Article Text

Abstract
Background While programmed cell death protein 1 (PD-1) and programmed death-ligand 1 (PD-L1) antagonists have improved the prognosis for many patients with melanoma, around 60% fail therapy. PVSRIPO is a non-neurovirulent rhinovirus:poliovirus chimera that facilitates an antitumor immune response following cell entry via the poliovirus receptor CD155, which is expressed on tumor and antigen-presenting cells. Preclinical studies show that oncolytic virus plus anti-PD-1 therapy leads to a greater antitumor response than either agent alone, warranting clinical investigation.
Methods An open-label phase I trial of intratumoral PVSRIPO in patients with unresectable melanoma (American Joint Committee on Cancer V.7 stage IIIB, IIIC, or IV) was performed. Eligible patients had disease progression on anti-PD-1 and V-raf murine sarcoma viral oncogene homolog B (BRAF)/mitogen activated protein kinase kinase (MEK) inhibitors (if BRAF mutant). The primary objective was to characterize the safety and tolerability of PVSRIPO. Twelve patients in four cohorts received a total of 1, 2 or 3 injections of PVSRIPO monotherapy, with 21 days between injections.
Results PVSRIPO injections were well tolerated with no serious adverse events (SAEs) or dose-limiting toxicities (DLTs) reported. All adverse events (AEs) were grade (G) 1 or G2 (G1 pruritus most common at 58%); all but two PVSRIPO-treatment related AEs were localized to the injected or adjacent lesions (n=1 G1 hot flash, n=1 G1 fatigue). Four out of 12 patients (33%) achieved an objective response per immune-related response criteria (two observations, 4 weeks apart), including 4/6 (67%) who received three injections. In the four patients with in-transit disease, a pathological complete response (pCR) was observed in two (50%) patients. Following study completion, 11/12 patients (92%) reinitiated immune checkpoint inhibitor-based therapy, and 6/12 patients (50%) remained without progression at a median follow-up time of 18 months.
Conclusion Intratumoral PVSRIPO was well tolerated. Despite the limited number of PVSRIPO treatments relative to the overall lesion burden (67% patients>5 lesions), intratumoral PVSRIPO showed promising antitumor activity, with pCR in injected as well as non-injected lesions in select patients.
Trial registration number NCT03712358
- melanoma
- oncolytic virotherapy
Data availability statement
All data relevant to the study are included in the article or uploaded as supplemental information.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Background
While the recent Food and Drug Administration approval of programmed cell death protein 1 (PD-1) and programmed death-ligand 1 (PD-L1) antagonist therapy has improved the prognosis for some patients with advanced melanoma, treatment comes with significant limitations.1–4 Primary resistance at the time of initial drug exposure occurs in around 60% of patients with melanoma, and of those who have an initial response, secondary resistance can then occur.4 There are multiple mechanisms that drive primary resistance to anti-PD-1 therapy, including insufficient intratumoral T-cell trafficking/function as well as descriptions of immune inhibitory signaling within the tumor microenvironment (TME).5 Efforts to overcome resistance, particularly the addition of anti-cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) directed therapy, increases response rates to around 60% but also results in a substantial increase in the risk of severe toxicity.6 One promising approach for patients with melanoma who lack immune-mediated antitumor responses to anti-PD-1 therapy is intratumoral delivery of agents designed to promote antigen presentation in the TME, which can revive or initiate antitumor responses.7 8
Intratumoral delivery of the oncolytic poliovirus PVSRIPO, the live attenuated, type I poliovirus (Sabin) vaccine carrying an internal ribosomal entry site (IRES) of human rhinovirus type 2,9 has shown initial promise in patients with recurrent glioblastoma multiforme.10 PVSRIPO enters cells via the poliovirus receptor CD155, which is widely expressed on neoplastic cells in many solid cancers,11 including melanoma,12 as well as on all antigen-presenting cells (APCs) of monocytic lineage.13 Accordingly, PVSRIPO elicits cytopathogenic damage and host innate type I/III interferon (IFN) responses in cancer cells14 and activates tumor-associated APC in the TME in preclinical models.15 16 The activated APCs produce sustained type I/III IFN dominant inflammation in the TME, which controls tumor growth in murine immunocompetent tumor models.15 16 The combined effects stemming from viral targeting of neoplastic cells and innate immune activation in the TME make intratumoral delivery of PVSRIPO a potentially effective strategy to promote sustained antitumor response in combination with systemic PD-1 therapy.7 15 17 Here, we present results of the initial phase I trial of intratumoral PVSRIPO in 12 patients with anti-PD-1 refractory, unresectable melanoma.
