Article Text

Abstract
Background Full application of cytokines as oncoimmunotherapeutics requires identification of optimal regimens. Our initial effort with intravenous bolus recombinant human interleukin-15 (rhIL-15) was limited by postinfusional reactions. Subcutaneous injection and continuous intravenous infusion for 10 days (CIV-10) provided rhIL-15 with less toxicity with CIV-10 giving the best increases in CD8+ lymphocytes and natural killer (NK) cells. To ease rhIL-15 administration, we shortened time of infusion. Treatment with rhIL-15 at a dose of 3–5 µg/kg as a 5-day continuous intravenous infusion (CIV-5) had no dose-limiting toxicities while effector cell stimulation was comparable to the CIV-10 regimen.
Methods Eleven patients with metastatic cancers were treated with rhIL-15 CIV-5, 3 µg (n=4), 4 µg (n=3), and 5 µg/kg/day (n=4) in a phase I dose-escalation study (April 6, 2012).
Results Impressive expansions of NK cells were seen at all dose levels (mean 34-fold), including CD56bright NK cells (mean 144-fold for 4 µg/kg), as well as an increase in CD8+ T cells (mean 3.38-fold). At 5 µg/kg/day, there were no dose-limiting toxicities but pulmonary capillary leak and slower patient recovery. This led to our choice of the 4 µg/kg as CIV-5 dose for further testing. Cytolytic capacity of CD56bright and CD56dim NK cells was increased by interleukin-15 assayed by antibody-dependent cellular cytotoxicity (ADCC), natural cytotoxicity and natural killer group 2D-mediated cytotoxicity. The best response was stable disease.
Conclusions IL-15 administered as CIV-5 substantially expanded NK cells with increased cytotoxic functions. Tumor-targeting monoclonal antibodies dependent on ADCC as their mechanism of action including alemtuzumab, obinutuzumab, avelumab, and mogamulizumab could benefit from those NK cell expansions and provide a promising therapeutic strategy.
Trial registration numbers NCT01572493, NCT03759184, NCT03905135, NCT04185220 and NCT02689453.
- cytokines
- natural killer T-cells
- therapies
- investigational
Data availability statement
Data are available upon reasonable request. Tabulated data will be posted on clinicaltrials.gov. Deidentified patient data and clinical trial protocol available by request from corresponding author.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Since the Food and Drug Administration approval of interleukin (IL)-2 and interferon alpha for the treatment of patients with cancer, diverse cytokines have been evaluated for the treatment of neoplasia.1–3 However, for cytokines to play a major role in cancer therapy, novel strategies for their use will have to be developed. Determination of the optimal treatment regimen is essential for the development of cytokines as cancer immunotherapeutics. IL-15, a pleiotropic cytokine that stimulates the differentiation and proliferation of natural killer (NK) and CD8 T cells, is among the cytokines showing promise.4–17 IL-15 signals through a heterotrimeric receptor including the common gamma chain (γc) shared with IL-2, IL-4, IL-7, IL-9 and IL-21, the beta chain shared with IL-2 and a unique IL-15Rα chain.4 14 15 IL-15Rα and IL-15 are produced in a coordinated fashion predominantly by activated monocytes and dendritic cells.4 14–16 Stimulation of these cells with type I or type II interferon through agonistic CD40 ligands or toll-like receptor agonists induces the synchronized expression of IL-15 and IL-15Rα.18–20 IL-15 is secreted only in small quantities and is mainly membrane bound under physiological conditions5 . IL-15 bound to IL-15Rα is presented at an immunological synapse in trans to cells that express IL-2/IL-15Rβ and γc but minimal IL-15Rα.5 21–23 Such IL-2/IL-15Rβ γc expressing target cells stimulated by the IL-15/IL-15Rα pair include NK and CD8+ T cells. In contrast to IL-2, IL-15 inhibited IL-2-mediated activation-induced cell death (AICD), less consistently activated Tregs and did not cause a significant capillary leak syndrome in mice or non-human primates.4 24 The ability of IL-15 to activate crucial effector T and NK cells, its inhibition of AICD and its capacity to maintain function of CD8 memory T cells provided the scientific basis for evaluation of IL-15 as a cancer immunotherapeutic. IL-15 demonstrated activity in syngeneic murine transgenic adenocarcinoma mouse prostate C2 prostatic cancer, Pmel-1, B16 melanoma and MC38 and CT26 colon carcinoma models.12 13 25–29
On the basis of these observations, an Escherichia coli recombinant DNA-based process was developed to provide clinical grade recombinant human interleukin-15 (rhIL-15) that ultimately produced a non-glycosylated, single-chain peptide of 150 amino acids with a calculated molecular weight of 12 901 Dc.
The different dose, dosing and formulation strategies employed with IL-15 can be compared in terms of the magnitude of expansion of total NK cells, in particular the CD56bright NK subset, and CD8+ T cells achieved with doses with acceptable toxicity. Postinfusional reactions limited dose escalation and immune activation in the first-in-human clinical trial of rhIL-15 given as a 30 min intravenous bolus (IVB) daily for 12 days.16 At the maximum tolerated dose (MTD) of 0.3 µg/kg/day, the maximum increase in total NK cells was twofold to threefold. Ten-day subcutaneous (Monday–Friday for two consecutive weeks) and 10-day continuous intravenous infusion (CIV) were better tolerated and produced greater stimulation of effector NK and CD8+ T cells than IVB.30 At the MTD of 2 µg/kg/day, there was a mean 10.8-fold increase maximum for NK cells, 37-fold for CD56bright NK cells and 3.3-fold for CD8+ T cells when rhIL-15 was administered subcutaneously. The third trial of rhIL-15 involved a continuous intravenous infusion for 10 days (CIV-10) with the MTD concluded to be 2 µg/kg/day.31 CIV-10 infusions of rhIL-15 resulted in the most dramatic increases in circulating effector cells—with a mean 38-fold increase in total NK cells, 358-fold increase in CD56bright NK cells and a 5.8-fold increase in CD8+ T cells.31 Lytic functions of both NK subsets were augmented. Stable disease was the best response in these trials, leading to the conclusion that for IL-15 to play a major role in cancer therapy, it would have to be used in combination trials. As NK cells are the main mediators of antibody-dependent cellular cytotoxicity (ADCC), combination trials will pair rhIL-15 with antitumor monoclonal antibodies which function through ADCC. In the present study, to increase patient and physician acceptance, a short course of IL-15 (5 days) was given as a CIV. We report the results of this 5-day continuous intravenous infusion (CIV-5) rhIL-15 regimen that had a safety profile and stimulation of effector cells comparable to CIV-10 and with doses up to 5 µg/kg/day without dose-limiting toxicities.
