Article Text

Abstract
Background Anti-programmed death-ligand 1 (αPD-L1) immunotherapy is approved to treat bladder cancer (BC) but is effective in <30% of patients. Interleukin (IL)-2/αIL-2 complexes (IL-2c) that preferentially target IL-2 receptor β (CD122) augment CD8+ antitumor T cells known to improve αPD-L1 efficacy. We hypothesized that the tumor microenvironment, including local immune cells in primary versus metastatic BC, differentially affects immunotherapy responses and that IL-2c effects could differ from, and thus complement αPD-L1.
Methods We studied mechanisms of IL-2c and αPD-L1 efficacy using PD-L1+ mouse BC cell lines MB49 and MBT-2 in orthotopic (bladder) and metastatic (lung) sites.
Results IL-2c reduced orthotopic tumor burden and extended survival in MB49 and MBT-2 BC models, similar to αPD-L1. Using antibody-mediated cell depletions and genetically T cell-deficient mice, we unexpectedly found that CD8+ T cells were not necessary for IL-2c efficacy against tumors in bladder, whereas γδ T cells, not reported to contribute to αPD-L1 efficacy, were indispensable for IL-2c efficacy there. αPD-L1 responsiveness in bladder required conventional T cells as expected, but not γδ T cells, altogether defining distinct mechanisms for IL-2c and αPD-L1 efficacy. γδ T cells did not improve IL-2c treatment of subcutaneously challenged BC or orthotopic (peritoneal) ovarian cancer, consistent with tissue-specific and/or tumor-specific γδ T cell contributions to IL-2c efficacy. IL-2c significantly altered bladder intratumoral γδ T cell content, activation status, and specific γδ T cell subsets with antitumor or protumor effector functions. Neither IL-2c nor αPD-L1 alone treated lung metastatic MB49 or MBT-2 BC, but their combination improved survival in both models. Combination treatment efficacy in lungs required CD8+ T cells but not γδ T cells.
Conclusions Mechanistic insights into differential IL-2c and αPD-L1 treatment and tissue-dependent effects could help develop rational combination treatment strategies to improve treatment efficacy in distinct cancers. These studies also provide insights into γδ T cell contributions to immunotherapy in bladder and engagement of adaptive immunity by IL-2c plus αPD-L1 to treat refractory lung metastases.
- drug evaluation
- preclinical
- immunotherapy
- lymphocyte activation
- tumor microenvironment
- urinary bladder neoplasms
Data availability statement
The datasets used and/or analyzed during the current study are available from the corresponding author upon reasonable request.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
- drug evaluation
- preclinical
- immunotherapy
- lymphocyte activation
- tumor microenvironment
- urinary bladder neoplasms
Background
Bladder cancer (BC) is a common malignancy with more than 80 000 new cases diagnosed in the United States annually.1 Tumors recur after treatment in approximately 50% of high-risk non-muscle invasive BC patients, and advanced or metastatic BC has low 5-year survival.2 3 Immune checkpoint blockade with anti-programmed death-ligand 1 (αPD-L1) and anti-programmed death-1 (αPD-1) antibodies are effective against muscle invasive and metastatic BC4 and the αPD-L1 antibody pembrolizumab was recently approved in non-metastatic, high risk non-muscle invasive BC. However, only 10%–30% of BC patients respond to immune checkpoint blockade and clinically useful biomarkers guiding treatment selection are still poorly defined.5 Immune checkpoint blockade outcomes vary widely in distinct cancers and is rarely curative outside specific settings,6 necessitating development of additional immunotherapeutic strategies.
High-dose interleukin-2 (IL-2) was approved for cancer immunotherapy in 1986, but is clinically limited by severe IL-2-induced toxicity and capacity to generate immunosuppressive T regulatory cells (Tregs).7 Deleterious Treg effects are fueled by IL-2 preferentially binding the high-affinity IL-2 receptor alpha subunit (IL-2Rα, CD25) on Tregs in lieu of the intermediate-affinity beta subunit (IL-2Rβ, CD122) preferentially used by antitumor CD8+ T cells.8
Selective IL-2 receptor stimulation with CD122-directed IL-2/αIL-2 complexes (IL-2c) improves antitumor immunity in many cancer models by augmenting CD8+ T cell effector cell functions while largely avoiding Treg expansion and/or reducing Treg activation and function.9 10 CD122-directed IL-2 therapy using distinct molecules is also promising in human BC trials, including when combined with αPD-1.11 Effects on other CD25+ and CD122+ cells including γδ T cells using targeted IL-2 therapies are unreported to our knowledge and recent reports suggest IL-2c can also act through non-IL-2 receptor expressing cells such as dendritic cells,12 necessitating improved understanding of specific treatment mechanisms in distinct tumor microenvironments.
Here, we define IL-2c and αPD-L1 immunotherapy effects in orthotopic primary (bladder) versus metastatic (lung) BC models. We show that the two agents have distinct mechanisms, including a novel γδ T cell requirement for IL-2c treatment efficacy, in a tissue-dependent manner. These studies provide significant insight into means to improve existing BC immunotherapies and suggest novel treatment strategies by elucidating specific immune contributions to BC treatment efficacy in primary and metastatic BC, including the utility of improving γδ T cell functions.
Methods
Mice
Wild-type C57BL/6J (BL6) and C3H/HeJ (C3H) mice were obtained from The Jackson Laboratory (Bar Harbor, Maine, USA). Genetic knock out mice on the BL6 background purchased from Jackson include: B6.129P2-Tcrbtm1Mom/J (TCRβKO), B6.129P2-Tcrdtm1Mom/J (TCRδKO), and B6.129S7-Rag1tm1Mom/J (RAG1KO). Mice were bred in our animal facility, given ad libitum water and food, and housed under specific pathogen-free conditions. All mice were at least 8 weeks old and age- and sex-matched for each experiment.
Tumor cell lines and cell culture
Mouse MB49 and MBT-2 BC cell lines were provided by R. Svatek. We developed the highly aggressive ID8 aggressive (ID8agg) subline expressing luciferase9 and generated luciferase-expressing MB49 using similar methods.
Cells were passaged <5 times prior to challenge and maintained in 5% fetal bovine serum (FBS)-containing DMEM (Dulbecco’s Modified Eagle Medium, MB49) or RPMI-1640 (Roswell Park Memorial Institute, MBT-2 and ID8agg), supplemented with 1% penicillin/streptomycin, 1% L-glutamate and 1% 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES).
In vivo challenges
Orthotopic MB49 and MBT-2 challenges were 8×104 or 1×106 cells in 50 µL Dulbecco’s Phosphate Buffered Saline (DPBS, Sigma Aldrich) into bladder, respectively, via indwelling urinary catheter in female mice under isoflurane anesthesia, as described.13 Bladder tumor growth was monitored in vivo by tumor bioluminescence and bladder weight at sacrifice. Subcutaneous tumor challenge was 2×105 MB49 cells in 100 µL DPBS in each hind flank of female mice (two tumors/mouse).14 Tumor growth was measured using Vernier calipers and volume was calculated as (length x width2)/2. Orthotopic challenge of ID8agg was 4×106 cells injected intraperitoneally into females.9 Lung tumors were generated via intravenous injection of 7×105 or 2.5×105 MB49 or MBT-2 cells, respectively,15 in 200 µL DPBS into male mice. Survival was defined as moribundity, spontaneous death, tumor volume ≥ 2000 mm3 (subcutaneous challenge), or significant weight loss from baseline (≥15% for bladder and ≥20% for lung tumors). To ensure uniform tumor size across all treatment groups, mice were occasionally excluded on first tumor measurement before randomization, if determined to be an outlier by Grubbs’ test.
In vivo tumor bioluminescence monitoring
Bioluminescent imaging (IVIS Lumina Imaging System; Perkin Elmer; Waltham, Massachusetts, USA) was used for treatment randomization and in vivo tumor monitoring as we reported.9 Briefly, mice were imaged 15 min after intraperitoneal injection of 3 mg PBS-dissolved d-luciferin K+ (Gold Biotechnology; St. Louis, Missouri, USA) with 1 min exposure, small binning, and F/stop=1. Identical regions of interest were drawn over each mouse and average radiance (photons/sec/cm2/sr) was quantified with Living Image software V.3.2.