Methods
Patient population
A full list of inclusion and exclusion criteria can be found on the clinicaltrials.gov website (https://clinicaltrials.gov/ct2/show/NCT03712358?cond=pvsripo&draw=2&rank=4). See online supplemental file 1 for the full clinical protocol.
Supplemental material
Briefly, patients were required to have histologically proven, unresectable, recurrent American Joint Committee on Cancer (AJCC) V.7, stage IIIB, IIIC, or IV melanoma, with three or fewer visceral metastases.18 All patients were required to have disease progression after anti-PD-1-based therapy. Patients whose tumors had V-raf murine sarcoma viral oncogene homolog B (BRAF)V600 mutations were required to have progressed on BRAF/mitogen-activated protein kinase kinase (MEK) inhibitors therapy as well. Prior intralesional therapies were allowed. Staging was determined by computed tomography (CT) of the chest, abdomen and pelvis, and/or whole-body positron emission tomography scan and magnetic resonance imaging (MRI) of the brain. Patients were administered trivalent inactivated IPOL® (Sanofi-Pasteur) at least 1 week prior to intratumoral injection of PVSRIPO and were required to have a positive serum antipoliovirus titer. To be treated, patients had to have at least one injectable cutaneous, subcutaneous or nodal melanoma lesion of ≥10 mm in longest diameter or multiple injectable melanoma lesions which in aggregate had a longest diameter of ≥10 mm.
Study oversight
This study was an open-label phase I trial of PVSRIPO in patients with anti-PD-1 refractory melanoma. Two authors have equity in Istari Oncology (MG and DDB).
Study drug
PVSRIPO is the live attenuated, type I poliovirus (Sabin) vaccine that was modified by exchange of its cognate IRES with the corresponding segment from human rhinovirus type 2.9 Dose-range finding and toxicology studies in non-human primates documented the absence of viral encephalomyelitis, poliomyelitis and meningitis with intracerebral injection of PVSRIPO up to a dose of 5×109 median tissue culture infective dose (TCID50).19 The dose of PVSRIPO for this trial was selected based on investigational new drug (IND)-directed toxicity studies and prior experience from a phase I dose-escalation study in adults with recurrent WHO G4 malignant glioma.10 In the phase I malignant glioma studies, patients received only one planned treatment of intratumoral PVSRIPO, although some patients received a second dose after amendments to the protocol.
PVSRIPO was manufactured at the Biopharmaceutical Development Program/National Cancer Institute-Frederick. The dose selected for this single institution trial was 1×108 TCID50 delivered via direct intratumoral injection. The desired aliquot was prepared at the intended volume (0.5 mL) and drawn into a delivery device (1 mL syringe). PVSRIPO was then injected into visible, palpable, or ultrasound-visualized lesions using a single insertion point and redirecting the needle within the lesion to disperse the volume equally within the entire lesion.
Study design
We initially enrolled three patient cohorts each consisting of three patients; a fourth cohort was later added to test an alternate dosing strategy of repeated injections into the same lesion. There was a minimum of 3 weeks between each cohort. Cohort 0 (n=3) received one full dose (1×108 TCID50) intratumoral injection of PVSRIPO into one lesion. Cohort 1 (n=3) received two total doses of PVSRIPO administered to two distinct lesions, with the first lesion treated on day 1 and the second on day 22. Cohort 2 (n=3) received three total doses of PVSRIPO administered to three separate lesions: the first lesion was treated on day 1, the second lesion on day 22, and the third lesion on day 43. Finally, during the study, an amendment allowed for an additional cohort of three patients to receive multiple injections into the same lesion. In this cohort (cohort 3), the same lesion was treated with three intratumoral injections, given on days 1, 22, and 43. If two or more patients in any cohort experienced a DLT, a detailed prespecified dose-reduction plan would be followed.
Primary source documentation of disease burden consisted of tumor measurements and digital photographs taken at baseline, prior to treatment. Up to five cutaneous lesions and up to five metastases (no more than two visceral organs) were chosen as index lesions for serial measurements throughout the study. Index lesions were selected on the basis of their size (lesions with longest bidimensionally perpendicular diameters; at least 10 mm in longest diameter or in aggregate if multiple superficial lesions) and suitability for accurate repeated measurements by imaging techniques and/or clinical exam. Index lesions were selected in a way that the number and distribution of index lesions could be considered representative of the subject’s overall disease status. Measurements were performed using imaging (CT, MRI, or ultrasound) or clinical exam (using a ruler or calipers), and the same technique was used consistently for lesions across time points. All other lesions, including any measurable lesions not chosen as index lesions as well as non-measurable lesions, were considered non-index lesions and were assessed qualitatively over the course of therapy but were not measured specifically.