In addition to providing a more patient and physician acceptable duration of 5 days, in figures 1–3, we provide major insights concerning the impact of IL-15 on the strong proliferation and phenotypical changes of NK cells induced by CIV-5. Our study includes the effects on the major activating and inhibitory receptors expressed by the NK subsets bright and CD56dim cells following IL-15 infusions. We provide data on IL-15 infusions augmenting cytokine production and cytokine function of both NK-cell subsets. Furthermore, we provide new data on the impact of IL-15 infusions on the induction, expansion, and effect of cytokine production on T-cell subsets.
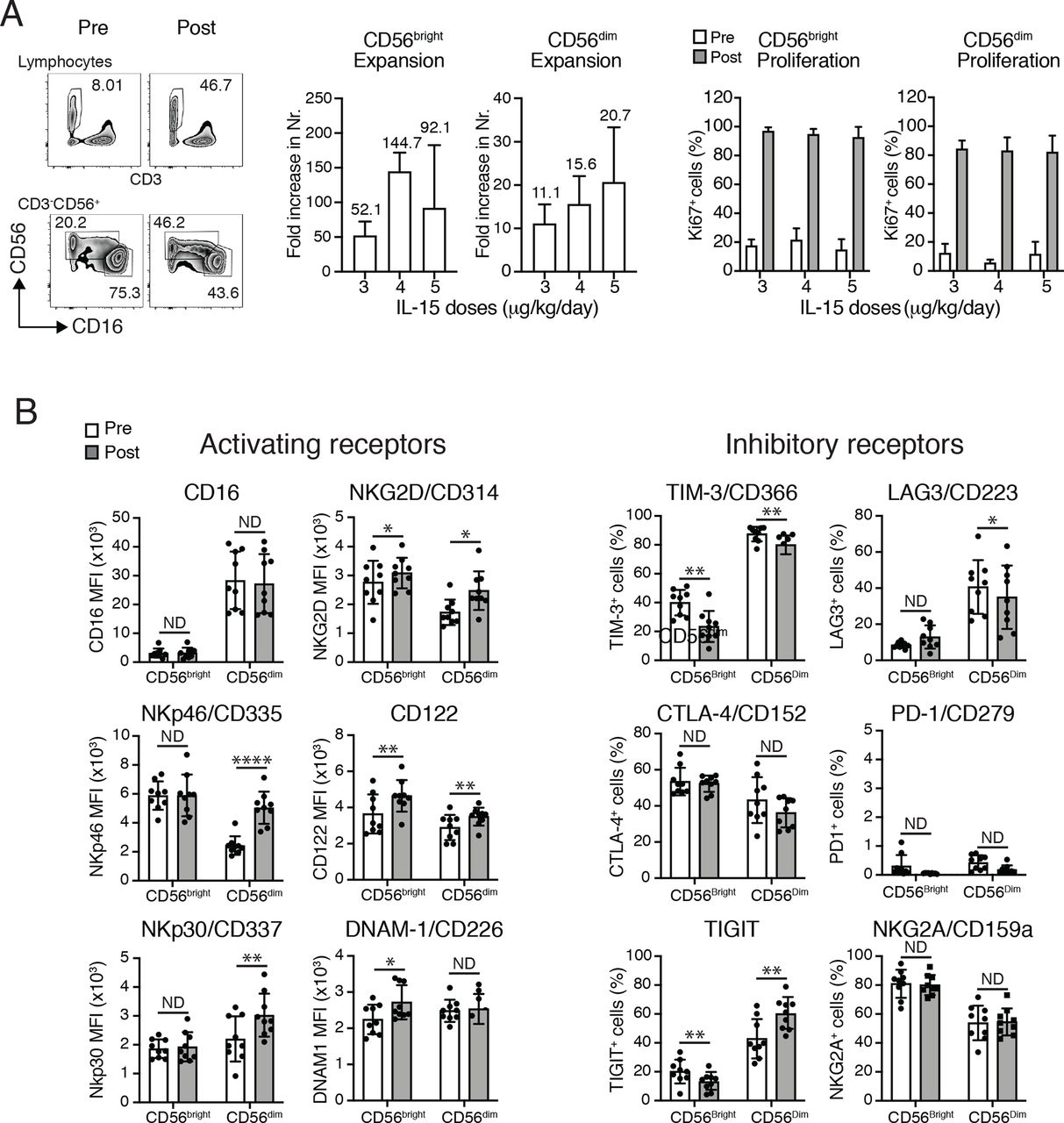
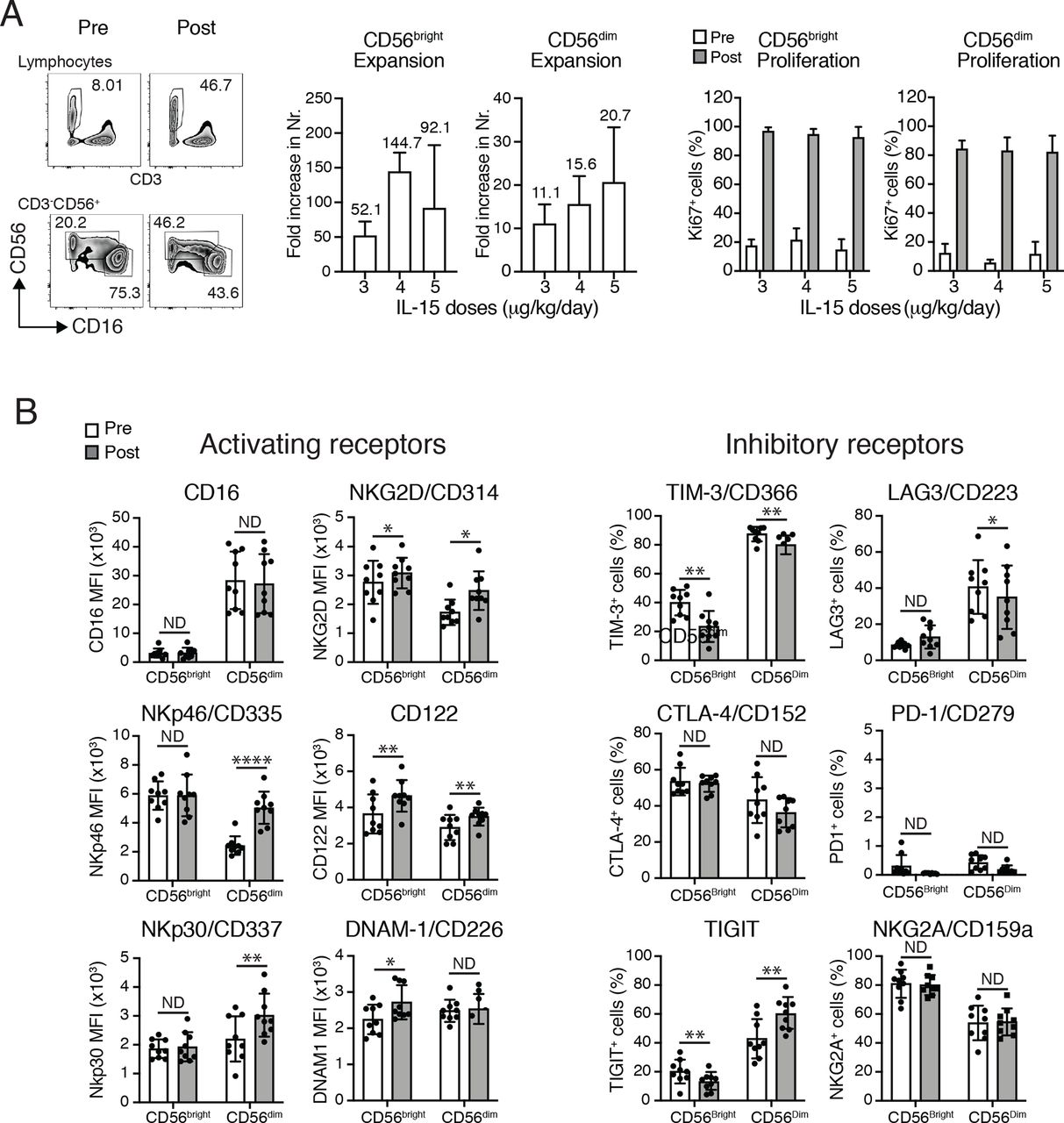
Strong proliferation and phenotypical changes of NK cells induced by CIV-5. (A,B) Analyses of the two major NK-cell subsets, CD56bright and CD56dim, comparing their percentages, numbers and phenotypes before and 3 days after the end of IL-15 infusions. (A) Left panels show an example of percentages of NK cells among PBMCs (top) and percentages of both NK subsets, CD56bright and CD56dimcells (bottom), prior to and after IL-15 infusions. Middle graphs depict fold increases of each NK subset following IL-15 treatment (three patients/dose). Right graphs display percentages of the proliferation marker Ki67 expressed among each NK subset prior to and after IL-15 infusions (three patients/dose). No statistical differences have been observed among the three different doses of IL-15. (B) Phenotypical changes of NK-cell subsets following IL-15 treatment. Left: graphs show MFI of the major activating receptors expressed by NK cells. Changes in expressions of those activating markers differed between NK subsets. Expressions of NKp46, NKp30, NKG2D and CD122 were increased on CD56dimcells. Augmentations in the surface expressions of NKG2D, CD122 and DNAX accessory molecule 1 (DNAM1) were observed on CD56bright cells. Right: graphs depict percentages among each NK subset expressing surface proteins involved in inhibition or exhaustion of NK cells. IL-15 appears to have opposite effects on percentages of cells expressing TIGIT with their decreases in CD56bright cells and their increases on CD56dim cells. Decreased percentages of TIM-3+ cells were detected in both NK subsets as well as LAG3+ among the CD56dim subset. Analyses were done using nine patients (three patients/dose). *P<0.05, **P<0.01, ****P<0.0001. IL, interleukin; LAG3+, lymphocyte-activation gene 3; MFI, mean fluorescence intensity; ND, no difference; NK, natural killer; NKG2D, natural killer group 2D; PBMC, peripheral blood mononuclear cell; TIGIT, T-cell immunoreceptor with Ig and ITIM domains; TIM-3+, T-cell immunoglobulin and mucin domain containing 3.