In vivo treatments
Carrier-free recombinant mouse IL-2 was purchased from Biolegend (San Diego, California, USA). αPD-L1 (clone 10F.9G2), αIL-2 (clone JES6-5H4), and isotype control antibodies (clone LTF-2 rat IgG2b and polyclonal Armenian hamster IgG) were purchased from BioXCell (Lebanon, New Hampshire, USA). IL-2c is 1.5 µg/mouse IL-2 complexed with 7.5 µg/mouse αIL-2 at an optimal 1:2 molar ratio in PBS at 37°C for 15–30 min,16 before intraperitoneal administration in 100 µL DPBS on days 7, 9, 11, and 13 after orthotopic or subcutaneous challenge, or days 8, 10, 12, and 14 after intravenous challenge. 100 µg/mouse αPD-L1 was given intraperitoneally on days 8, 13, and 18 for orthotopic and subcutaneous challenge, or days 9, 14, and 19 for intravenous challenge. Relevant isotype controls were used in all experiments.
In vivo cell depletions
Anti-γδ TCR (clone UC7-13D5) was purchased from Biolegend. αCD8 (clone 2.43) and isotype control antibodies were purchased from BioXCell. Antibodies were given intraperitoneally to deplete relevant cells every 3 days until the end of the tumor treatment regimen. Doses/mouse: 250 µg αCD8 and 75 µg anti-γδ TCR. Confirmation of depletion efficiency is shown in online supplemental figures S1 and S2.
Supplemental material
Carcinogen-induced BC
Wild-type BL6 and TCRδKO male mice were given 0.05% N-Butyl-N-(4-hydroxybutyl)nitrosamine in drinking water for 4 months to induce BC as reported,17 then sacrificed to assess bladder weight and cancer prevalence.
Flow cytometry
Mice were sacrificed by cervical dislocation after induction of deep isoflurane anesthesia. Bladder tumors were dissected and placed in a 60 mm x 15 mm petri dish and minced with a scalpel blade. Cells were incubated for 45–60 min in 3 mL serum-free RPMI-1640 with 0.25 mg/mL DNAse I and 1.65 mg/mL collagenase type IV (both Sigma Aldrich; St. Louis, Missouri, USA) and passed through a 70 µm filter to generate single cell suspensions. Then, 5×106 cells were transferred to 96-well plates and samples with <5×106 cells from the same group were pooled to ensure uniform cell counts for all replicates.
Dead cells were excluded by LIVE/DEAD Fixable Blue Dead Cell Stain Kit for ultraviolet excitation (Thermo Fisher Scientific; Waltham, Massachusetts, USA). Non-specific labeling was preblocked with anti-CD16/32 at 1:100 dilution (clone 93, Biolegend). Cells were stained for surface antigens by incubating at 4°C for 30–45 min with antibodies from Biolegend: αCD3 (17A2), αCD11b (M1/70), αPD-1 (29F.1A12), αCD45 (30-F11), αCD8 (53–6.7), αDNAM-1 (10E5), and αCD27 (LG.3A10); Thermo: αNKG2A (16A11) and αTCR gamma/delta (GL-3); and BD (San Jose, California, USA): αLy-6G (1A8), αCD4 (GK1.5), αCD69 (H1.2F3), αCD122 (TM-β1), αVγ2 (UL3-10A6), αVγ1.1/Cr4 (2.11), αVδ4 (GL2), αLy-6C (AL-21) and αCD25 (PC61), all at manufacturer recommended dilutions. Of note, while the Garman γ chain nomenclature is used by the antibody manufacturers, the Heilig and Tonegawa nomenclature is used throughout the text.18
For intracellular staining, cells were fixed and permeabilized with a FoxP3/transcription factor buffer kit (eBioscience; San Diego, California, USA) according to manufacturer instructions, and incubated at 4°C for 45 min. Cells were then stimulated with Cell Activator Cocktail (Biolegend) containing phorbol 12‐myristate 13‐acetate, ionomycin, and brefeldin A at 2 µL cocktail/mL CR10 medium (RPMI-1640 with 10% FBS, L-glutamine, sodium pyruvate, non-essential amino acids, penicillin/streptomycin and HEPES buffer) for 6 hours in a 37°C incubator. After stimulation, intracellular staining was performed by incubating cells at 4°C for 30–45 min with antibodies from Biolegend: αTumor necrosis factor (TNF)-α (MP6-XT22), αEomes (W17001A), and αIL-17A (TC11-18H10.1); Thermo: αGranzyme B (NGZB), αFoxP3 (FJK-16s), and αT-bet (4B10) and BD: αInterferon (IFN)-γ (XMG1.2), all at manufacturer recommended dilutions in 1X FoxP3 permeabilization buffer. Surface and intracellular staining also included Brilliant Stain Buffer Plus (BD) at the manufacturer recommended dilution. Absolute cell numbers were determined by multiplying the cell ratio per live, singlet cells in each flow sample by total cell numbers in the sample. Flow data were acquired on a Cytek Aurora flow cytometer (Cytek Biosciences; Freemont, California, USA) and analyzed using FlowJo software (BD) V.10.7.1.
Statistical and data analyses
Data were analyzed and graphed with GraphPad Prism V.9.0.0. Data with error bars represent the mean±SEM. To compare two means, we used an unpaired t-test. Three or more means were compared with one-way analysis of variance (ANOVA) and post hoc Sidak’s test. Tumor growth curves were compared by two-way ANOVA, analyzed for overall treatment effect, followed by post hoc Sidak’s test of discrete time points. Log-rank test was used to compare Kaplan-Maier survival curves. Tumor prevalence was compared using a two-tailed Mann-Whitney U test. Occasionally, data sets with suspected outliers were identified by Grubbs’ test (used only once for a given data set) and removed from analysis. For all analyses, significance was based on a multiplicity corrected α of 0.05.
Results
Single agent IL-2c or αPD-L1 effectively treats orthotopic BC
We studied BC immunotherapy using the established orthotopic mouse BC cell lines MB49 and MBT-2 on BL6 and C3H genetic backgrounds, respectively.19 Single agent IL-2c reduced tumor growth and bladder weight (figure 1A,B) and improved survival (figure 1C,D) after orthotopic challenge with either MB49 or MBT-2. As both tumors express PD-L1,20 21 we tested αPD-L1 for comparison, which also significantly reduced tumor growth in vivo (figure 1A), reduced bladder weight (figure 1B) and improved survival (figure 1C,D) in both models.
