of 16,617 genes was extracted, log2 transformed and scaled to have zero mean and unit variance, and normalized to reference 1 target distribution using FSQN (feature-specific quantile normalization, https://github.com/jenniferfranks/FSQN) to remove cross-platform batch effects before performing MDS. Together we have 6 M0, 15 M1 and 8 M2 macrophage reference samples.
Our RNA sequencing fastq files were aligned to Mouse Genome mm10 using STAR V.2.5.3a, and then quantified by genes to mm10—Ensembl Transcripts release 100 on Partek Flow platform. The expression of 17,891 genes was log2 transformed and scaled to have zero mean and unit variance, and normalized to reference 1 target distribution using FSQN.
MDS was performed with cmdscale function in R3.6.0, and the number of retained dimensions was set to 3 to cluster the M0/M1/M2 macrophage samples, then visualized in 3D dimension using R package ggfortify (V.0.4.10).
High-throughput screen
The FDA-approved oncology drugs set including 147 current approved anti-cancer small molecules was obtained from the Division of Cancer Treatment & Diagnosis at the National Cancer Institute (NCI). For the high-throughput screen, individual anti-cancer small molecule compounds were incubated with 0.03×106 MDA-MB-231 cells expressing GFP-luciferase and 0.06×106 macrophages in the presence or absence of CD47 blockades (anti-CD47 or anti-Sirpα antibodies) in 96-well plates for 24 hours. Thereafter, luciferin was added into the plate and luminescence signal was measured with Cytation 3. Wells containing 10 µM drug and 231 cells were used as a control for calculating phagocytosis rate (phagocytosis rate=0%). The normalized phagocytosis index was calculated as the ratio of phagocytosis rate induced by drug treatment to that by DMSO.
Cytokine/chemokine analysis
BMDM cells were seeded in 24-well plates at a density of 5×105 cells per well and incubated with DMSO (control) or cabazitaxel (10 µM). Cell culture medium without BMDM cells (negative control), DMSO-treated BMDM conditioned medium and cabazitaxel-treated BMDM conditioned medium were collected and centrifuged. The supernatants were collected and submitted to Eve Technologies for Mouse Cytokine Array/Chemokine Array 44-plex discovery arrays which were analyzed using Millipore MILLIPLEX mouse cytokine/chemokine kit.
Statistical analysis and graphing software
All data were statistically analyzed and graphed using Microsoft Excel or GraphPad Prism V.8. Unpaired t-test or ANOVA test was employed to detect the significant differences between groups. Overall survival was evaluated with the Kaplan-Meier method and log-rank test was used for comparisons between groups. The quantitative data in the study were expressed as the mean±SD. P values below 0.05 were considered statistically significant differences.
Results
CD47 is a therapeutic target in TNBC
We first evaluated whether CD47 can be harnessed as a valid therapeutic target for inducing PrCR of TNBC. Successful PrCR-based therapies, involving a blockade of CD47, rely on abundant macrophage infiltration in TNBC tumors as well as CD47 expression on TNBC cells. First, we analyzed RNA sequencing data of four cohorts of TNBC patient specimens using the CIBERSORT analysis, which allowed us to quantify different immune cell populations in the tumor microenvironment. We found that all of the TNBC tumor specimen cohorts examined were highly infiltrated with TAMs, T cells, and plasma cells, with TAMs representing the most abundant immune cell population (figure 1A). Next, we analyzed the gene expression landscape of tumor samples of patients with breast cancer, including both TNBC and non-TNBC. We found that CD47 was highly expressed in breast tumors and was more highly expressed in TNBCs than non-TNBCs (figure 1B). When compared with PD-L1 transcripts, a higher number of CD47 transcripts were detected in TNBCs (figure 1C). We then examined several representative human and mouse TNBC preclinical cell models, including MDA-MB-231, MDA-MB-453, MDA-MB-468 and 4T1. We were able to confirm CD47 protein expression on all cell lines, but PD-L1 expression was not detected on MDA-MB-453 and MDA-MB-468 (figure 1D; online supplemental figure 1A). Interestingly, when compared with CD47 expression on hematopoietic cancers such as lymphoma (figure 1D), TNBC expression of CD47 was relatively lower.

CD47 is a therapeutic target in triple-negative breast cancer (TNBC). (A) Inferred composition of 10 immune cell subsets in TNBC biopsies. The gene signature datasets were obtained from Gene Expression Omnibus (GEO) database. The results were analyzed using CIBERSORT. (B) A comparison of CD47 gene expression between non-TNBC (n=974) and TNBC (n=123), basing on TCGA Breast Cancer (BRCA). Whiskers represent min–max values. ***p<0.001 (t-test). (C) A comparison of gene expression of PD-L1 and CD47 on TNBC (n=123), basing on TCGA Breast Cancer (BRCA). Whiskers represent min–max values. ***p<0.001 (t-test). (D) Representative histogram plots showing CD47 expression on NHL cell lines (Raji and Mac-1) and TNBC cell lines (MDA-MB-231, MDA-MB-453, MDA-MB-468 and 4T1). Anti-hCD47 (clone B6H12) or anti-mCD47 (clone miap301) were used for human (Raji, Mac-1, MDA-MB-231, MDA-MB-453 and MDA-MB-468) or mouse (4T1) lines. (E) An in vitro flow cytometry–based phagocytosis assay comparing the efficiency of blocking CD47 with an anti-CD47 antibody between TNBC cell lines (MDA-MB-231, MDA-MB-453 and MDA-MB-468) and NHL cell lines (Raji and Mac-1). Mouse M0 bone marrow–derived macrophages (BMDMs) were used for the assay. Phagocytosis was normalized to the maximal response in the experiments. (F) An in vitro flow cytometry–based phagocytosis assay comparing the efficiency of blocking CD47 by knocking down CD47 gene expression between TNBC cell lines (MDA-MB-231, MDA-MB-453 and MDA-MB-468) and NHL cell lines (Raji and Mac-1). Mouse M0 BMDMs were used for the assay. Phagocytosis was normalized to the maximal response in the experiments. (G and H) In vivo tumor engraftment assay of Raji and MDA-MB-231 cells. Mice engrafted with Raji or MDA-MB-231 cells were treated with vehicle or CD47-blocking antibody. Tumor engraftment and growth were measured by bioluminescence imaging. p=0.0101 (Raji) or 0.3471 (MDA-MB-231) (t-test).