Il-15 infusions augment cytokine production and cytotoxic functions of both NK-cell subsets. (A) IL-15 treatments intensify cytokine productions by NK subsets. Top: depicts a schematic view and bottom graphs detail analyses of the major changes in cytokine productions by following intracellular detections of IFN-γ, TNF-α and GM-CSF by each NK subset when PBMCs were stimulated with IL-12 and IL-18. After treatments, two different subpopulations among CD56bright subset emerged: one subpopulation was able to secrete the three cytokines, whereas the second subpopulation coproduced IFN-γ and GM-CSF. For the CD56dim subset, only percentages of cells releasing IFN-γ increased after IL-15 infusions. (B) IL-15 treatments increase cytotoxic functions of both NK subsets. Left graphs compare lytic activities of same numbers of total NK cells prior to and after treatment at an effector: target ratio of 5:1 against three different target cells involving various activating receptors. Middle graphs (CD56bright NK cells) and right graphs (CD56dim NK cells) depict degranulation assessed via CD107 detection and intracellular IFN-γ production induced by stimulation with the same target cells at an effector: target ratio of 2:1. In the CD56bright NK subset, IL-15 treatments supported cells having abilities to degranulate and produce IFN-γ at the same time, whereas in CD56dim NK subset cells gained mainly in degranulation on target recognition. (C) IL-15 infusions increase target-induced degranulation despite TIGIT-mediated inhibition. Top histogram displays TIGIT ligand expression, CD155, on K562 and Raji target cells. Middle and bottom graphs compare percentages of CD107 upregulation among TIGIT+ and TIGIT− CD56dim NK subsets on CD155+K562 (middle graph) and CD155− aCD20Raji (bottom graph) stimulations prior and after IL-15 treatments. Although TIGIT+ CD56dim subset had lower abilities to degranulate than TIGIT− CD56dim cells when target cells express CD155, their CD107 percentages augmented after IL-15 infusions. Analyses were done using six patients (two patients/dose of IL-15 tested). *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001. GM-CSF, granulocyte-macrophage colony-stimulating factor; IFN-γ, interferon gamma; IL, interleukin; ND, no difference; PBMC, peripheral blood mononuclear cell; TIGIT, T-cell immunoreceptor with Ig and ITIM domains; TNF-α, tumor necrosis factor alpha.