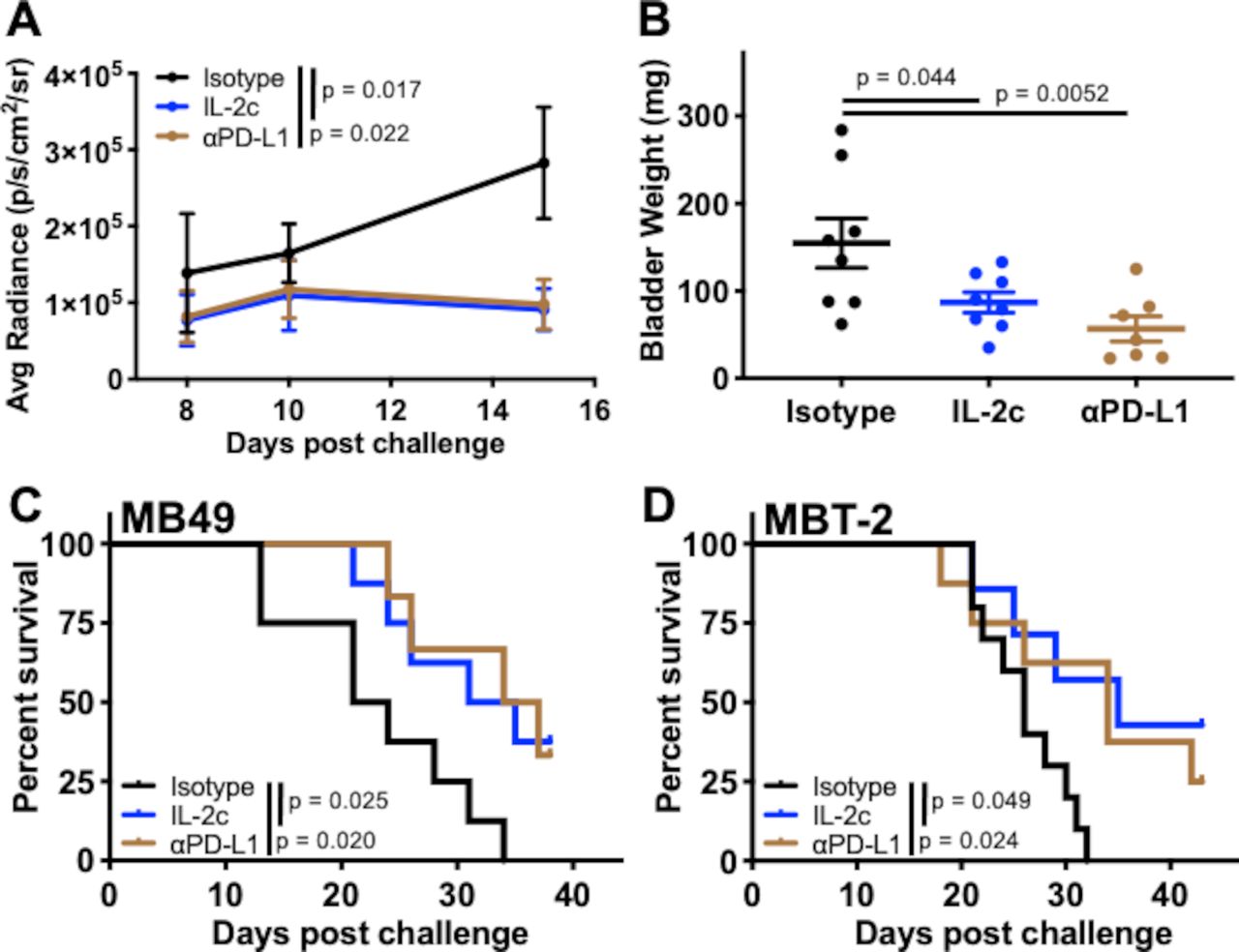
Single agent IL-2c or αPD-L1 effectively treats orthotopic bladder cancer. Wild-type BL6 (A-C) or C3H (D) female mice were challenged orthotopically with (A–C) 8×104 MB49 or (D) 1×106 MBT-2 cells and treated with IL-2c on days 7, 9, 11 and 13, 100 µg αPD-L1 on days 8, 13 and 18, or isotype control (100 µg/mouse). (A) Tumor bioluminescence corresponding with pre-treatment, mid-treatment, and post-treatment timepoints. N=8/group p value, two-way ANOVA of day 15 signal. (B) Mice sacrificed on day 21. N=7–8/group, p value, one-way ANOVA. Mouse survival in orthotopic (C) MB49 (N=6–8/group) or (D) MBT-2 (N=7–10/group) challenge, p value, log-rank. ANOVA, analysis of variance; IL-2, interleukin-2; PD-L1, programmed death-ligand 1.
IL-2c treats orthotopic BC independent of CD8+ T cells
Improving CD8+ T cell antitumor immunity is a major mechanism of many cancer immunotherapies,22 including the IL-2c used here.23 To assess the role of CD8+ T cells in IL-2c treatment of orthotopic BC, we administered an anti-CD8 antibody concurrently with IL-2c, which thoroughly depleted bladder CD8+ T cells (online supplemental figure S1). Surprisingly though, tumor growth (figure 2A) and survival (figure 2B) of mice receiving IL-2c ± αCD8 was indistinguishable. Flow cytometric analysis of MB49 bladder tumors confirmed CD8+ T cell content and showed that IL-2c did not significantly improve CD8+ T cell frequency, number, activation status or IFN-γ production over control treatment (figure 2C–G). In support, IL-2c significantly improved survival in orthotopic MB49 challenge into TCRβKO mice lacking all conventional T cells (figure 2H). By striking contrast, but as expected based on known CD8+ T cell requirements for αPD-L1 efficacy,24 αPD-L1 was ineffective in treating orthotopic BC in TCRβKO mice (figure 2I). These data suggest that CD8+ T cells are not essential for IL-2c-mediated treatment of orthotopic BC, contrasting with the critical role of CD8+ T cells in the efficacy of αPD-L1 and other cancer immunotherapies.25

IL-2c treats orthotopic bladder cancer in the absence of CD8+ T cells. Wild-type (A–G) or TCRβKO (H, I) female mice were challenged orthotopically with 8×104 MB49 cells and treated with (A–H) IL-2c on days 7, 9, 11, and 13 ± (A, B) 250 µg αCD8 on days 6, 9, 12, and 15, (I) 100 µg αPD-L1 on days 8, 13, and 18, or (C-I) isotype control. (A) Tumor bioluminescence corresponding with pre-treatment, mid-treatment and post-treatment timepoints. N=9–10/group, p value, two-way ANOVA of day 14 signal. (B) Mouse survival. N=9–10/group, p value, log-rank. Flow cytometric analysis of CD8+ T cell frequency (C), number (D), activation status (E), and IFN-γ production (F, G). N=6–8/group, p value, unpaired t-test. (H, I) Mouse survival. N=6–8/group, p value, log-rank. ANOVA, analysis of variance; IFN, interferon; IL-2, interleukin-2; PD-L1, programmed death-ligand 1; MFI, mean fluorescence intensity.
γδ T cells are essential for IL-2c treatment efficacy in orthotopic BC
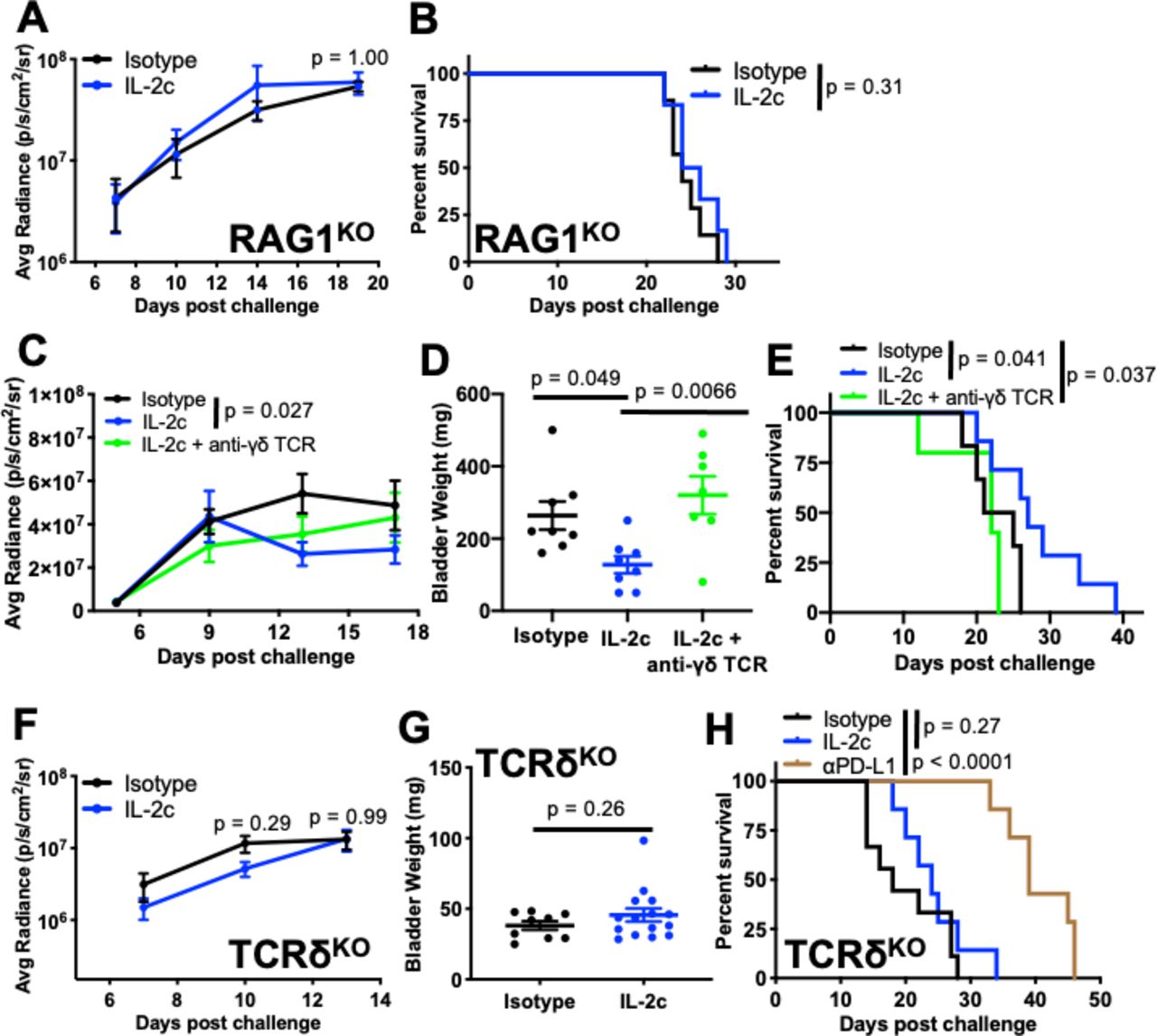
IL-2c is well known to activate protective, antitumor natural killer (NK) cell immunity.10 However, IL-2c failed to treat orthotopic MB49 in RAG1KO mice (figure 3A,B) that have functional NK cells but lack γδ and conventional T cells, compared with IL-2c efficacy in TCRβKO mice (figure 2H) that have functional γδ T and NK cells but lack conventional T cells. These data suggest that NK cells are dispensable for IL-2c efficacy against orthotopic MB49, or that NK cells also require γδ T cells, conventional T cells, or RAG26 to mediate IL-2c treatment efficacy in this setting.