Response was determined according to immune-related response criteria (irRC) and repeated assessments of index lesion tumor measurements over time as well as qualitative assessment of non-index lesions.20 Tumor measurement data in combination with photographic records of lesions obtained prior to intervention were used to establish baseline measurements, and measurements were repeated at predetermined time points: days 22, 43, and 84 after initial PVSRIPO injection.
Confirmation of disease response to PVSRIPO at 4 weeks was planned according to irRC. However, after PVSRIPO treatment, patients who were not having clinical benefit, as determined by the principal investigator (PI), were taken off study at earlier time points (before the 4-week confirmation) so patients could pursue additional therapies. After patients were off study, patient medical records were reviewed to capture any poststudy therapies as well as progression free and overall survival. Additional patient characteristics for manuscript preparation were obtained by PI chart review.
Biologic analyses
Immunohistochemistry for the poliovirus receptor (CD155) on pretreatment tumor tissue was performed in a Clinical Laboratory Improvement Amendment-certified laboratory, and CD155 expression was assessed and quantified by a trained dermatopathologist (figure 1).11 Stool samples were collected on patients 1–5 on post-treatment days 10, 22, 43, and 63 after PVSRIPO injection if they remained on study to test for viral shedding. Because the first five patients had no evidence of viral shedding in the stool, an amendment was approved for only one stool collection and analysis at day 10 after first injection for the remaining patients. Patient 6 had stool collected on days 10 and 21 due to timing of the amendment and patients 7–12 had stool collected only on day 10 from first PVSRIPO injection.

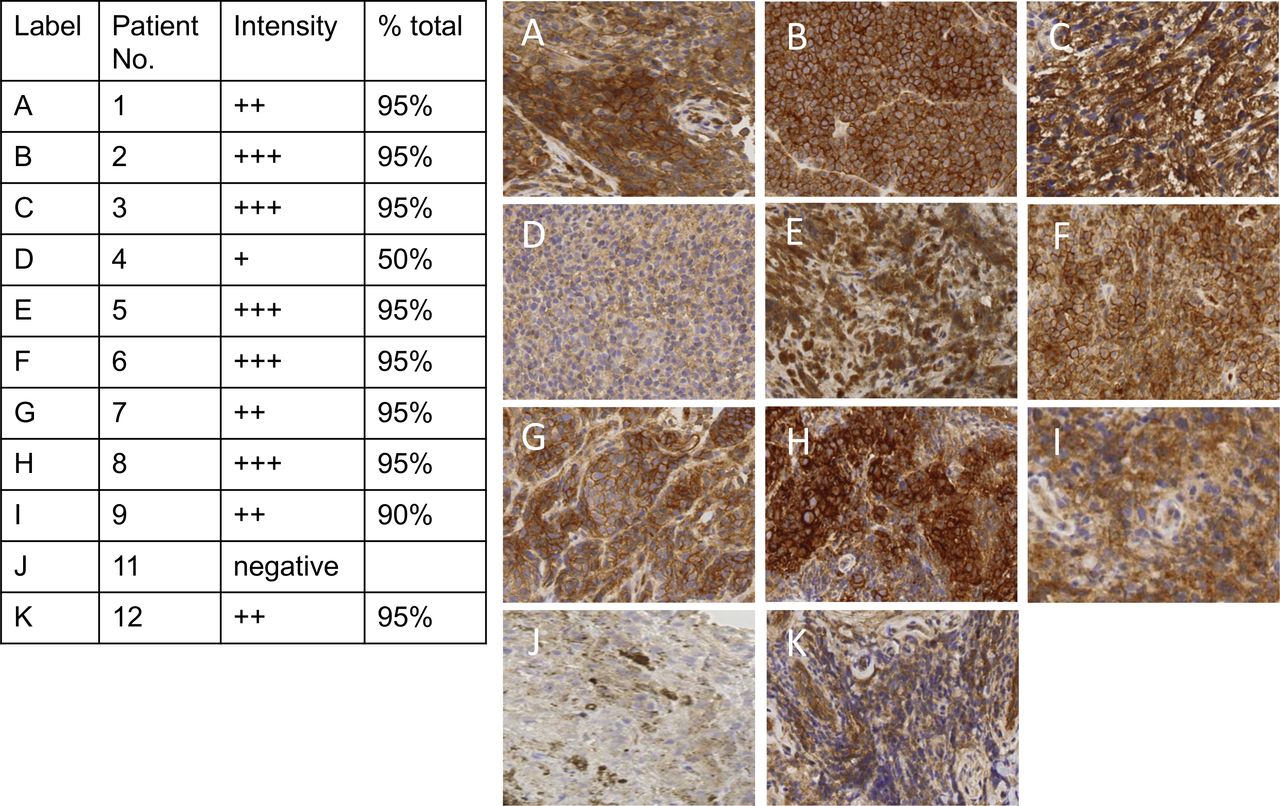
CD155 expression in melanoma tissue. Immunohistochemistry staining for the poliovirus receptor (CD155) was conducted on pretreatment tumor tissue for patients with available tumor biopsies. No tissue was available for patient 10. In patient 11 (J), the sample consisted of abundant macrophages; image shows the magnified region of the small region of viable tumor from a punch biopsy, which is negative for CD155. The brown staining in patient 11’s tumor is melanoma pigmentation and not CD155 staining. The intensity of the stain as reported by a pathologist (+lowest intensity to +++highest intensity) is listed in the table and the percent viable tumor on the slide staining positive is reported as % total.