IL-15 infusions induce expansions and affect cytokine production in T-cell subsets. (A) Fold increases, proliferations with detection of Ki-67 in CD8+ cells, CD4+ cells and γδ T cells obtained after analyses of samples frozen prior and 3 days after the end of IL-15 infusions. No statistical differences were observed among the three different doses of IL-15. (B) Comparison of TNF-α, IFN-γ and IL-2 production in each T-cell subset prior and after treatments following PMA/ionomycin stimulation. No change was observed within γδ T cells. Both CD8+ and CD4+ T cells gained in their abilities to secrete TNF-α. * P<0.05, ** P<0.01, *** P<0.001. IFN-γ, interferon gamma; IL, interleukin; ND, no difference; PMA, Phorbol 12-myristate 13-acetate; TNF-α, tumor necrosis factor alpha.
Materials and Methods
Additional information is provided in online supplemental file 1.
Supplemental material
Recombinant human interleukin-15
rhIL-15 was produced in E. coli as previously reported.16
Patients and study design
Patients with advanced metastatic solid tumors for whom standard curative or palliative treatments did not exist were enrolled in this single-center, non-randomized, phase I trial to determine the safety and toxicity of IL-15 (online supplemental table 1). As inpatients, patients received rhIL-15 by CIV for five consecutive days. IL-15 cycles were repeated every 3 weeks. The study used a 3+3 interpatient dose-escalation design administering doses at 3, 4 and 5 µg/kg/day. Treatments continued until tumor regression or two cycles if the best response was stable disease. Evaluation of the MTD was limited to the first cycle, but overall tolerability of the regimen was considered in the selection of the dose for further study (see online supplemental methods).
Supplemental material
Investigational treatment
rhIL-15 was produced under current good manufacturing practice conditions in an E. coli expression system, as described previously.16
The rhIL-15 was delivered intravenously by infusion or ambulatory pump for five consecutive days (120 hours). The bags containing IL-15 were changed daily. The rhIL-15 was diluted to a concentration of 1 µg/mL with 0.1% human serum albumin to improve stability and was administered by CIV at doses of 3, 4, and 5 µg/kg/day for 5 days.
Clinical assessment
Patients had regular measurement of vital signs, oral intake and output, physical examination, and daily chemistry and hematology laboratories. We performed staging CT scans and/or other appropriate radiographical procedures to evaluate their disease after the even-numbered cycles. Antitumor responses were assessed by RECIST V.1.1 criteria.32
Correlative laboratory analyses
Fluorescence-activated cell sorting (FACS) analysis
Blood samples were taken on day 1, just prior to treatment, and on day 8, which was the expected time of maximum lymphocytosis. Peripheral blood mononuclear cells (PBMCs) were purified via Ficoll centrifugation and viably frozen at 15×106 cells per vial. After thawing, PBMCs were rested overnight at 37°C in RPMI/10% human AB serum (MediaTech).
Antibodies used are listed in online supplemental table 2. PBMCs were blocked with human TruStain FcX BioLegend for 15 min at room temperature and stained with antibodies recognizing surface proteins (45 min at 4°C). Cells were permeabilized with Transcription Factor Fixation Permeabilization (eBioscience) to stain Ki-67. Stains for intracellular proteins were done with the provided buffers (30 min RT). Analyses were done with FACS Verse. Choices for gating strategies to distinguish NK CD56bright and CD56dim NK-cell subsets are described in online supplemental figure 4.
Supplemental material
Ex vivo activations
For cytokine production analyses, thawed and rested PBMCs were stimulated with PMA (5 ng/mL, Sigma) plus ionomycin (500 nmol/L, Sigma), or with human IL-12 (10 ng/mL, PeproTech) plus human IL-18 (50 ng/mL, R&D Systems). Alternatively, PBMCs were exposed to the target cell lines K562 (ATCC CCL-243), MHC class I chain-related protein A (C1R-MICA) MHC class I related stress-inducible surface glycoprotein (kindly provided by Dr Veronika Groh-Spies, Fred Hutchinson Cancer Research Center) or to anti-CD20-coated Raji (ATCC CCL-86) cells that had been preincubated with 10 µg/mL of the antibody (30 min at 4°C) and washed. Stimulation and coincubations were done for 5 hours at 37°C with brefeldin A present during the last 4 hours.
Cytotoxicity assays
Cytotoxicity assays were performed as described previously.33 K562, anti-CD20-coated Raji, and C1R-MICA were used as target cells and uncoated Raji as control cells. Target and control cells were labeled at different carboxyfluorescein succinimidyl ester concentrations (1 vs 0.1 μmol/L, Invitrogen). Target and control cells were mixed at equal numbers and added to various numbers of NK cells. Incubations were done in the presence of human IL-15 (2 ng/mL, 18 hours at 37°C). The ratios of surviving propidium iodide-negative target and control cells with or without NK-cell exposures were determined by FACS and used to calculate the percentages of target cell lysis with the formula 100−TNK/CNK÷TCON/CCON×100 (where T is target; C is control; NK is NK-cell presence; and CON is NK-cell absence).
Statistical analyses
Prism seven was used. P values were calculated using the paired t-test to compare cytometry analyses of patient samples before and after treatments.