We then performed two layers of experiments to investigate PrCR induced by CD47 blockade: in vitro phagocytosis assays to examine direct PrCR of cancer cells by macrophages and in vivo tumor engraftment assays to examine the anti-cancer effect of CD47 blockade in tumor development. Bone marrow progenitor cells and monocytes are the main sources for macrophages in the tumors.19 Previous studies measuring binding affinity between CD47 and Sirpα demonstrated that Sirpα molecules on macrophages from BALB/c or NSG mice display comparable or stronger binding to human CD47, as compared with human Sirpα (Kd for binding of human CD47 to human, BALB/c, NSG or C57BL/6 Sirpα are 290, 308, 2.5, 5×1010 nM, respectively),53–55 highlighting BMDMs from these strains as sound research models for assessing the response of cancer cells to PrCR. In this study, we used BMDMs from BALB/c mice for in vitro studies and mice with BALB/c background for evaluation of the efficacy of CD47 blockade in vivo. An additional macrophage model, human peripheral blood monocyte-derived macrophages, was also included for evaluating our findings. On blockade of CD47, whether via antibody or via CRISPR/Cas9-mediated gene editing (online supplemental figure 1B), all the TNBC cell lines examined became prone to macrophage recognition and phagocytosis (figure 1E,F). Consistent with previous studies, a higher phagocytosis rate was observed with CD47-blocking antibody compared with that induced by CD47KD, which is due to an additional effect by Fc-region of the antibody.26 56 57 However, when quantifying PrCR efficiency, we found that TNBC cells were generally less susceptible to macrophage PrCR, as compared with lymphoma cells (figure 1E,F). When we examined the anti-cancer effects of CD47 blockade–induced PrCR in in vivo xenotransplantation models with RAG2−/−, γc−/− mice (T-, B- and NK-deficient, with functional phagocytes maintained23), we found that the response of MDA-MB-231 (TNBC) cells to CD47-blocking antibody was not as significant as that of Raji cells (non-Hodgkin’s lymphoma) (figure 1G,H).
High-throughput screens with FDA-approved anti-cancer small molecule compounds identify cabazitaxel as a PrCR-promoting agent
We reasoned that PrCR-based therapy may be effectively applied to TNBC through increasing the susceptibility of TNBC cells to PrCR, or promoting macrophages toward a more phagocytosis-capable phenotype. It follows that therapeutic agents eliciting these particular effects may be combined with a CD47 blockade to yield a superior efficacy of macrophage-mediated TNBC elimination. The FDA has so far approved more than 100 anti-cancer small molecule compounds as chemotherapy or target therapy agents. In addition to their cytotoxicity, some of these anti-cancer drugs were reported to have immunomodulatory effects,58 59 but whether these small molecule compounds can be combined with PrCR-based therapy for TNBC has not been explored. To this end, we were motivated to investigate such a possibility by screening a library of 147 FDA-approved anti-cancer small molecule compounds obtained from the NCI. We established a high-throughput screening system in which macrophages and MDA-MB-231 cells were co-cultured, and CD47-blocking or SIRPα-blocking antibodies were used to induce PrCR (figure 2A,B). PrCR efficacy was evaluated by quantifying the number of cancer cells surviving PrCR. MDA-MB-231 cells were equipped with a GFP-luciferase reporter, such that the surviving cells can be precisely quantified by measuring luminescence upon the addition of luciferin. Results from screens with CD47-blocking and SIRPα-blocking antibodies showed a strong correlation with each other, confirming that the enhancement or attenuation of cancer cell survival resulting from the addition of the small molecular compounds were due to the inhibition or promotion of PrCR (figure 2C). Cabazitaxel, a chemotherapeutic agent approved by FDA for prostate cancer treatment, was identified as the most effective agent that dramatically enhanced the efficacy of CD47 blockade–induced PrCR of MDA-MB-231 cells (figure 2B). Cabazitaxel is a taxane which inhibits cell proliferation by promoting microtubule polymerization and thus inhibiting mitosis. Cabazitaxel demonstrated a decreased binding to P-glycoprotein, a frequent cause for resistance to other taxanes,39 and excellent intratumoral penetration ability, as shown in preclinical studies and clinical trials.60 61 Therefore, we decided to verify our screening results by examining the effects of cabazitaxel in enhancing PrCR in a dose-dependent manner on multiple TNBC lines. Cabazitaxel was able to significantly promote CD47-blocking antibody–induced PrCR at a concentration of 1.25 µM or higher for MDA-MB-231, MDA-MB-453 and MDA-MB-468 cells (figure 2D,E). Interestingly, at the concentrations in which PrCR was strongly promoted, cabazitaxel only demonstrated minor cytotoxicity toward MDA-MB-231 cells (figure 2D).