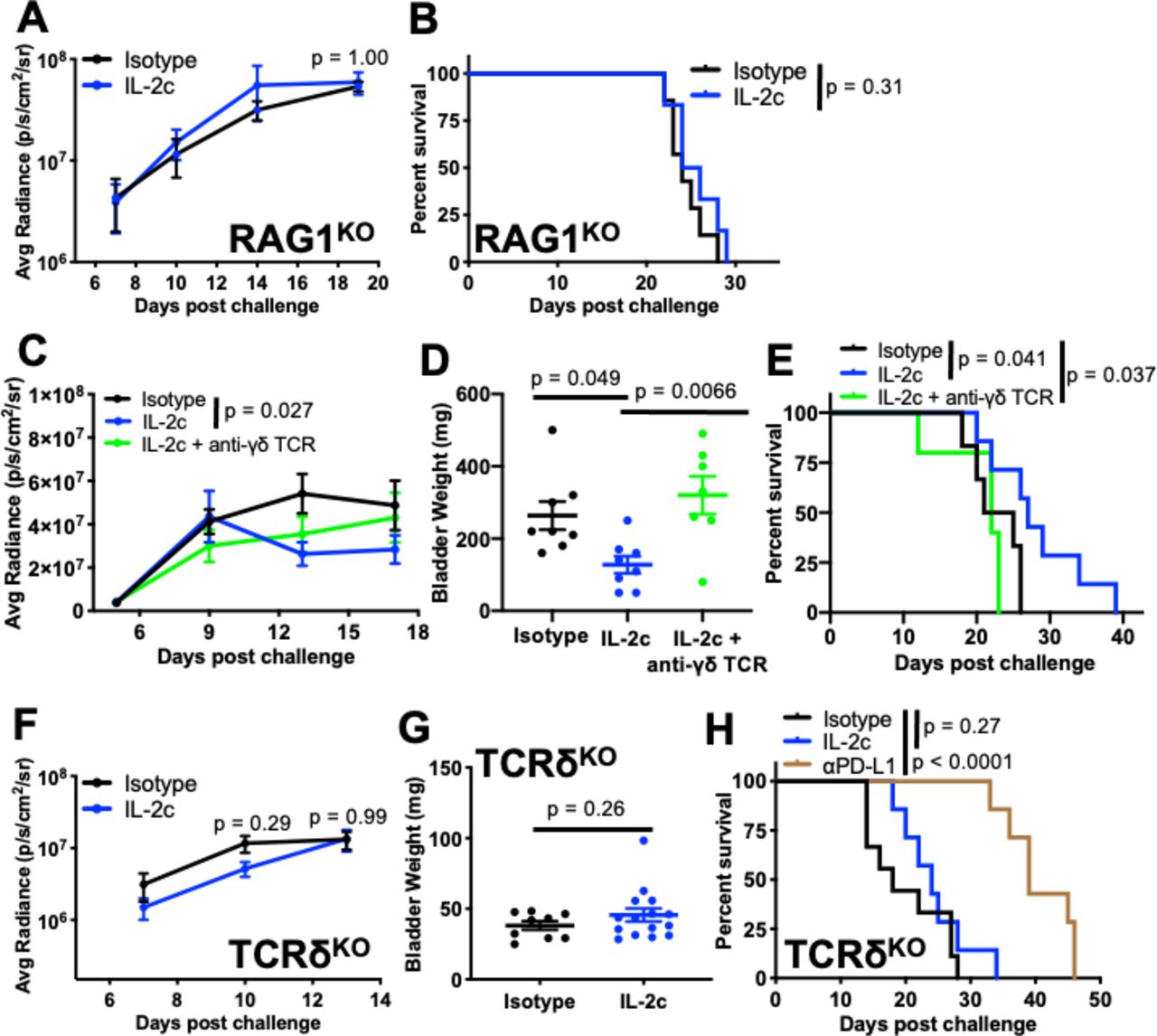
γδ T cells are essential for IL-2c treatment efficacy in orthotopic bladder cancer. RAG1KO (A,B), wild-type (C-E), or TCRδKO (F-H) female mice were challenged orthotopically with 8×104 MB49 cells and treated with IL-2c on days 7, 9, 11, and 13 ± (C-E) 75 µg anti-γδ TCR on days 6, 9, 12, 15, and 18 or isotype control. (A) Tumor bioluminescence corresponding with pre-treatment, mid-treatment, and post-treatment timepoints. N=6–7/group, p value, two-way ANOVA of day 19 signal. (B) Mouse survival. N=6–7/group, p value, log-rank. (C) Tumor bioluminescence corresponding with pre-treatment, mid-treatment, and post-treatment timepoints. N=7–8/group, p value, two-way ANOVA of day 13 signal (Isotype vs IL-2c). (D) Mice from (C) sacrificed on day 20. N=7–8/group, p value, one-way ANOVA. (E) Mouse survival. N=5–7/group, p value, log-rank. (F) Tumor bioluminescence on day 7, 10, and 13 corresponding with pre-treatment, mid-treatment, and post-treatment timepoints. N=9–15/group, p value, two-way ANOVA of day 10 and 13 signal. (G) Mice from (F) sacrificed on day 14. N=9–15/group, p value, unpaired t-test. (H) Mouse survival with isotype control, IL-2c (as above), or 100 µg αPD-L1 on days 8, 13, and 18. N=7–9/group, p value, log-rank. ANOVA, analysis of variance; IL-2, interleukin-2; PD-L1, programmed death-ligand 1; TCR, T cell receptor.