Results
Between December 12, 2016, and May 8, 2018, 11 patients with refractory solid cancers were treated as inpatients with the CIV-5 regimen. The most common diagnosis was colorectal cancer; the median age was 60 years, with four male and seven female patients (online supplemental table 1). Of the 11 patients, four were treated at 3 µg/kg, three at 4 µg/kg and four at 5 µg/kg/day without deaths or dose-limiting toxicities. One patient each at 3 and 5 µg/kg/day dose levels did not complete the first cycle due to adverse events (AEs) unrelated to CIV-5 treatment, which subsequently resolved (non-steroidal, anti-inflammatory drug-induced syndrome of inappropriate antidiuretic hormone secretion and suspected septic or crystal arthritis, respectively) (table 1). These two patients were replaced. The most common AEs were similar to those observed for the CIV-10 regimen and included fever, chills, fatigue, nausea, transient liver function abnormalities, anemia, and thrombocytopenia (table 1). Patients receiving 5 µg/kg/day had transient decreases in oxygenation from pulmonary capillary leak syndrome. The 4 µg/kg dose level was the optimal treatment regimen used in subsequent trials.
Clinical adverse events and laboratory a(bnormalities possibly, probably or definitely related to research (5-day CIV cohort) excluding single grade 1 events
Clinical response to treatment
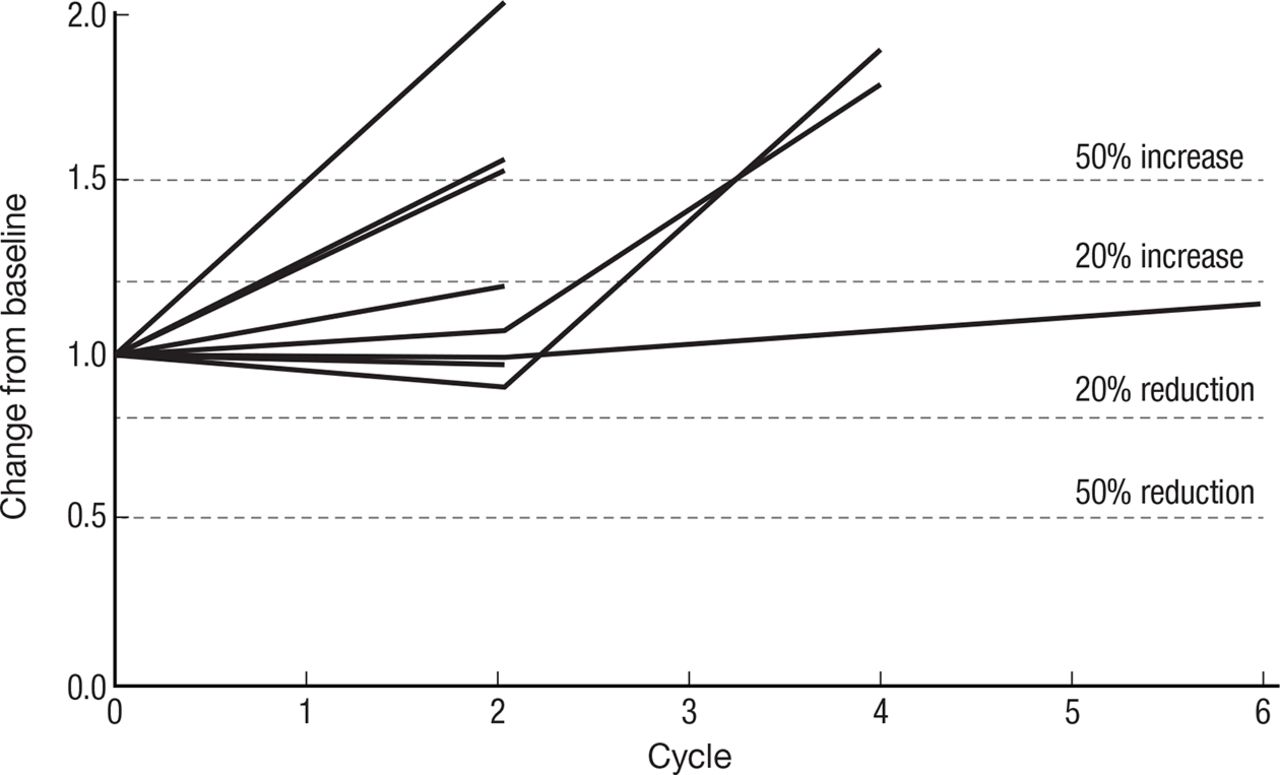
Spider plot (figure 4) of 8 of 11 patients’ response to therapy as assessed by RECIST V.1.1 criteria: stable disease was the best observed response. Three patients fulfilled the criteria of ≥15% decrease in measurable disease required to receive treatment beyond cycle 2.