High-throughput screens with FDA-approved anti-cancer small molecule compounds identify cabazitaxel as a PrCR-promoting agent. (A) A schematic showing the experimental design of the high-throughput screen. (B) PrCR-based high-throughput screens of 147 FDA-approved anti-cancer small molecule compounds. Cells were treated with antibodies blocking CD47–Sirpα interaction (anti-CD47 or anti-Sirpα) and subjected to luminescence-based phagocytosis assay. Phagocytosis was normalized to DMSO control. Spots represent individual compounds. (C) The correlation of phagocytosis change between screens with anti-CD47 and anti-Sirpα antibodies. Spots represent individual compounds. (D) Representative bioluminescence images of the luminescence-based phagocytosis assay measuring surviving cancer cells, with MDA-MB-231 cells as the target cells. Mouse M0 bone marrow–derived macrophages (BMDMs) were used for the assay. Cabazitaxel (Cab) was used at doses from 0 to 10 µM. (E) An in vitro luminescence-based phagocytosis assay measuring surviving cancer cells, with MDA-MB-231, MDA-MB-453 or MDA-MB-468 cells as the target cells. Mouse M0 BMDMs were used for the assay. Cells were treated with CD47-blocking antibodies and various concentrations of cabazitaxel (0 µM, 1.25 µM, 2.5 µM, 5 µM, 10 µM). Phagocytosis was normalized to the maximal response in the experiments. Each group was compared with the control group (0 µM cabazitaxel). **p<0.01, ***p<0.001 (one-way ANOVA test). Error bars represent SD.
Cabazitaxel promotes PrCR by directly activating macrophages and independently of its cytotoxic effects toward TNBC cells
Given that cabazitaxel promoted PrCR of TNBC cells without directly inducing cell death, we further hypothesized that cabazitaxel functioned by either inhibiting anti-PrCR mechanisms of TNBC cells or by potentiating the phagocytic capacity of macrophages. To test this hypothesis, we devised two treatment protocols for a phagocytosis assay: (1) cabazitaxel pre-treated BMDMs co-cultured with untreated MDA-MB-231 cells or (2) cabazitaxel pre-treated MDA-MB-231 cells co-cultured with untreated BMDMs. We showed that the pre-treatment of MDA-MB-231 cells showed minor effects on PrCR, while the pre-treatment of BMDMs significantly induced PrCR without impacting their cell viability, suggesting that cabazitaxel played a role in directly activating macrophages (figure 3A,B; online supplemental figure 2). Macrophages pre-treated with cabazitaxel were evaluated in multiple TNBC cell models and demonstrated an enhanced phagocytic ability against all models examined, when CD47 was blocked with blocking antibodies or gene knockdown (figure 3C,D; online supplemental figure 3A–E). Interestingly, docetaxel, another taxane which promotes microtubule polymerization (figure 2B), and colchicine (online supplemental figure 3F), a taxane antagonist which induces microtubule disassembly, demonstrated no effect on PrCR, suggesting the manipulation of microtubules in macrophages is not the main cause for their enhanced phagocytic ability. This unique effect was also observed in human peripheral blood monocyte-derived macrophages, in which their pre-treatment with cabazitaxel resulted in similar enhancement of CD47 blockade–induced PrCR of TNBC cells (figure 3E), indicating the occurrence of cabazitaxel-mediated enhancement of PrCR in human macrophages.

Cabazitaxel promotes PrCR by directly activating macrophages and independently of its cytotoxic effects toward triple-negative breast cancer (TNBC) cells. (A) An in vitro luminescence-based phagocytosis assay measuring surviving cancer cells with either TNBC cells or mouse M0 bone marrow–derived macrophages (BMDMs) pre-treated with cabazitaxel. TNBC cells or BMDMs were pre-treated with various concentrations of cabazitaxel before cocultured with untreated BMDMs or TNBC cells respectively. Phagocytosis assay was performed in the presence of CD47-blocking antibodies. Phagocytosis was normalized to the maximal response in the experiments. Each group was compared with the control group (0 µM cabazitaxel). *p<0.05, **p<0.01, ***p<0.001 (one-way ANOVA test). Error bars represent SD. (B) Representative bioluminescence images of the luminescence-based phagocytosis assay measuring surviving cancer cells, with MDA-MB-231 cells as the target cells. Mouse M0 BMDMs pre-treated with 10 µM cabazitaxel or DMSO were used for the assay. (C) An in vitro flow cytometry–based phagocytosis assay, with MDA-MB-231, MDA-MB-453 or MDA-MB-468 cells as target cells. Mouse M0 BMDMs pre-treated with DMSO or cabazitaxel were used for the assay. Phagocytosis assay was performed in the presence of CD47-blocking antibodies. Phagocytosis was normalized to the maximal response in the experiments. Each group was compared with the control group (0 µM cabazitaxel). *p<0.05, **p<0.01, ***p<0.001 (one-way ANOVA test). Error bars represent SD. (D) Representative fluorescence microscopic images of a phagocytosis assay, with CD47KD MDA-MB-231 cells as target cells. Mouse M0 BMDMs (red) were stained with CellTrace Far Red, while CD47KD MDA-MB-231 (green) were labeled with CellTrace Calcein Green. Arrows indicated the macrophages (double colors) that phagocytosed cancer cells. (E) An in vitro luminescence-based phagocytosis assay measuring surviving cancer cells, with MDA-MB-231 cells as target cells. Human peripheral blood monocytic cell–derived macrophages were used for the assay. Phagocytosis assay was performed in the presence of CD47-blocking antibodies. Phagocytosis was normalized to the maximal response in the experiments. Each group was compared with the control group (0 µM cabazitaxel). **p<0.01, ***p<0.001 (one-way ANOVA test). Error bars represent SD.