Statistical analysis
The primary objective was to characterize the safety and tolerability of PVSRIPO in patients with AJCC V.7 unresectable stage IIIB, IIIC, or IV melanoma, as determined by the proportion of patients experiencing DLT.18 Exploratory objectives were to describe response rates of injected and non-injected lesions by following measurements of lesions every 3 weeks and to determine pathological response from tumor biopsies after treatment with PVSRIPO. Patients were continuously monitored for toxic effects during the study using Common Terminology Criteria for Adverse Events V.4.03. DLT was defined as any G4 or higher non-hematologial or hematological toxicity probably, possibly or definitively related to PVSRIPO with the exception of vitiligo. All 12 patients who received at least one injection of PVSRIPO were included in the safety analyses. All toxicities were tabulated by type and grade.
Results
Twelve patients in four cohorts were enrolled and treated over 17 months between November 2018 and March 2020 (table 1). The median age was 66 years (range 38–80); 50% (n=6) had stage IV disease with 50% (n=6) having stage IIIC melanoma. Seven of the 12 patients (58%) were considered to have primary resistance to anti-PD-1 therapy, while 4 (33%) were considered to have secondary resistance, as classified according to the Society for Immunotherapy of Cancer (SITC) taskforce consensus.4 One patient (patient 3) was felt to have had inadequate exposure, due to limited treatment duration secondary to rapid disease progression, to determine type of anti-PD-1 resistance. Additional patient characteristics of patients enrolled and treated are summarized in table 1.
Patient characteristics and treatment response
All patients received all PVSRIPO injections as planned. Injected lesions included subcutaneous metastases (n=8), superficial palpable lymph nodes (n=3), and one external iliac lymph node injected using ultrasound guidance. Only two patients on the study had M1b disease, and no visceral lesions were injected. The median time from patient consent to study end for all patients (n=12) during which safety events were captured was 75 days. The median time after the last injection to study end, during which safety events were captured, was 39 days for patients receiving one injection (n=3), 21 days for patients receiving two injections (n=3), and 31.5 days for the six patients who received three injections.
Treatment was well tolerated with no SAEs or DLTs. Table 2 lists the frequency of AEs, stratified by all AEs reported during the study period, and treatment-related AEs attributed as possibly, probably or definitely related to PVSRIPO treatment. Only G1 and G2 toxicities were observed with no G3 or higher toxicities. G1 localized pruritus was the most common toxicity, seen in 58% (8/12) of patients. Viral shedding in stool was not detected in any patient.
Adverse Events (AEs)
Expression of the poliovirus receptor CD155 on tumor cells was determined in 10 of the 11 patients with available pretreatment tumor biopsies (figure 1). No tumor tissue was available for patient 10. Patient 11 had tumor that was negative for CD155 expression. However, the tumor biopsy for patient 11 had just a small area of viable tumor, such that minimal tumor was present on the slide to test for CD155. However, there was an abundance of pigment-laden macrophages on the slide for patient 11 which did express CD155.