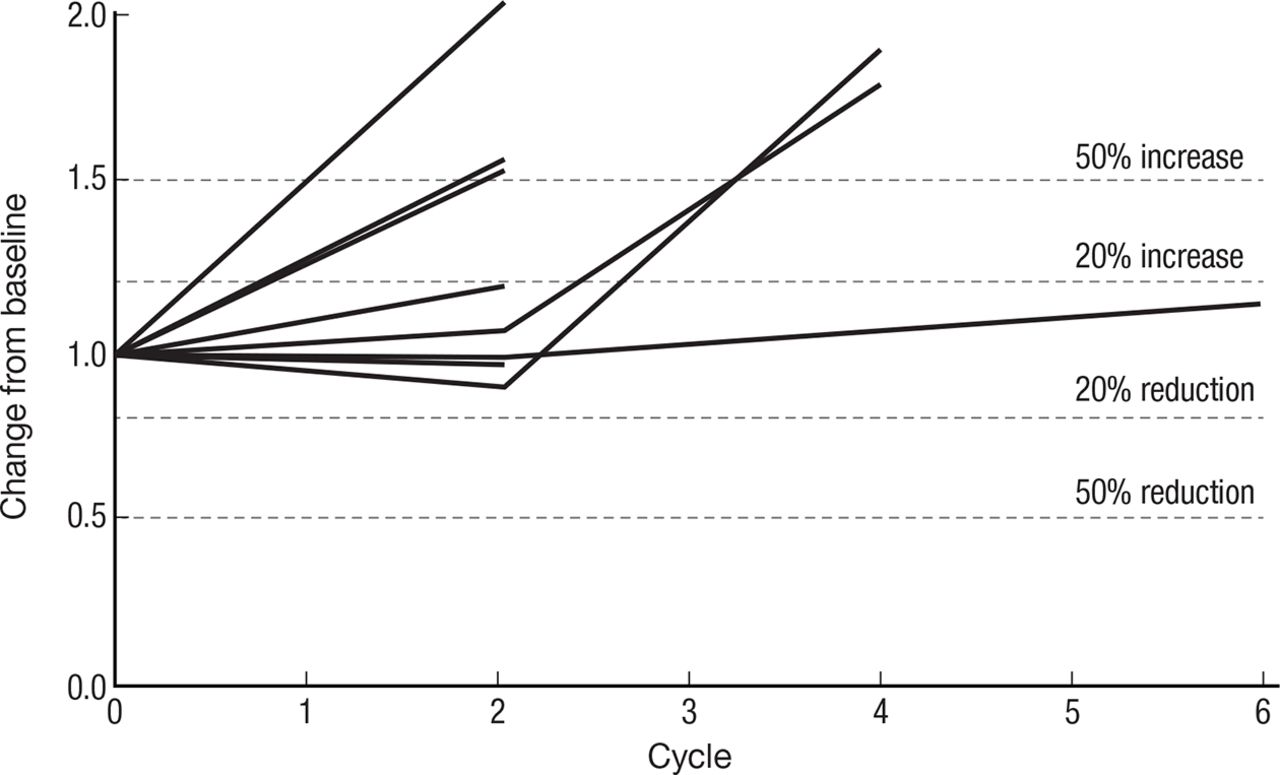
Spider plot of CIV IL-15 treatment spider plot of 8 of 11 treated patients who had at least one restaging CT scan, showing changes from baseline in tumor burden (Y-axis) measured as the product of the longest diameters of solid metastatic target lesions of >1 cm on high-resolution CT scans (shortest diameter for lymph nodes) assessed at the end of every CIV rhIL-15 cycle (X-axis). Above the 20% increase, dashed line indicates progressive disease by RECIST criteria and below 30% reduction indicates partial response. Patients who had stable disease after their first two cycles of treatment continued to be restaged at regular intervals even though their treatment had been stopped. CIV, continuous intravenous infusion; IL, interleukin.
Lymphocyte response to treatment
Previously, we demonstrated that IVB treatment with rhIL-15 resulted in acute lymphocytopenia, followed by hyperproliferation of cells with an increase in NK and CD8+ T cells. Here we show that the administration of rhIL-15 by CIV had an even greater impact on CD8+ T and NK, particularly CD56bright NK-cell expansions (figure 5 and table 2). As expected, all patients experienced generalized acute lymphocytopenia at the initiation of treatment. Following this, the blood lymphocytes numbers returned to baseline with evidence of high levels of proliferation lasting through the end of treatment. Changes were maximal 3 days following treatment cessation but were still dramatic 10 days later.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Increase in lymphocytes predominantly NK-cell count during continuous infusion of rhIL-15 IL-15 was administered at progressively increasing doses of 3, 4 and 5 µg/kg/day by 5-day CIV infusion to patients with metastatic malignancy. Three days following termination of the treatment, there was a dramatic 34-fold mean increase in the number of circulating lymphocytes, predominantly NK-cell counts, and in the 4 µg/kg/day cohort 144-fold increase in the number of circulating CD56bright NK cells. IL, interleukin; NK, natural killer; rhIL, recombinant human interleukin-15.
IL-15 clinical trials in patients with metastatic malignancy
Assessment of anti-rhIL-15 antibodies
Patients were assessed for the development of anti-IL-15 antibodies in serum during each treatment cycle. Immunogenicity timepoints were pre-treatment and day 21. Testing was extended to cycle 3, day 43 for one patient. None of the serum samples obtained from the 11 patients treated in this trial showed evidence of antibodies against IL-15. Antibodies to IL-15 were not observed on any of the previous studies involving intravenous IL-15, whereas such antibodies developed in a proportion of patients receiving more than three cycles of subcutaneous IL-15.30 31
Inflammatory cytokine production
By 8 hours after the onset of infusion interferon gamma (IFN-γ) increased 60-fold and IL-6 increased 30-fold (online supplemental figure 2). By day 6, soluble IL-2Rα increased 10-fold, IL-18 to 4-fold, IL-1β to 2-fold and tumor necrosis factor alpha (TNF-α) to 8-fold (online supplemental figure 3).
Supplemental material
Supplemental material
Expansions of the different lymphocyte subsets following CIV-5 administration
The 5-day CIV IL-15 infusions induced a maximum increase in total numbers of circulating NK cells of 21-fold to 44-fold (mean 34-fold). Detailed analyses of the two major NK subsets showed higher effects in the expansions of the CD56bright NK subset with an increase from 52.1-fold to 144.7-fold compared with an increase of the CD56dim NK subset from 11.1-fold to 20.7-fold (figure 1A). Among T lymphocytes, γδ T cells were the most affected by IL-15 infusion with an increase in total numbers from 4.6-fold to 7.7-fold, followed by CD8+ T cells with increase expansions from 2.5-fold to 4.1-fold (figure 3A). Treatments had lesser effects on the CD4+ T-cell subset that increased in total numbers by 1.2-fold to 2.4-fold. Those discrepancies between expansions of the different lymphocytes subsets were supported by evaluation of expressions of the proliferation marker, Ki-67, showing that between 80% and 90% of NK cells, 70% of γδ T cells, 45% of CD8+ T cells and 25% of CD4+ cells had proliferated 3 days following cessation of treatment (figure 1A). There was no statistical difference observed among the three different doses of IL-15 (figures 1–3).