Cabazitaxel significantly enhances the efficacy of blocking CD47 in inhibiting TNBC tumor development and metastasis
Next, we evaluated the therapeutic potential of combining cabazitaxel and CD47 blockade, which we termed “enhanced PrCR therapy”, for TNBC treatment using several different preclinical mouse models. First, MDA-MB-231 cells were injected into the mammary fat pads of RAG2−/−, γc−/− mice which entirely lack T, B and NK cells, but maintain functional macrophages. A monoclonal anti-CD47 antibody was used to block CD47–Sirpα interaction and induce a PrCR response. The CD47-blocking antibody and/or cabazitaxel were administered to the mice engrafted with MDA-MB-231 cells. We showed that neither CD47-blocking antibody nor cabazitaxel alone was able to significantly affect the tumor progression, whereas a combination of these two dramatically inhibited tumor development (figure 4A,B; online supplemental figure 4A), suggesting a strong synergism between these two agents. Next, syngeneic mouse TNBC models were used to evaluate the efficacy of the enhanced PrCR therapy. CD47 blockade was achieved by suppression via CRISPR/Cas9-mediated gene editing (online supplemental figure 4B). Cas9 and non-targeting sgRNA or CD47 targeting sgRNA were transduced into a mouse TNBC line, 4T1. Control knockdown (CtrlKD) or CD47 knockdown (CD47KD) 4T1 cells were injected into the mammary fat pads of immunocompetent BALB/c mice. Treatment with cabazitaxel elicited minor effects on CtrlKD tumors but was able to significantly inhibit tumors developed by CD47KD 4T1 cells, which lack protection from PrCR conferred by CD47 (figure 4C,D; online supplemental figure 4C), reinforcing that cabazitaxel enhanced CD47 blockade–mediated anti-cancer activity. Depletion of TAMs by clodronate liposomes showed no effects on the burden of CtrlKD 4T1 tumors, but largely abrogated the inhibition of tumor growth in CD47KD 4T1 tumors treated with cabazitaxel (figure 4E; online supplemental figure 4D–F), confirming that TAMs were the critical immune components responsible for the diminished tumor growth by the enhanced PrCR therapy. Lastly, our enhanced PrCR therapy was evaluated in a lung metastatic model. 4T1 cells were intravenously injected to BALB/c mice, which were treated with cabazitaxel after 4T1 metastases were established in the lungs. CD47KD 4T1 cells demonstrated a slightly compromised ability to colonize the lung and thus improved mice survival (figure 4F–H; online supplemental figure 4G,H). Treatment with cabazitaxel had no significant effect on the tumor progression and lung colonization in mice engrafted with 4T1 cells (figure 4F–H); however, a combination of cabazitaxel and CD47 suppression nearly completely eradicated lung colonization of 4T1 cells on days 17 and 20 on engraftment, and therefore significantly improved the survival of the mice when compared with mice transplanted with CtrlKD 4T1 cells, from an average of 17.4 days to 40.9 days (figure 4F–H). Taken together, our data indicate that cabazitaxel treatment enabled dramatically stronger in vivo anti-tumor activity mediated by CD47 blockades in both immunocompromised and immunocompetent settings.

Cabazitaxel significantly enhances the efficacy of blocking CD47 in inhibiting triple-negative breast cancer tumor development and metastasis. (A, B) Growth of tumors developed by MDA-MB-231 cells in RAG2−/− γc−/− mice. Mice orthotopically engrafted with MDA-MB-231 cells were treated with PBS, anti-CD47 antibody, cabazitaxel or a combination of anti-CD47 antibody and cabazitaxel. Tumor growth was measured by bioluminescence imaging. (A) Tumor growth curve, ***p<0.001 (log-linear regression analysis). (B) Tumor burden, ***p<0.001 (one-way ANOVA test). Error bars represent SD. (C, D) Growth of tumors developed by 4T1 cells in BALB/c mice. Mice orthotopically engrafted with CtrlKD or CD47KD 4T1 cells were treated with vehicle or cabazitaxel. Tumor sizes were measured at indicated dates. (C) Tumor growth curve, *0.01<p<0.05, ***p<0.001 (log-linear regression analysis). (D) Tumor burden, *p<0.05, **p<0.01, ***p<0.001 (one-way ANOVA test). Error bars represent SD. (E) Growth of tumors developed by 4T1 cells in BALB/c mice. Mice were orthotopically engrafted with CtrlKD 4T1 and treated with vehicle (Ctrl), or were orthotopically engrafted with CD47KD 4T1 cells and treated with cabazitaxel (Combo). The mice were treated with control liposomes or clodronate liposomes, and tumor sizes were measured at indicated dates. (F, G) Metastasis of 4T1 cells in BALB/c mice. CtrlKD or CD47KD 4T1 cells were intravenously injected to the mice. Colonization and growth of 4T1 cells in the lungs were measured by bioluminescence imaging. (F) Growth curve of lung metastases, ***p<0.001 (log-linear regression analysis). (G) Tumor burden, *p<0.05, **p<0.01, ***p<0.001 (one-way ANOVA test). Error bars represent SD. (H) Survival analysis of metastasis of 4T1 cells in BALB/c mice, **p<0.01, ***p<0.001 (log-rank (Mantel-Cox) test).