Of 12 patients, 4 (33%) achieved an objective response per irRC; these four patients were all in cohort 2 or 3 and thus received three injections of PVSRIPO as listed in table 1. Of the four patients with objective response, two (patients 8 and 9) had three separate lesions injected (each lesion once), while two (patients 11 and 12) had one lesion injected three times. Table 3 describes the types of lesions injected and response in injected and non-injected lesions for these four patients evaluable by irRC. Among the four patients with in-transit disease (patients 5, 6, 9, and 12), we observed pathological complete responses (pCRs) (ie, no viable tumor detected in injected and non-injected lesions biopsied) in two patients. In 67% (8/12) of patients, no clinical benefit from PVSRIPO was observed (as assessed by the PI of the clinical study). These eight patients who did not appear to have a clinical benefit after PVSRIPO were taken off study early and were therefore unable to have disease progression confirmed by irRC criteria because they received additional therapies before the 4-week confirmation period (table 1). A history plot of prior and subsequent therapies as well as timing of these therapies is shown in figure 2.
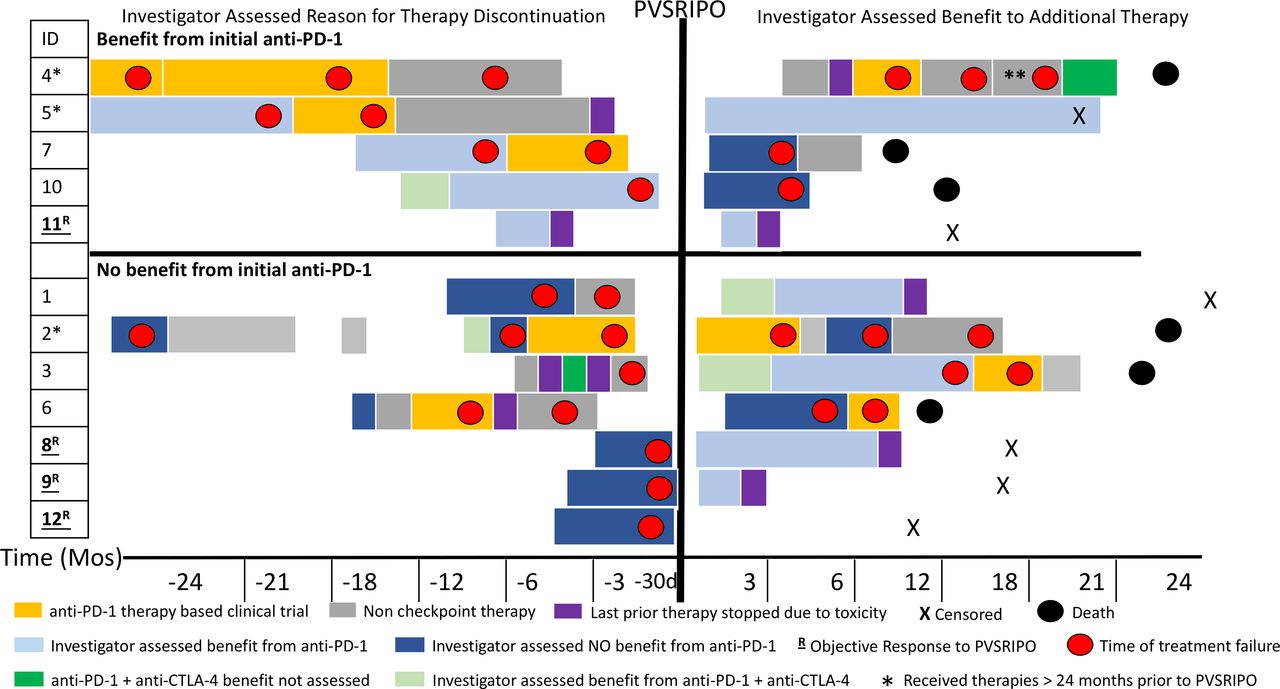
Event history plot. Visual depiction of patients’ therapies prior to and after PVSRIPO. Patients are grouped on the basis of physician-assessed benefit of initial anti-PD-1 therapy, noted on the left side of the panel. Left top panel shows patients who had an initial benefit from anti-PD-1 (programmed cell death protein 1) therapy while left bottom panel shows patients who did not have initial benefit from anti-PD-1 therapy. The X-axis shows time in months before PVSRIPO and then months after PVSRIPO except where d is indicated. The duration of treatment is indicated by length of the block,;red dots indicate time of disease progression. Light blue is assessed benefit to PD-1; dark blue is no benefit to PD-1; dark green is PD-1 plus anti-CTLA-4 (cytotoxic T -lymphocyte-associated protein 4) therapy for which response could not be assessed (limited time or limited exposure); light green is benefit from PD-1 plus anti-CTLA-4 therapy; and purple indicates therapy stopped due to toxicity. Patients were censored (X) and deceased (black circle). Non-checkpoint therapies included BRAF (v-raf murine sarcoma viral oncogene homolog B1)/MEK (mitogen-activated protein kinase kinase) inhibition, radiation, T-VEC, and surgery. Orange indicates other clinical trials in which patients were enrolled that included anti-PD-1 as part of the experimental regimen. The single asterisk (patients 4, 5, and 2) indicates patients who received therapies for the treatment of metastatic melanoma greater than 24 months prior to PVSRIPO. The double asterisk indicates patient 4 was on anti-PD-1-based therapy plus chemotherapy. Patient number in the trial is listed under ID; patients underlined and bolded with R are those who had an objective response to PVSRIPO. d, day; PD-1, programmed cell death protein 1; T-VEC, talimogene laherparepvec.