Phenotypical changes of NK-cell subpopulations after CIV
To examine whether IL-15 treatments induce phenotypical changes that could affect gain and/or loss in functions in NK cells, we used two distinct panels of antibodies targeting the main receptors known to drive activation and inhibition or exhaustion of NK cells. Expression of those receptors were evaluated on both NK CD56bright and CD56dim NK-cell subsets prior to and 3 days after cessation of IL-15 infusions using PBMCs obtained from nine patients (three patients for each of the three doses of IL-15 tested, figure 1B). Looking at the mean fluorescence intensity of the major activating receptors expressed by NK cells (figure 1B, left), we observed changes within the CD56dim population with increases in expression of NKp46, NKp30, natural killer group 2D (NKG2D) and CD122. Augmentation of cell-surface expression of NKG2D (CD314), CD122 and DNAM-1 (CD226) was detected in CD56bright cells. Levels of CD16 expression were not affected by treatment of both NK subsets. None of the activating receptors studied showed decreased expressions after IL-15 infusions. Surface proteins involved in inhibition or exhaustion of NK cells were presented as percentages of each NK subset (figure 1B, right). After IL-15 infusions, the percentages of T-cell immunoreceptor with Ig and ITIM domains (TIGIT) cells appeared to be differentially regulated between the two NK subpopulations since their proportions were decreased among CD56bright cells and increased within CD56dim cells. IL-15 treatments notably decreased percentages of T-cell immunoglobulin and mucin domain containing 3+ cells in both NK subsets and of lymphocyte-activation gene 3+ cells in CD56dim subpopulation. Proportions of cytotoxic T-lymphocyte-associated protein (CTLA-4)+ and NKG2A+ were not affected by IL-15 infusions in both NK-cell subpopulations. Together, those data suggest that IL-15 treatments favor NK-cell activation since infusions lead mainly to upregulation of receptors involved in NK-cell activation. There were no dose-dependent differences in the outcomes observed.
IL-15 infusions enhanced cytokine production by NK, CD8+ and CD4+ T cells
To monitor effects of IL-15 infusions on cytokine secretions by NK cells, we used ex vivo stimulation with IL-12 and IL-18 to detect intracellular production of IFN-γ, TNF-α and granulocyte-macrophage colony-stimulating factor (GM-CSF) in both NK subsets (figure 2A). Both NK subpopulations produced mainly IFN-γ prior to treatment. After infusions, percentages of CD56bright secreting IFN-γ increased from 50% to 80% in two distinct populations; one subpopulation secreted all three cytokines, IFNγ, TNF-α and GM-CSF increasing from below 2% to 25% after treatment, whereas the second population, augmenting from below 5% to 30% after CIV-5, produced only IFN-γ and GM-CSF. For the CD56dim subset, only percentages of cells producing IFN-γ alone increased after IL-15 infusions from 35% to 70%.
T-cell subpopulations (CD4+, CD8+, and γδ) were assayed for TNF-α, IFN-γ, and IL-2 production following PMA/ionomycin stimulation (figure 3B). Both CD8+ and CD4+ T cells gained mainly in their abilities to produce TNF-α. No change was observed for the γδ T cells.
Il-15 administration increased the cytotoxic functions of both NK-cell subsets
The IL-15 infusions improved cytotoxic functions of total NK cells when lytic activities were compared among the same numbers of total NK cells pretreatment and post-treatment at an effector to target ratio of 5:1 using three different target cells, involving for their killing different activating receptors: C1R-MICA (figure 2B, top row), K562 (figure 2B, middle) and anti-CD20-coated Raji cells (figure 2B, bottom) recognized by NK cells via NKG2D, natural cytotoxic receptors (NKp30 and NKp46) and CD16-mediated ADCC, respectively. Degranulation via CD107 detection and IFN-γ production were assessed in CD56bright (figure 2B, middle column), and in CD56dim (right column) subsets after stimulation with the same target cells described previously at an effector to target ratio of 2:1. Within the CD56bright NK cells, CIV-5 promoted cells that had the abilities to degranulate and produce IFN-α at the same time, whereas CD56dim NK cells gained mainly in degranulation on target recognition.
Degranulation via CD107 detection and IFN-γ production were simultaneously assessed in CD56bright (figure 2B, middle column) and in CD56dim (figure 2B, right column) subsets after stimulation with the same target cells described earlier at an effector to target ratio of 2:1. In response to the three targets, we observed that CIV-5 promoted CD56bright NK subset to degranulate and produce IFN-γ at the same time, whereas CD56dim NK-cell subset gained mainly in degranulation. Phenotypical analyses revealed increased percentages of cells expressing TIGIT within the CD56dim NK subpopulation after IL-15 treatment. Since TIGIT is known to attenuate cytotoxicity of NK cells, we compared degranulation abilities of TIGIT+ and TIGIT− among CD56dim subsets prior and after CIV-5 on stimulation with target cells expressing or not TIGIT ligand, CD155 (using CD155+ K562 cells vs CD155− Raji cells, figure 2C). Percentages of CD107+ among TIGIT+ CD56dim subsets were lower than among TIGIT− CD56dim subsets following stimulation with CD155+ target cells. However, degranulation was augmented after IL-15 infusions despite TIGIT-mediated inhibition.
Taken together, the short 5-day course of rhIL-15 given as a continuous infusion led to massive expansion of NK-cell numbers. After the termination of IL-15 infusions, both NK-cell subsets showed vigorous proliferation, increased cytokine production abilities and effective cytotoxic functions involving the major activating receptors. Augmentation in total numbers of CD8+ T cells and NK cells with gain in their cytotoxic functions supports ongoing trials of CIV-5 IL-15 with anticancer monoclonal antibodies to increase their efficacy via ADCC as antitumor antibodies.