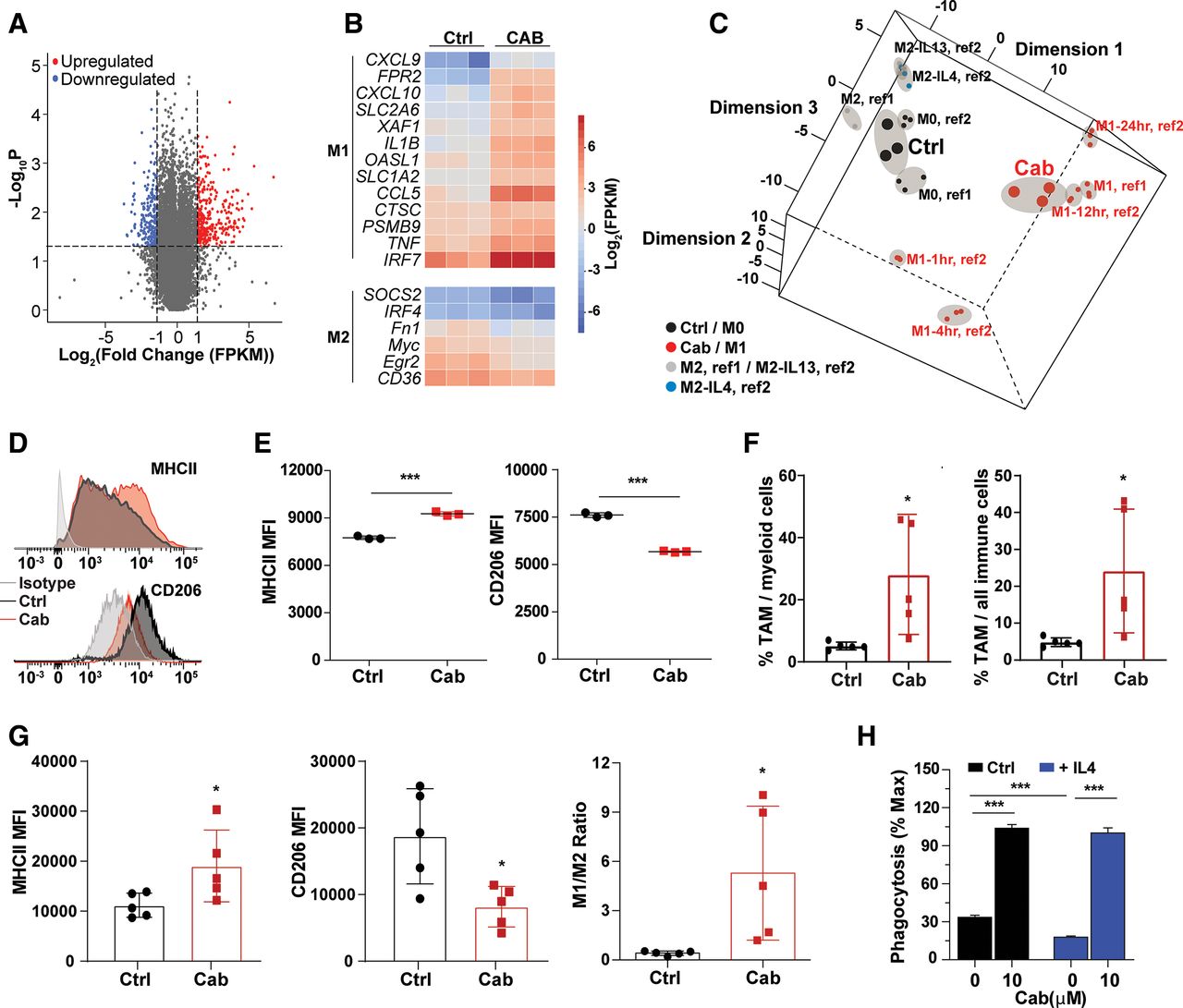
Cabazitaxel induces differentiation of macrophages toward M1-like state
Next, we sought to understand the molecular mechanisms of how cabazitaxel promoted the phagocytic ability of macrophages toward TNBC cells. We showed that the cell surface expression of Sirpα or calreticulin on macrophages, known receptors for transducing “don’t eat me” signals or targeting “eat me” signals on cancer cells,57 62 was not affected by cabazitaxel treatment (online supplemental figure 5A, left and right). We decided to dissect the gene expression profiling in macrophages reprogrammed by cabazitaxel, using RNA sequencing. First, total RNA was extracted from vehicle-treated or cabazitaxel-treated BMDMs and subjected to RNA sequencing. A total of 452 genes were significantly upregulated and 491 genes were significantly downregulated by at least twofold (figure 5A). We then examined genes related to polarization of macrophages. TAMs are broadly classified into two groups—classically activated macrophages (M1) and alternatively activated macrophages (M2) based on their polarization states, although not always mutually exclusive.63 We compared the statistically significant changes in gene expression profiling of cabazitaxel-treated macrophages with gene signatures of M1-like and M2-like human monocyte-derived and mouse bone marrow–derived macrophages reported in literature.51 52 64 We uncovered an upregulation of genes associated with M1-like polarization, and a downregulation of genes associated with M2-like polarization on cabazitaxel-treated BMDMs, suggesting cabazitaxel treatment elicited differentiation of BMDMs toward M1-like phenotypes (figure 5B). We then performed MDS analysis,65 using previously identified M0, M1 (polarization induced by LPS+IFNγ), M2 (polarization induced by IL4 or IL13) transcriptional signatures as references.51 52 Control BMDMs were clustered to M0 macrophages and on cabazitaxel treatment, the transcriptional cluster of macrophages shifted toward a M1-like signature (figure 5C). By flow cytometry analysis, we showed that on cabazitaxel-treated mouse BMDMs (figure 5D, top and bottom) and human PBMC-derived macrophages (figure 5E, left and right), a treatment led to M1-like surface profiling with increased MHCII expression and decreased CD206 expression. In addition, when analyzing TAMs isolated from breast tumors developed by 4T1 cells in BALB/c mice, we found the proportion of TAMs among myeloid or total immune cells in the tumors significantly increased in cabazitaxel-treated mice (figure 5F, left and right). Consistently, we showed an increase of MHCII and a decrease of CD206 expression on TAMs (figure 5G, left and middle; online supplemental figure 5B), as well as a significantly increased ratio of M1-like TAMs to M2-like TAMs (figure 5G, right), in the 4T1 tumors from BALB/C mice receiving cabazitaxel treatment, as compared with the mice treated with vehicles. The polarization of BMDMs by IL4 to M2-like macrophages, which phenotypically resemble TAMs, decreased their phagocytic ability (figure 5H). Importantly, treatment of M2-like polarized BMDMs with cabazitaxel was sufficient to restore PrCR against TNBC cells (figure 5H). Significantly, cabazitaxel-induced increased ratio of M1-like to M2-like TAMs may have profound therapeutic impact on human patients as we found with TCGA data that the increased ratio serves as an independent prognostic factor favoring patient survival in TNBC (online supplemental figure 5C).