We then studied γδ T cells as they express the IL-2c target (CD122)27 and can kill orthotopic BC.28 We administered IL-2c plus a γδ T cell depleting antibody to wild-type mice bearing orthotopic MB49 BC (online supplemental figure S2A), which completely abrogated IL-2c treatment efficacy as measured by bioluminescence (Fig. 3C), bladder weight (figure 3D), and mouse survival (figure 3E). In confirmation, we found difference in tumor growth (figure 3F), bladder weight (figure 3G), or mouse survival (figure 3H) in control versus IL-2c-treated TCRδKO mice lacking γδ T cells. By contrast, αPD-L1 efficacy against orthotopic MB49 challenge was unaffected in TCRδKO mice (figure 3H). Survival of isotype-treated wild-type and TCRδKO mice challenged with MB49 was comparable (figure 3E vs 3H) and we found no difference in BC size or prevalence between wild-type and TCRδKO mice receiving the carcinogen N-Butyl-N-(4-hydroxybutyl)nitrosamine to induce BC (online supplemental figure S2B,C), suggesting that γδ T cell-mediated BC immunity is not critical for endogenous BC immune surveillance but can be mobilized by IL-2c for effective anti-BC immunotherapy. These data firmly establish that IL-2c and αPD-L1 have distinct mechanisms of action against orthotopic BC, which could be exploited in distinct scenarios, and suggest that these agents could cooperate to treat BC.
IL-2c treatment effects on γδ T cells are tissue-specific
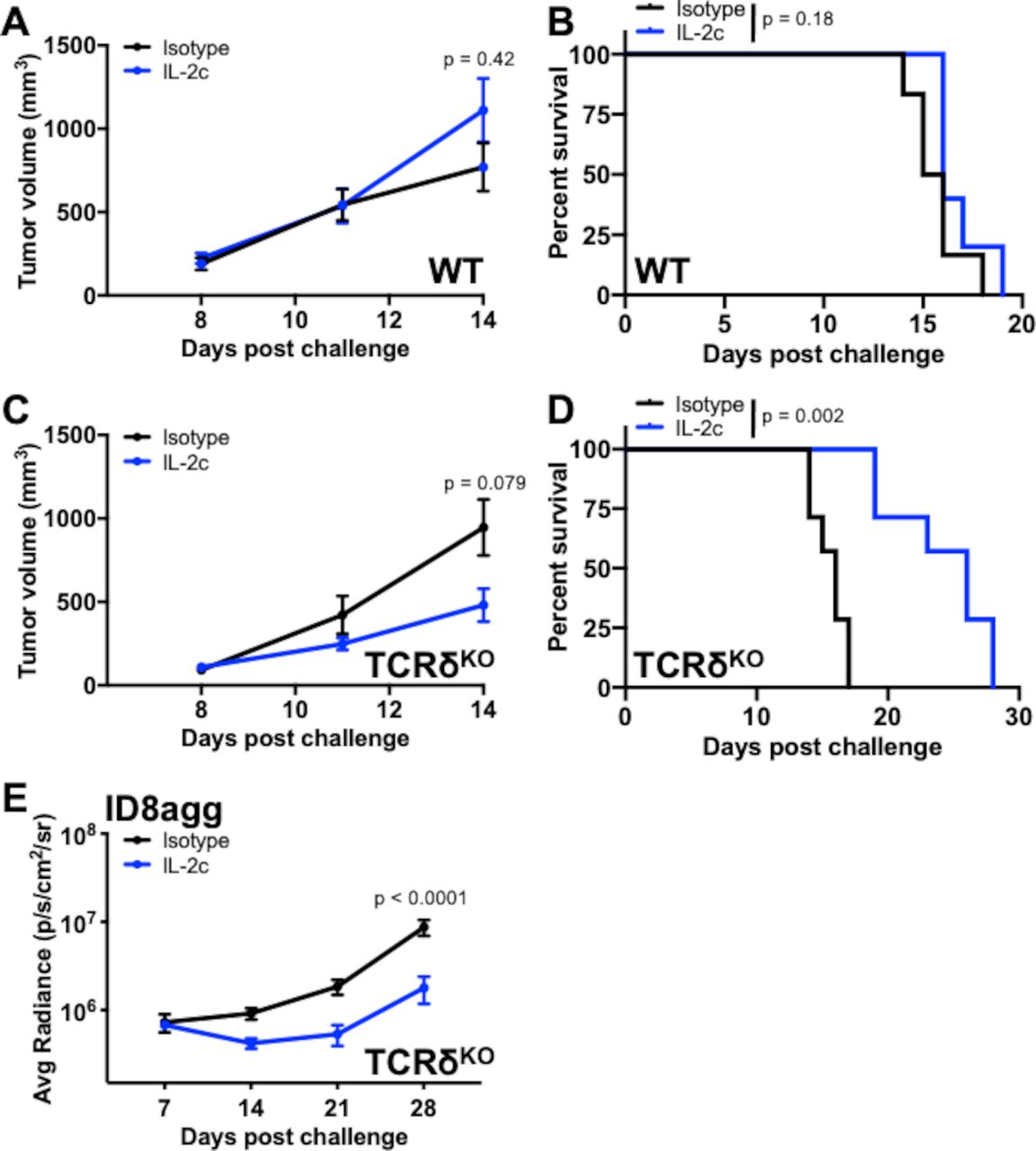
The tumor microenvironment plays an important role in BC pathogenesis and immunotherapy responsiveness29–31, leading us to explore IL-2c effects outside the bladder tumor microenvironment. We challenged MB49 cells subcutaneously into wild-type mice and found that IL-2c did not slow tumor growth (figure 4A) or extend survival (figure 4B), in direct contrast to our findings in orthotopic MB49 challenge (figure 1). As γδ T cells have tissue-specific properties,32 we challenged TCRδKO mice with subcutaneous MB49, treated with IL-2c or isotype control and observed a trend toward reduced tumor growth (figure 4C) and a dramatic improvement in survival with IL-2c versus control-treated mice (figure 4D). These data suggest differences in skin versus bladder γδ T cells in immunotherapy treatment mechanisms, including that γδ T cells could impede IL-2c efficacy against subcutaneous MB49, a difference not based on tumor type. To test an alternative orthotopic tumor and anatomic compartment, we challenged TCRδKO mice intraperitoneally with ID8agg mouse ovarian carcinoma cells. ID8agg tumors in TCRδKO mice were also sensitive to IL-2c (figure 4E), as we previously reported in wild-type mice,9 consistent with tissue- and/or tumor-specific γδ T cell effects of IL-2c.

IL-2c treatment effects on γδ T cells are tissue-specific. WT (A, B) or TCRδKO (C, D) female mice were challenged subcutaneously with 2×105 MB49 cells/flank (two tumors/mouse) and treated with IL-2c on days 8, 10, 12, and 14, or isotype control (100 µg/mouse). (A) Mouse tumor volume. N=5–6 mice and 10–12 tumors/group, p value, two-way ANOVA of day 14 tumor volume. (B) Mouse survival of (A). N=5–6 mice/group, p value, log-rank. (C) Mouse tumor volume. N=7 mice and 14 tumors/group, p value, two-way ANOVA of day 14 tumor volume. (D) Mouse survival of (C). N=7 mice/group, p value, log-rank. (E) Bioluminescent signal of female mice orthotopically (intraperitoneally) challenged with 4×106 ID8agg cells and treated with isotype control or IL-2c on days 7, 9, 11, and 13. N=11 mice/group, p value, two-way ANOVA of day 28 signal. ANOVA, analysis of variance; IL-2, interleukin-2; PD-L1, programmed death-ligand 1; WT, wild-type.