Response details in patients evaluable by irRC
At time of last follow-up for all patients (range 10–25 months), 11 of 12 patients (92%) treated with PVSRIPO had gone on to receive additional immune checkpoint inhibitor therapy. Subsequent therapies after PVSRIPO are listed in table 1 and depicted in figure 2. Six patients died during the follow-up period related to progressive metastatic melanoma. At the time of data cut-off (median 18 months), 6 of 12 patients (50%) currently remain alive without evidence of further disease progression (table 1). Below are clinical details for two patients with confirmed responses to PVSRIPO.
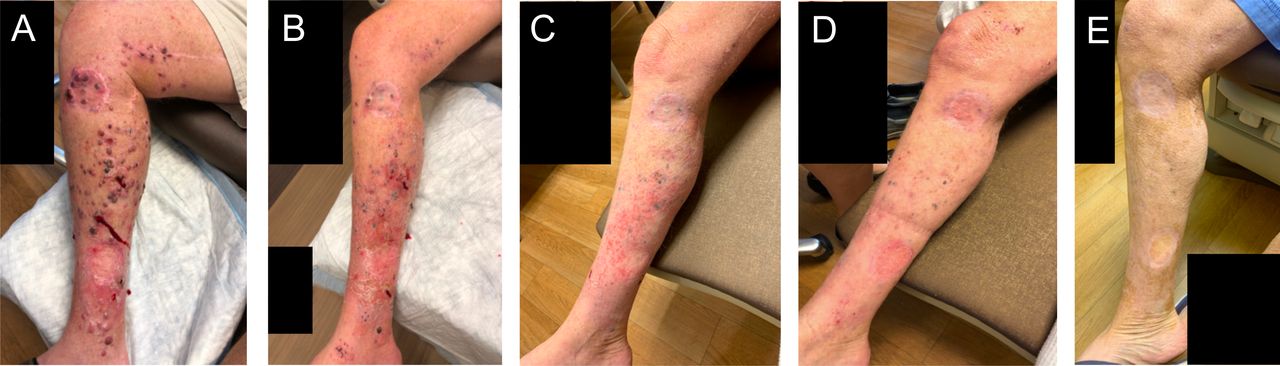
Patient 9 presented with extensive in-transit disease of the right lower extremity without any other metastatic disease as shown in figure 3A. He had previously received four cycles of nivolumab with clinical progression and an extensive whole-body rash, which was worst on the chest and extremities. The patient was enrolled to cohort 2 and received three PVSRIPO injections into three different lesions on the lower extremity 15 days after the patient’s last cycle of nivolumab. Nine days after the first injection, both the injected and non-injected lesions appeared to regress as shown in figure 3B. The patient’s lesions continued to decrease in size consistent with an immune-related response criteria-confirmed partial response (irPR). On day 63 after the first PVSRIPO injection (figure 3C), pathological analysis of biopsies taken of both an injected lesion and a non-injected lesion showed melanophages only, without viable tumor, consistent with pCR. Following study completion, the patient then went on to receive an additional cycle of nivolumab but then discontinued nivolumab therapy, as the rash returned; the patient declined additional treatment. At the time of last follow-up, 16 months after the first injection of PVSRIPO, the patient continues to have scattered, flat, pigmented lesions remaining which have been stable (figure 3D,E); his clinical picture is consistent with treated disease and there is no other metastatic disease on serial whole-body imaging.

Patient 9 clinical photographs. (A) Pre-PVSRIPO, (B) 9 days after first PVSRIPO injection, (C) 63 days after first PVSRIPO injection, (D) 5 months after first PVSRIPO injection, and (E) 12 months after first PVSRIPO injection.