Cabazitaxel induces differentiation of macrophages toward M1-like state. (A) Volcano plot showing differentially expressed genes in DMSO-treated and cabazitaxel-treated bone marrow–derived macrophages (BMDMs). Genes with a p value <0.05 and a fold change FC >2 were considered as significantly upregulated or downregulated. BMDMs were generated from three individual BALB/c mice for triplicated samples. (B) A heatmap of gene expression profile by RNA sequencing showing the upregulation of M1-like gene signature and downregulation of M2-like gene signature in cabazitaxel-treated BMDMs, as compared with DMSO-treated BMDMs. (C) Multidimensional scaling (MDS) analysis of RNAseq results from DMSO-treated and cabazitaxel-treated BMDMs. Two references (ref1 and ref2) were used for MDS analysis in which M1 and M2 polarization were induced. In ref2, M1 was induced by LPS+IFNɤ for different time points (1, 4, 12, 24 hours) and M2 was induced by IL4 or IL13. The dots representing DMSO-treated and cabazitaxel-treated BMDMs were presented with bigger size than the dots representing the references. Replicates of the same sample are circled in a shade. (D) Representative FACS plots showing the expression of MHCII (top) and CD206 (bottom) on BMDMs stimulated with cabazitaxel or DMSO (control). (E) Mean fluorescent intensity (MFI) of MHCII (left) and CD206 (right) expression on human peripheral blood monocytic cell–derived macrophages stimulated with cabazitaxel or DMSO (control). ***p<0.001 (t-test). (F) The proportion of F4/80+tumor-associated macrophages (TAMs) in CD11b+ myeloid cells, and the proportion of F4/80+CD11b+tumor-associated macrophages in CD45+ immune cells, in tumors developed by 4T1 cells in BALB/c mice. Mice were treated with vehicle or cabazitaxel. *p<0.05 (t-test). Error bars represent SD. (G) MFI of MHCII (left) and CD206 (middle) expression, and the ratio of M1:M2 TAMs (right) in tumors developed by 4T1 cells in BALB/c mice. Mice were treated with vehicle or cabazitaxel. *p<0.05 (t-test). (H) An in vitro luminescence-based phagocytosis assay, with MDA-MB-231 cells as the target cells. Mouse M0 BMDMs were stimulated with control medium or IL4 (10 ng/mL, to induced M2 polarization) for 48 hours and then treated with DMSO or cabazitaxel. Phagocytosis assay was performed in the presence of CD47-blocking antibodies. Phagocytosis was normalized to the maximal response in the experiments. ***p<0.001 (one-way ANOVA test). Error bars represent SD.
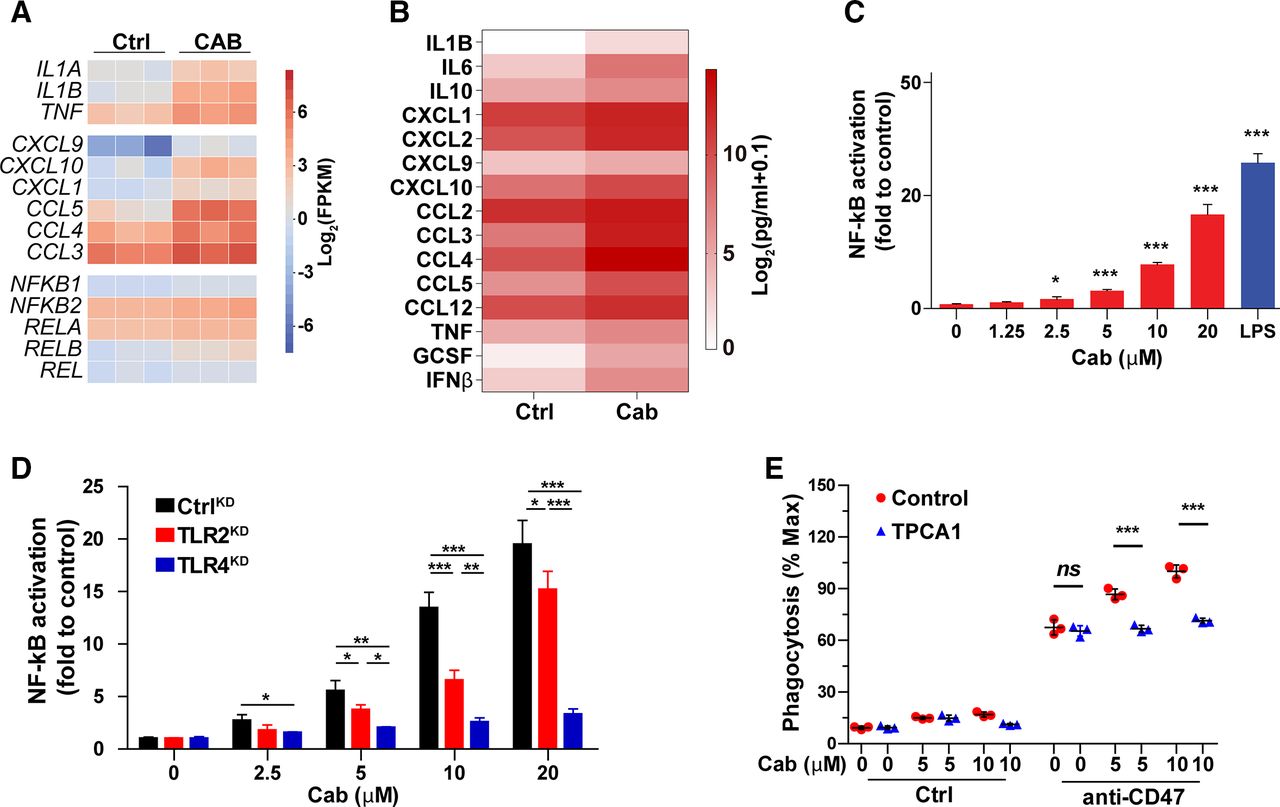
Activation of NF-kB signaling is critical for cabazitaxel-induced enhancement of PrCR
RNA sequencing of the vehicle-treated or cabazitaxel-treated BMDMs revealed that among the pathways regulating macrophage polarization, NF-kB signaling was significantly activated in cabazitaxel-treated macrophages. Particularly, NF-kB targeted transcription factors, cytokines and chemokines were upregulated, as well as the expression of NF-kB family members (figure 6A). Considering the roles cytokine and chemokine molecules play in facilitating macrophage recruitment and target cell recognition and interaction, we then examined the secreted cytokine and chemokine profiling at the protein level. Consistent with our discoveries in RNAseq analysis, upon cabazitaxel treatment, a significant enhanced production of TNF, IL1B, IL6, IFNβ, GCSF, CCL3, CCL4, CCL5, CXCL1, CXCL9 and CLCL10 by macrophages were observed, which are encoded by downstream targeted genes in the NF-kB pathway (figure 6B; online supplemental figure 5D). In addition, activation of NF-kB signaling was confirmed in macrophages with a NF-kB reporter system. A mouse monocyte/macrophage cell line RAW264.7 was equipped with a NF-kB reporter whose activation can drive the expression of luciferase and can be qualified by the luminescence signals. Cabazitaxel treatment drove the activation of NF-kB signaling, as indicated by the luminescence (figure 6C). Gene set enrichment analysis (GSEA) of the RNA sequencing results demonstrated the activation of innate immunity-related signaling pathways in cabazitaxel-treated macrophages, with Toll-like receptor (TLR) signaling being the most enriched pathway (online supplemental figure 6A–D). TLRs play crucial