IL-2c enhances antitumor Tγδ1 effects and reduces protumor Tγδ17 effects in MB49 bladder tumors
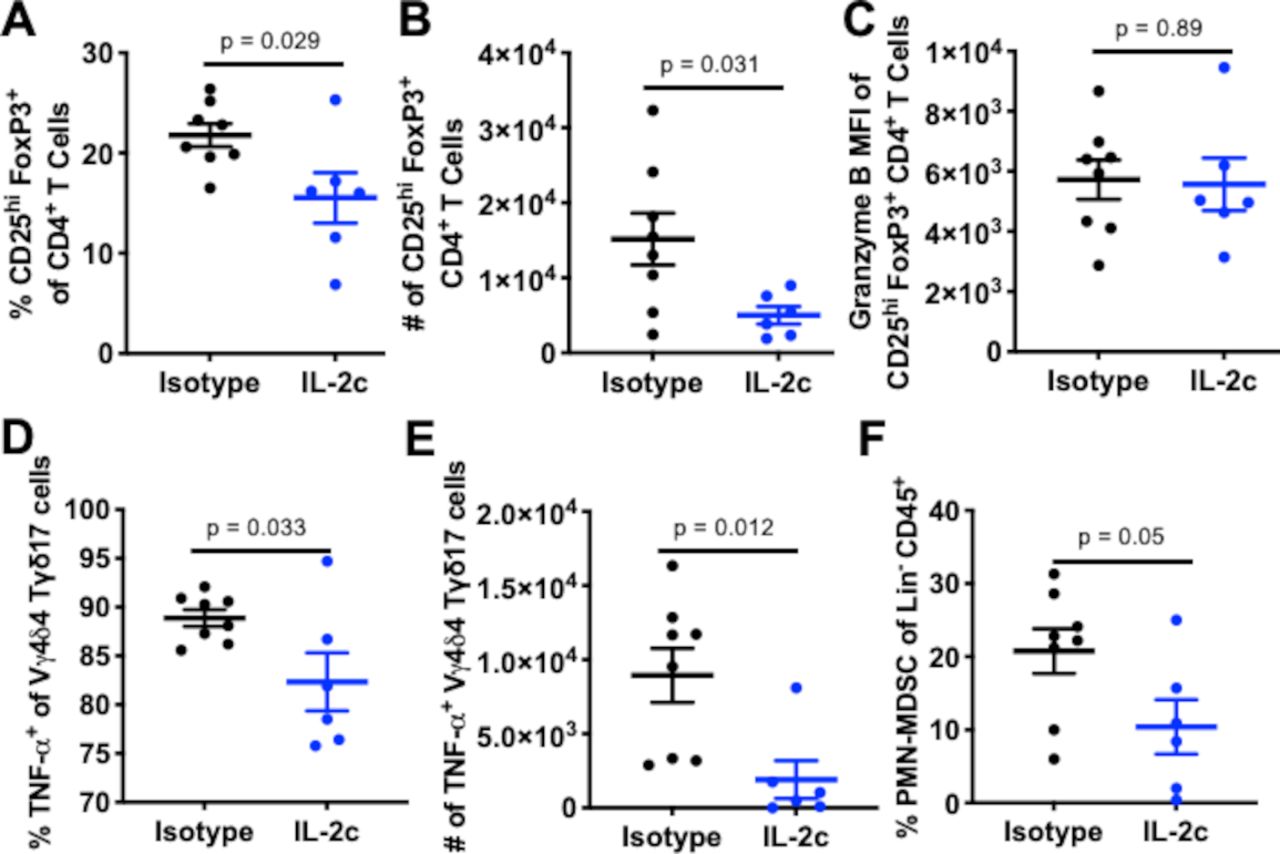
Murine γδ T cell subsets can be functionally divided into Tγδ1 and Tγδ17 subsets that produce IFN-γ or IL-17A (herein IL-17), respectively.33 Tγδ1 cells express CD27 and mediate antitumor immunity in many mouse tumor models, whereas a pro-tumorigenic role is attributed to Tγδ17 cells, which lack CD27 expression.34 Tγδ1 cells also express CD122 (IL-2Rβ) whereas Tγδ17 cells express CD25 (IL-2Rα), resulting in preferential IL-2 binding and downstream signaling by Tγδ17 cells versus Tγδ1 cells.35 We observed increased total intratumoral γδ T cell prevalence (figure 5A), CD27+ T-bet+ γδ T cells (figure 5B), and a significant increase in CD27+ Vγ1+ cells with increased DNAM-1 expression (figure 5C), consistent with an activated Tγδ1 immune phenotype. We also found increased numbers of IL-17- γδ T cells expressing CD69 and Eomes (figure 5D) that had increased DNAM-1 and reduced expression of the inhibitory receptor NKG2A (figure 5E,F). Collectively, these populations represent functionally active Tγδ1 cells based on their CD27, Vγ1, Eomes, CD69, and DNAM-1 expression with lack of IL-17 production.36

IL-2c enhances antitumor Tγδ1 and reduces protumor Tγδ17 effects in bladder. Wild-type female mice were challenged orthotopically with 8×104 MB49, treated with IL-2c on days 7, 9, 11, and 13 or isotype control and sacrificed on day 16. Flow cytometric analysis of (A–F) Tγδ1 and (G, H) Tγδ17 populations. (A–H) N=6–8/group, p value, unpaired t-test. IL-2, interleukin-2; MFI, mean fluorescence intensity.
The loss of IL-2c efficacy with γδ T cell depletion or genetic knock out is likely due to loss of beneficial Tγδ1 effects. However, we also considered that IL-2c could reduce deleterious Tγδ17 cells, defined as CD27- CD25+ IL-17+ γδ T cells.37 Prior studies of CD27- Tγδ17 cells have also shown that compared with CD27+ γδ T cells, high-density CD25+ on Tγδ17 leads to more robust STAT5 signaling in response to IL-235 suggesting that this γδ T cell population could be sensitive to loss of CD25 signals from IL-2c. IL-2c reduced the prevalence of Tγδ17 cells in orthotopic BC (figure 5G), which are known to segregate functionally into Vγ4 or Vγ6 populations in mice.38 We found Vγ4+ cells to be the predominant Tγδ17 subtype in orthotopic MB49 BC (online supplemental figure S3A). Vγ4 chains commonly pair with Vδ434 and in IL-2c treated tumors, we found a significant reduction in Vγ4δ4 prevalence (figure 5H) and a trend to decreased numbers (online supplemental figure S3B). This Tγδ17 subset has known pro-tumorigenic properties39 and expressed CD25 in MB49 challenge.
IL-2c reduces bladder Tregs without inducing fragility and alters myeloid-derived suppressor cell content
To assess other candidate immune-suppressive cells that could be altered by IL-2c, we undertook initial studies of Tregs and myeloid-derived suppressor cells (MDSCs), both of which are detrimental in human BC40 41 and we previously reported that Tregs affect MB49 BC growth in bladder more than lung.42 In our studies of IL-2c mechanisms in ovarian cancer, we found that IL-2c induces fragile Tregs with reduced suppressive function which are identified by maintained FoxP3 expression, and upregulated IFN-γ, T-bet, and PD-1.9 However, in wild-type mice bearing orthotopic MB49, IL-2c reduced Treg frequency (figure 6A) and number (figure 6B) but did not affect functional molecules (figure 6C) or induce a fragile phenotype (online supplemental figure S4A–D). Similar to γδ T cell effects, these data demonstrate a tissue- and/or tumor-specific IL-2c effect on Tregs and suggest that reduced Treg effects could contribute to IL-2c treatment efficacy.

IL-2c reduces bladder Tregs without inducing fragility and alters bladder PMN-MDSC content. Wild-type female mice were challenged orthotopically with 8×104 MB49 cells and treated with IL-2c on days 7, 9, 11, and 13 or isotype control (100 µg/mouse) and sacrificed on day 16. (A–C) Flow cytometric analysis of Treg frequency (A), number (B) and function (C). (D, E) Flow cytometric analysis of TNF-α-producing Vγ4δ4 cell frequency (D) and number (E). (F) Flow cytometric analysis of polymorphonuclear myeloid derived suppressor cells (PMN-MDSC) frequency, defined as CD11b+ Ly6G+ Ly6Clo cells. (A–F) N=6–8/group, p value, unpaired t-test. IL-2, interleukin-2; Lin, lineage; PMN-MDSC, polymorphonuclear myeloid-derived suppressor cell; TNF, tumor necrosis factor; MFI, mean fluorescence intensity.
Tγδ17 cell production of IL-17 and TNF-α can recruit and promote polymorphonuclear MDSC (PMN-MDSC) expansion in human colorectal carcinoma.43 Interestingly, we found that IL-2c reduced the frequency (figure 6D) and number (figure 6E) of bladder TNF-α+ Vγ4δ4 cells, with a concomitant reduction in PMN-MDSC frequency (figure 6F). Intratumoral monocytic MDSC (M-MDSC) content was unchanged by IL-2c (online supplemental figure S4E), suggesting that IL-2c treatment could include a specific reduction of PMN-MDSCs, possibly via IL-17 or TNF-α effects that could include IL-2c-induced TNF-α production by innate lymphoid cells recently described in a subcutaneous melanoma model,12 an area requiring further investigation.