Discussion
IL-15 plays a pivotal stimulatory role in the differentiation and proliferation of NK and CD8+ T cells. Preclinical data demonstrated considerable potential of IL-15 as an immunotherapeutic for cancer; by translating these observations into the clinic, over 170 clinical trials have been initiated in the treatment of cancer with different IL-15 preparations.15 Compared with other treatment regimens or formulations of IL-15, the CIV-5 or CIV-10 regimens resulted in the greatest increase in circulating effector cells (table 2).15 16 30 34–36 There have been extensive efforts to develop potent long-acting IL-15 agonists, including ALT-803 (N-803, IL-15, N72D linked to the sushi domain of IL-15Rα IgG Fc fusion protein).34–36 However, due to pharmacokinetic disadvantages these preparations do not supply sufficient steady state serum levels of IL-15 that are consistent with their less significant effects on NK and CD8+ T-cell expansions. A large proportion of subcutaneous injected drug may not reach the systemic circulation due to local consumption by both activated αβ and γδ T cells at the site of injection, resulting in characteristic injection site reactions which can be up to 30 cm in diameter.34 There is with ALT-803 only a bioavailability of 3% indicating ~97% of the dose not reaching the systemic circulation. This represents a significant drawback despite the ease of administration and the weekly treatment schedule. As a consequence of the low bioavailability, ALT-803 at 10 µg/kg/day by subcutaneous injection yielded only a twofold increase in the number of CD8+ T cells and only a fivefold and sixfold increase in the number of NK and CD56bright NK cells, respectively.34 This, as noted in table 2, contrasts with the 3.8-fold increase in the number of CD8+ T cells, 34-fold increase in the number of NK cells, and up to 144-fold increase in the number of CD56bright NK cells observed by CIV-5 of IL-15. CIV-5 gave comparable efficacy with CIV-10. It would clearly be of value to evaluate whether yet shorter infusions with CIV-1, CIV-2 or CIV-3 are efficacious since this would dramatically increase patient and physician acceptability and facilitate the studies, especially if multiple cycles are envisioned. Thus, IL-15 by CIV-5 or CIV-10 was the most successful among the different IL-15 preparations in inducing the maximal increase in the numbers of CD8+ T cells, NK cells and CD56bright NK cells. Felices and coworkers, studying purified NK cells in vitro suggested that continuous treatment with IL-15 exhausts such purified NK cells resulting in decreased viability and a cell cycle arrest gene expression pattern.37 Furthermore, they proposed that these findings should inform IL-15 dosing strategies. Frutoso and Mortier,17 and Frutoso and coworkers38 provide excellent reviews of ‘NK cell hyporesponsiveness: more is not always better’. They outline circumstances where providing additional IL-15 could actually be counterproductive and could lead to NK-cell hyporesponsiveness. Furthermore, our group demonstrated hyporesponsiveness when there are repeated cycles of IL-15 in less than 3-week intervals. Our clinical trials using continuous IL-15 infusions in vivo demonstrated that continuous IL-15 treatments led to massive NK-cell proliferations as shown with Ki-67 expression in more than 80% of NK cells at termination of 5-day IL-15 CIV. Both NK subsets after treatment gained in expressions of some activating receptors. Moreover, lytic capacities of both NK subsets after IL-15 infusions were effectively increased in killing of target cells recognized by three major types of activating receptors triggering NK cytotoxic activities. Finally, both CIV trials showed that NK cells significantly gained in cytokine release functions. In conclusion, our data support the view that continuous IL-15 treatments in vivo for 5 or 10 days result in extensive expansions and gain of functions of NK cells rather than their exhaustion. The discrepancy between our studies and the work of Felices and coworkers may reflect overgrowth and decreases in nutriments in the ‘in vitro setup’ of purified NK cells versus our examination of NK cells continuously stimulated with fresh IL-15 in vivo. Additionally, other factors are absent in culture, in particular interaction between NK cells and other cell types, such as monocytes/macrophages and dendritic cells known to support NK functions.27
Although diverse forms of IL-15 monotherapy augment the number of circulating NK and CD8+ T cells, monotherapy trials with any of the treatment strategies/or agents did not demonstrate clinical activity, with the sole exception being postallogeneic stem cell transplant patients who possessed an underlying graft versus leukemic capacity.34 There are a variety of explanations possible for this lack of efficacy, including induction of immunological checkpoints and lack of significant targeting of these effector cells to sites of tumor. An attractive strategy that exploits the NK target cell expansion would be to use IL-15 in association with anticancer monoclonal antibodies to target the NK cells to the tumor, to increase ADCC and effector action on cancer cells. IL-15 preparations have been reported to be of value in combination with in vivo administered monoclonal antibodies.39–43 Our preclinical experiments with IL-15 and rituximab in a syngeneic mouse model of the EL4 lymphoma transfected with human CD20 and alemtuzumab (CAMPATH-1H) in a xenograft model of human adult T-cell leukemia27 showed that IL-15 enhanced therapeutic efficacy of both antibodies.
In conclusion, the short duration CIV-5 rhIL-15 regimen and its safety profile may make outpatient administration via an ambulatory infusion pump feasible. The massive expansion of NK cells and increases in CD8+ T cells it produced were much greater than with other non-CIV IL-15 regimens. Since NK cells are key mediators of ADCC, we have opened trials administering CIV IL-15 with antibodies, obinutuzumab (NCT03759184), avelumab (NCT03905135), mogamulizumab (NCT04185220) and alemtuzumab (NCT02689453) that may benefit from IL-15-induced expansion and gain in functions of NK cells to augment their antitumor ADCC and antitumor efficacy.
Supplemental material
Data availability statement
Data are available upon reasonable request. Tabulated data will be posted on clinicaltrials.gov. Deidentified patient data and clinical trial protocol available by request from corresponding author.
Ethics statements
Ethics approval
The study was approved by the institutional review board of the National Cancer Institute and was performed in accordance with the ethical guidelines of the Declaration of Helsinki, Ethical Principles of Medical Research. All patients signed a written informed consent to participate in the clinical trial.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
SPD and MDM contributed equally.
TAW and KCC contributed equally.
Contributors Conception and design: KCC, MDM, SD, and TAW. Acquisition of data: SD, KCC, MDM, TAF, SP, BRB, and MNP. Analysis and interpretation of data: SD, MDM, KCC, TAW, JHS, BRB, and JRM. Provision of study materials or patients: KCC, JH-A, LPP, and MDM. All authors reviewed and approved the manuscript for publication prior to submission.
Funding This study was supported by the Intramural Research Program of the National Cancer Institute, National Institutes of Health.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.