roles in activating NF-kB signaling and eliciting innate immune response.66 To further understand the molecular mechanisms by which cabazitaxel stimulated NF-kB signaling in macrophages, we used the CRISPR/Cas9 system to suppress the expression of TLRs in macrophages to examine their possible involvement. We showed that knockdown of the expression of TLR2 and TLR4 largely blocked NF-kB signaling induced by cabazitaxel, suggesting these receptors played critical roles in mediating PrCR-promoting effects of cabazitaxel in macrophages (figure 6D; online supplemental figure 6E). Treatment of macrophages with NF-kB inhibitor TPCA1 which blocked IkB kinases attenuated phagocytosis and reversed cabazitaxel’s effects in promoting phagocytosis, indicating that NF-kB signaling is critical for cabazitaxel-mediated enhancement of PrCR (figure 6E; online supplemental figure 6F,G).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Activation of NF-kB signaling is critical for cabazitaxel-induced enhancement of PrCR. (A) A heatmap of gene expression prolife by RNA sequencing showing the upregulation of genes regulated or relevant to NF-kB signaling in cabazitaxel-treated bone marrow–derived macrophages (BMDMs), as compared with DMSO-treated BMDMs. (B) Measurement of cytokine and chemokine secretion profiling of BMDMs treated with DMSO (control) or cabazitaxel by a Luminex assay. Scale bars indicate log2(pg/mL+0.1). (C) Activation of NF-kB signaling pathway, as measured by a NF-kB reporter system. RAW 264.7 macrophages were stimulated with various concentrations of cabazitaxel or 1.25 ng/mL LPS (positive control). Unstimulated RAW 264.7 cells acted as a negative control. Each group was compared with the control group (0 µM cabazitaxel). *p<0.05, ***p<0.001 (one-way ANOVA test). Error bars represent SD. (D) Activation of NF-kB signaling pathway, as measured by a NF-kB reporter system. CtrlKD, TLR2KD or TLR4KD RAW 264.7 macrophages were stimulated with various concentrations of cabazitaxel. Unstimulated RAW 264.7 cells acted as a negative control. Each group was compared with the control group (0 µM cabazitaxel). *p<0.05, **p<0.01, ***p<0.001 (one-way ANOVA test). Error bars represent SD. (E) An in vitro flow cytometry–based phagocytosis assay, with MDA-MB-231 cells as target cells. Mouse M0 BMDMs were pre-treated with various concentrations of cabazitaxel in absence or presence of TPCA-1 or CD47-blocking antibodies. Phagocytosis was normalized to the maximal response in the experiments. ***p<0.001 (one-way ANOVA test). Error bars represent SD.
Discussion
During cancer progression, conveying inhibitory signals to the immune system improves the chances of malignant cells in surviving immunosurveillance, leading to the dominance of cancer cells that possess or have acquired immune evasive mechanisms in the tumors.67 The identification of immune checkpoints and invention of agents targeting signals that inhibit an immune response have revolutionized the treatment of multiple types of human cancers, including those that are very aggressive and advanced.10 68
A combination of atezolizumab and nab-paclitaxel led to significantly improved overall survival in patients with PD-L1+TNBC, demonstrating the efficacy of immunotherapy in treating this subgroup of aggressive breast cancers.3 13 However, the recent Impassion 131 study (NCT03125902) revealed that paclitaxel in combination with atezolizumab failed to prolong progression-free survival of patients with TNBC when comparing with paclitaxel plus placebo,69 underscoring the need to better understand the cancer and immune system interactions. Nevertheless, the only FDA-approved immunotherapy for TNBC, atezolizumab, holds potential for improving patient prognosis or even a cure for a group of patients with TNBC where other therapies have been rendered ineffective and unsuccessful. Critical factors that may contribute to determining the overall efficacy of atezolizumab for TNBC include the expression levels of PD-L1 in tumors, lymphocytic tumor infiltration and tumor mutational burden.13 16 17 Although some PD-L1 cancers also showed a response to atezolizumab, this may be partially attributed to the highly heterogeneous nature of TNBC tumors.