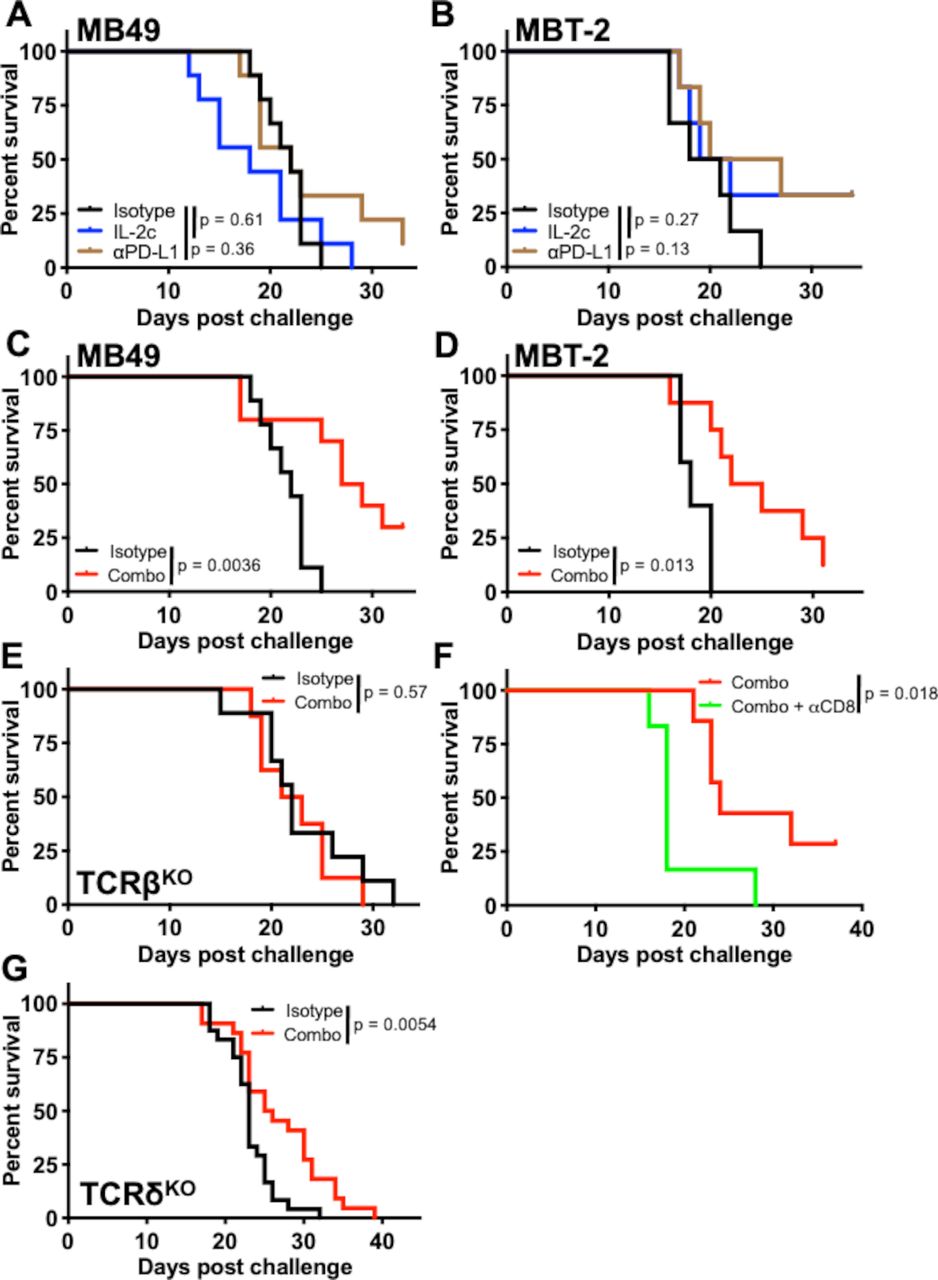
Neither IL-2c nor αPD-L1 alone treat BC lung metastases, but their combination is effective
We next generated lung metastases that are relevant to BC44 by intravenous tumor cell injection. Strikingly, but consistent with tissue-specific effects, neither single agent IL-2c nor αPD-L1 was effective against lung metastatic MB49 or MBT-2 (figure 7A,B). By contrast, their combination provided a significant survival benefit in both models (figure 7C,D).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
IL-2c and αPD-L1 combine to treat bladder cancer lung metastases. Male mice were challenged intravenously with 7×105 MB49 (A,C,E-G) or 2.5×105 MBT-2 (B,D) cells and treated with (A,B) IL-2c on days 8, 10, 12, and 14, 100 µg αPD-L1 on days 9, 14, and 19, (C–H) a combination of both, or isotype controls. (A) Wild-type mouse survival in intravenous MB49 challenge. N=9–10 mice/group, p value, log-rank. (B) Wild-type mouse survival in intravenous MBT-2 challenge. N=6 mice/group, p value, log-rank. (C) Wild-type mouse survival in intravenous MB49 challenge. N=10 mice/group, p value, log-rank. (D) Wild-type mouse survival in intravenous MBT-2 challenge. N=5–8 mice/group, p value, log-rank. (E) TCRβKO mouse survival in intravenous MB49 challenge. N=8–9 mice/group, p value, log-rank. (F) Wild-type mouse survival in intravenous MB49 challenge treated with IL-2c + αPD-L1 (as above) ± 250 µg αCD8 on days 7, 10, 13, 16, and 19. N=6–7 mice/group, p value, log-rank. (G) TCRδKO mouse survival in intravenous MB49 challenge. Combined results of 2 independent experiments. N=11–13 mice/group in each experiment. p value, log-rank. IL-2, interleukin-2; PD-L1, programmed death-ligand 1.
We investigated if treatment mechanisms for successful immunotherapy treatment of BC lung tumors were similar to bladder. Combination treatment of lung metastatic MB49 in TCRβKO mice was ineffective (figure 7E), suggesting a role for conventional T cells against lung metastasis in this model. TCRβKO mice had a similar median survival as wild-type mice (figure 7E vs figure 7C), consistent with our prior report that Tregs impact MB49 BC in bladder more than lung42 and suggesting that CD8+ T cells are responsible for combination treatment efficacy in lung metastatic BC. Accordingly, combination therapy plus a CD8+ T cell depleting antibody in mice bearing lung metastatic MB49, abrogated the combination therapy survival benefit (figure 7F). Studies of γδ T cells in lung metastases are limited.45 Because of γδ T cell requirements for IL-2c efficacy in bladder and differing effects in skin (figures 3 and 4), we evaluated γδ T cell effects in lung metastatic MB49 and found that IL-2c plus αPD-L1 still improved survival in TCRδKO mice (figure 7G), with isotype controls having a similar median survival as wild-type mice (figure 7G vs figure 7C). Collectively, these data establish tissue-specific effects of individual immunotherapy agents and demonstrate that effective treatment strategies can engage different immune subsets in bladder and lungs to treat BC tumors.
Discussion
BC is highly immunogenic and boasts United States Food and Drug Administration approvals for five immune checkpoint blockade agents and bacillus Calmette-Guérin (BCG) as distinct immunotherapy agents to treat non-muscle invasive, muscle invasive, and metastatic disease.46 However, typically under 30% of BC patients respond to immune checkpoint blockade and BCG suffers from high relapse rates, necessitating further insights to improve these therapies, as well as look for alternative or complementary treatment strategies.47 48
Because the tumor microenvironment is an important determinant of BC immunotherapy success,29–31 we used orthotopic mouse BC models to assess immune responses to immune checkpoint blockade with αPD-L1 and showed effective immune checkpoint blockade treatment in two orthotopic BC models as a baseline for comparison versus IL-2 complexes made by incubating recombinant IL-2 with the CD122-directing αIL-2 antibody clone JES5H416 (IL-2c). We previously described distinct IL-2c treatment mechanisms in mouse melanoma and ovarian cancer models9 and here we found IL-2c to be efficacious for orthotopic BC via mechanisms distinct from those tumors and strikingly distinct from αPD-L1 immune checkpoint blockade, including previously unreported γδ T cell requirements.