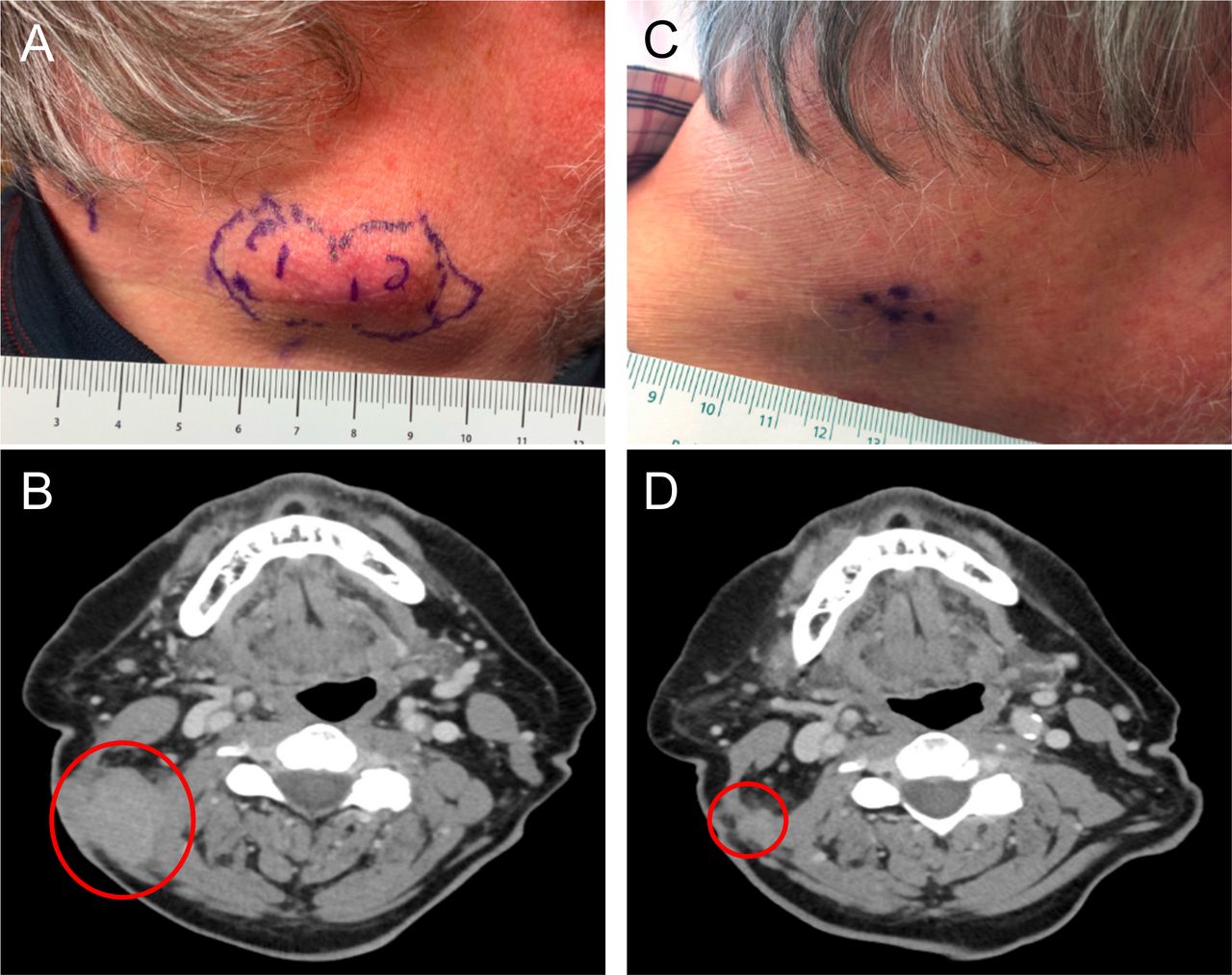
Patient 8 presented with melanoma of unknown primary with dermal and regional lymph node metastases. Prior to enrolling on study, he was noted to have clinical progression after initially receiving three cycles of nivolumab and then switching to three cycles of pembrolizumab due to an infusion reaction (figure 4A,B). The patient was then enrolled in the current study in cohort 2 and received three injections of PVSRIPO into three different lesions. Approximately 10 days after the first PVSRIPO injection, the injected lesion appeared to be regressing. At day 63, the patient had >75% reduction in all index lesions as shown in the photos and CT scans in figure 4C,D. The irPR was confirmed 3 weeks later with CT scan. Because the patient had residual disease, he was taken off study before the 4-week confirmation of irPR in order to resume systemic therapy. The patient subsequently received four cycles of pembrolizumab and remains without progression 17 months from PVSRIPO treatment.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Patient 8 clinical photographs and corresponding CT scans. (A) Photograph of lesions pre-PVSRIPO treatment; the subcutaneous lesions to be injected are outlined in purple. (B) Pre-PVSRIPO treatment CT scan; the red circle outlines subcutaneous lesions. (C) Photograph of lesions 63 days after first PVSRIPO injection. (D) CT scan 63 days after first PVSRIPO injection; the red circle outlines remaining subcutaneous lesions.