The fact that a large group of patients with TNBC resistant to chemotherapy showed little to no response to atezolizumab–chemotherapy combination treatment3 highlighted an urgent need for the development of additional effective treatment approaches. Given that TNBC tumors have a more ubiquitous upregulation of CD47 and are highly infiltrated by macrophages, this makes PrCR-based therapy a promising new class of immunotherapy for TNBC. More importantly, PrCR relies on TAMs that are abundant in the tumor microenvironment, and its execution is via mechanisms distinct from PDL1-PD1 signaling pathways; therefore, an induction of PrCR may yield effective therapies that complement or synergize with T-cell immune checkpoint inhibitors.
Recent progress from clinical trials demonstrated significant clinical benefits of using a CD47-blocking antibody on patients with non-Hodgkin’s lymphoma with no clinically significant safety risks, demonstrating the potential for the restoration of macrophage-mediated PrCR as an effective cancer immunotherapy.32 Efficient activation of PrCR is critical for therapies exploiting the tumoricidal role of macrophages.22 25 33 34 Targeting “don’t eat me” signals on cancer cells remains the most effective approach for inducing PrCR, and CD47 is thus far the only “don’t eat me” signal whose therapeutic potential has been evaluated in a clinical setting for multiple cancers.25 31–34 Blockade of the negative signaling transduced by the CD47–SIRPα axis re-establishes macrophage-mediated immunosurveillance.23–25 70 71 Few studies, however, have addressed therapeutic strategies for the treatment of TNBC by targeting TAMs, which are especially highly enriched in TNBC tumors.
Here, we show that the blockade of CD47 induced PrCR of TNBC cells, but its efficacy needs to be improved to meet the demands for an effective therapy. In addition to this, previous studies indicated that blocking antibodies need to reach a local concentration threshold in order to effectively activate PrCR,72 which is often difficult to achieve, especially in solid tumors. Therefore, agents that can penetrate tumor tissues to directly activate TAMs are attractive candidates for improving the efficacy and feasibility of PrCR induction. Using anti-cancer small molecule compounds with a proven tumor-penetrating ability becomes an attractive strategy for promoting PrCR directly. Here, we demonstrate that the chemotherapeutic agent cabazitaxel can be reprofiled as an immune adjuvant to directly enhance PrCR ability of macrophages. Cabazitaxel was approved by FDA as a chemotherapy for treating prostate cancers and exerts its cytotoxicity by stabilizing tubulin in the microtubule to inhibit cell division.39 Surprisingly, our results demonstrated that the anti-cancer effects of cabazitaxel in TNBC resulted from the activation of macrophages rather than direct cytotoxicity toward TNBC cells. Cabazitaxel induces polarization of macrophages toward a M1 state, as demonstrated by the activation of TLR/NF-kB pathways and enhanced expression of pro-inflammatory cytokines. Interestingly, paclitaxel has been shown to be a TLR4 ligand which directly binds to myeloid differentiation factor 2 (MD-2), a TLR4 accessory protein, and activates downstream signaling.73 Further investigation may be performed to dissect the molecular mechanisms by which cabazitaxel interacts with and activates TLRs. Importantly, M2-polarized macrophages can be converted to be M1-like by using cabazitaxel to acquire a significantly improved phagocytic ability, revealing a plastic and inducible nature of intrinsic phagocytic machinery and its regulator programs. A recent phase II clinical trial demonstrated that cabazitaxel is active (disease control rate of almost of 60%) with a manageable toxicity profile with HER2-negative metastatic breast cancer,74 further suggesting the feasibility of combining cabazitaxel with CD47 blockades in TNBC treatment.
Taken together, our study revealed the significant therapeutic potential of combining cabazitaxel and CD47 blockade, and uncovered a promising strategy for targeting TAMs for the treatment of TNBC, including metastatic TNBC. Next, further analysis at the single-cell level may be performed to characterize TAM subpopulations and identify populations that are sensitive to cabazitaxel, with the most potential for phagocytosis on administration of treatment, which may serve as a predictive parameter for selecting patients with TNBC most likely to benefit from the enhanced PrCR therapy.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
XC, BL and JC contributed equally.
Contributors XC: Project conceptualization, validation, experimental design, investigation, methodology, writing-original draft, writing-review and editing. BL: Project conceptualization, validation, experimental design, investigation, methodology. JC: Project conceptualization, validation, experimental design, investigation, methodology. JD: Experimental design, investigation, methodology. SC: Experimental design, investigation, methodology. EGG: Experimental design, scientific input. BX: Experimental design, scientific input. LT: Experimental design, scientific input. SM: Experimental design, scientific input. MR: Experimental design, scientific input. CQ: Scientific input, supervision. JY: Scientific input, supervision. STR: Scientific input, supervision. YW: Project conceptualization, validation, investigation, methodology, bioinformatics and RNAseq data analysis, writing-original draft, writing-review and editing. MF: Project conceptualization, supervision, funding acquisition, experimental design, investigation, project administration, writing-original draft, writing-review and editing.
Funding We thank the excellent technical support of Core Facilities at City of Hope, including the Analytical Cytometry, Light Microscopy, Animal Resource Center and Small Animal Imaging Core Facilities supported by the National Cancer Institute of the National Institutes of Health under award number P30CA033572. This work was supported by the National Cancer Institute of the National Institutes of Health K99/R00 Pathway to Independence Award K99CA201075/R00CA201075 (to MF) and CA247550 (to JY), the National Institute of Allergy and Infectious Diseases of the National Institutes of Health AI129582 (to JY), the Leukemia & Lymphoma Society 1364-19 (to JY), the Damon Runyon-Dale F. Frey Award for Breakthrough Scientists DFS-22-16 (to MF), the V Foundation for Cancer Research V Scholar Award (V2018-012) (MF), and the startup research funding from City of Hope (to MF). Funds from an anonymous donor helped accelerate these studies.
Competing interests MF declares patent applications pertaining to stimulating TLR/BTK signaling to promote CRT in macrophages assigned to the Stanford University and equity and/or consulting with Forty Seven, Inc.
Patient consent for publication Not required.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available on reasonable request.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.