Most studies of the bladder immune landscape in early stage BC have been following BCG immunotherapy.49 CD122 (to which IL-2c directs IL-2) is expressed on conventional T (CD4+ and CD8+), NK, and γδ T cells, all of which are important in localized human BC treatment with BCG.50
We explored immune cell contributions to IL-2c and αPD-L1 treatment success in mouse BC models and found that surprisingly, conventional T cells, specifically CD8+ T cells, were dispensable for IL-2c treatment effects, despite their necessity for αPD-L1 efficacy here, as also shown by others.24 As CD8+ T cell content in BC is a favorable prognostic indicator,25 the relative independence of IL-2c efficacy on CD8+ T cells in orthotopic BC and reliance on γδ T cells that do not affect αPD-L1 efficacy suggests that these agents could combine clinically for improved treatment efficacy. In support, we found that neither IL-2c nor αPD-L1 alone was effective in lung metastatic BC, whereas their combination was effective.
IL-2c requirements for γδ T cells were noted in orthotopic but not subcutaneous BC, and were dispensable for treating orthotopic ovarian cancer in the peritoneal cavity. It appears that γδ T cells could be detrimental to IL-2c treatment efficacy of subcutaneous MB49. Thus, γδ T cell effects in IL-2c treatment are tissue-selective, consistent with known γδ T cell biology and could also be tumor specific, which requires additional investigations. In this regard, if specific scenarios are identified in which γδ T cells impede IL-2c treatment efficacy, its efficacy could be improved by depleting γδ T cells or their functions in specific anatomic compartments, outcomes not achievable in humans with currently available agents.
In bladder, IL-2c promoted frequency, numbers, and activation of beneficial Tγδ1 cells and reduced frequency of detrimental Tγδ17 cells. Tγδ1 cells express CD122 whereas Tγδ17 cells express CD25, which likely contributes to outcomes using this CD122-selective agent. These studies also include, to our knowledge, the first report of specific γδ T cell subsets in murine bladder, including what appears to be an antitumor Vγ1 and protumor Vγ4 (specifically Vγ4δ4) γδ T cell subset. Unlike our prior studies in ovarian cancer and melanoma,9 IL-2c did not induce a fragile Treg phenotype, further consistent with tissue-specific or tumor-specific IL-2c effects but specific Treg contributions to IL-2c treatment efficacy in BC remain to be fully defined.
Granulocytic but not M-MDSC number is elevated in the circulation of human BC patients51 and PMN-MDSCs predominate over M-MDSCs in BC patient peripheral blood52 and bladder tissue.53 We found that IL-2c reduced orthotopic PMN-MDSC frequency, which could be from an IL-17/TNF-α signaling effect based on prior reports,43 and could specifically include TNF-α from innate lymphoid cells, a novel IL-2c effect just reported.12 MDSC-mediated immunosuppression of BC immunotherapy responses merit additional investigation and may be linked to other beneficial IL-2c effects. A Tγδ17-MDSC immunosuppressive axis had been previously identified in human colorectal carcinoma,43 which could be disrupted by IL-2c treatment based on our data. Further, RAG1KO mice have functional innate lymphoid cells that contributed to IL-2c efficacy against subcutaneous B16 melanoma challenge,12 whereas IL-2c did not treat subcutaneous MB49 in these studies, further supporting tissue- and/or tumor-specific IL-2c effects requiring additional investigations.
Of great interest was the lack of conventional T cell activation and participation in IL-2c efficacy in orthotopic BC, despite αPD-L1 activation of these cells and their requirement for αPD-L1 treatment efficacy, as well as contributions to other cancers as described.25 Potential explanations include specific attributes of CD8+ T cells in bladder, contributions from specific immunosuppressive factors such as Tregs and MDSCs, and specialized dendritic cell effects as have been noted with IL-2c in subcutaneous tumors,9 areas requiring further study. Identification of such factors could be exploited to improve IL-2c, αPD-L1, and other immunotherapies.
Optimal initial immunotherapy of metastatic BC should reduce tumor burden in both primary and metastatic sites.54 Evaluating the ability of BC immunotherapies to treat metastatic tumors or reduce metastases from the primary site requires a detailed understanding of tissue-specific immunity in the local BC microenvironment. In this regard, neither IL-2c nor αPD-L1 alone effectively treated BC lung metastases, whereas their combination was highly effective and required CD8+ T cells. This combinatorial efficacy could arise from additive effects of individually ineffective single agents or from contributions from distinct mechanisms of these agents in lung as we found in bladder treatments, requiring further investigation. Our metastatic models relied on intravenous tumor challenge, omitting several critical steps in spontaneous metastasis. In mice surviving treatment of orthotopic BC (single agent of combination) we occasionally found macroscopic metastases, including focal lung metastases, suggesting that additional strategies to prevent metastatic tumor spread will be required for optimal treatment efficacy. As metastatic BC is clinically challenging to treat, additional studies of innate and adaptive immune contributions to immunotherapy of metastatic BC in lung and other metastatic sites are required.
Collectively, these data show important new effects of CD122-targeted BC immunotherapy in orthotopic and metastatic disease that complement known mechanisms for existing BC immunotherapies. IL-2c-mediated promotion of antitumor immunity makes it a promising candidate for BC trials and for combinations with αPD-L1, where we showed improved treatment efficacy in melanoma and ovarian cancer models, with induction of durable, protective immunity.9 Whether other directed IL-2 molecules now in cancer clinical trials have similar γδ T cell activating properties also merits investigation.
Supplemental material
Data availability statement
The datasets used and/or analyzed during the current study are available from the corresponding author upon reasonable request.
Ethics statements
Ethics approval
All animal studies were approved by the UT Health San Antonio Institutional Animal Care and Use Committee and each experiment was conducted in accordance with the standards required by the UT Health San Antonio Department of Laboratory Animal Resources.
Acknowledgments
Graphical abstract created using BioRender.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Twitter @SvatekRob
Contributors RMR: conceptualization, data curation, formal analysis, funding acquisition, investigation, methodology, validation, visualization, writing—original draft, writing—review and editing. YD: data curation, formal analysis, investigation, methodology, visualization and writing—review and editing. DZ: conceptualization, investigation, methodology. NJ: investigation, methodology, resources. NM: data curation, investigation, methodology, resources. KW: investigation, methodology, resources. HBG: investigation, methodology, validation. ASP: investigation, resources. AK: investigation. CZ: investigation, validation. MG: investigation. AVRK: investigation and methodology. OB: conceptualization, writing - review and editing. JRC-G: conceptualization, writing—review and editing. RS: conceptualization, data curation, formal analysis, funding acquisition, resources, supervision, and writing—review and editing. TC: conceptualization, data curation, formal analysis, funding acquisition, methodology, project administration, resources, software, supervision, writing—original draft, writing—review and editing.
Funding R. Reyes (NIH T32GM113896, NIH/NCATS TL1 TR002647, NIA T32 AG 021890), YD (CPRIT Research Training Award (RP 170345) and Ovarian Cancer Research Alliance Ann and Sol Schreiber Mentored Investigator Award), NM (CPRIT Research Training Award (RP170345)), MG (NIH T32GM113896, NIH/NCATS TL1 TR002647), AVRK (NIH F30CA239390), RS (8KL2 TR000118, K23, P30 CA054174, Roger L. And Laura D. Zeller Charitable Foundation Chair in Urologic Cancer, CDMRP CA170270/P1P2), T. Curiel (CA054174, CA205965, CDMRP, The Owens Foundation, The Skinner endowment, The Barker endowment, P30 CA054174). Flow cytometry data was generated in the UT Health San Antonio Flow Cytometry Shared Resource Facility, which is supported by UT Health San Antonio, the Mays Cancer Center P30 Cancer Center Support Grant (NIH-NCI P30 CA054174) and the National Center for Advancing Translational Sciences, National Institutes of Health, through Grant UL1 TR002645.
Competing interests OB holds a patent on IL-2c.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.