Three patients (patient 8, 9, and 11) with objective responses to PVSRIPO were taken off study and reinitiated on anti-PD-1 therapy as part of standard of care. Patient 12 (immune related stable disease (irSD) and pCR) did not resume anti-PD-1 therapy. Currently, all four of the patients with objective responses to PVSRIPO remain without progression as listed in table 1 and depicted in figure 2. Of the eight patients with no clinical benefit after PVSRIPO (as assessed by clinical study PI), all received additional treatment after going off-study. Subsequent treatments for eight of these patients consisted of checkpoint inhibitor therapy, including anti-PD-1 therapy (n=4), anti-PD-1 plus anti-CTLA-4/ipilimumab (n=2), and anti-PD-1 plus other investigational agents (n=2). Of those eight patients, two currently remain without disease progression at a median follow-up of 22.5 months (figure 2). After receiving checkpoint inhibitor therapy, additional treatments pursued by the eight patients who did not have objective responses from PVSRIPO included targeted therapy (n=2), radiation (n=2), talimogene laherparepvec (n=2), chemotherapy (n=1) or additional clinical trials for other investigational agents (n=2) (table 1). In total, 6 of 12 (50%) patients treated in the trial currently remain without progression at the median follow-up 18 months.
Discussion
This study demonstrated that the intratumoral injection of PVSRIPO is feasible and safe in patients with unresectable stage IIIB/C and limited stage IV melanoma. The safety profile of PVSRIPO compares favorably with other intralesional therapies, with the most common AE being G1 localized pruritus occurring in approximately 60% of patients; importantly no G3 or higher AEs were observed in this study.21 Additionally, early signs of clinical activity were seen, as 4 of 12 patients had an objective response, despite the limited number of PVSRIPO treatments relative to the overall lesion burden (67% patients>5 lesions).
Oncolytic viruses are increasingly being used for the treatment of many cancers.7 22 23 Given these encouraging responses and the relatively favorable safety profile of local tumor injections, the development of additional, mechanistically refined recombinant viruses for clinical use in melanoma is ongoing.22 24 PVSRIPO targets a single host cell receptor that is widely upregulated in solid neoplasias, CD155.11 Indeed, 10 of 11 patients with available tumor had expression of CD155 confirmed in this study (figure 1). The patient with negative CD155 expression (patient 11) had only a small area of viable tumor obtained from the punch biopsy. However, there were abundant pigment-laden macrophages on the pre-PVSRIPO tumor biopsy for patient 11. These pigmented macrophages may have signified ongoing effects from prior anti-PD-1 therapy, even though the last dose of anti-PD-1 therapy was 90 days prior to PVSRIPO.25 Additionally lack of CD155 expression on tumor cells has been associated with responsiveness to anti-PD-1 therapy.12 26 However, the abundance of CD155 expressing macrophages may have also contributed to response from PVSRIPO given that in preclinical models, PVSRIPO causes non-lethal infection of tumor associated macrophages,15 16 and emerging data show this may be the primary mechanism associated with PVSRIPO’s antitumor response.16
Despite encouraging results from anti-PD-1 therapy for patients with metastatic melanoma, the majority of patients do not respond to anti-PD-1 therapy initially or subsequently develop acquired resistance.27 Dual checkpoint inhibitor therapy with anti-CTLA-4 plus anti-PD-1 results in higher response rates, but carries substantial toxicity, with a high percentage of patients experiencing clinically significantG3/G4 AEs.1 Given resistance and toxicity, additional therapeutic options are clearly needed which balance efficacy and risk. One promising strategy to overcome resistance to anti-PD-1 is to consider combination approaches with intratumoral oncolytic virus therapy.7 28 Encouraging early data suggest the potential for synergism between intratumoral oncolytic virus therapy in combination with systemic therapy, whereby oncolytic virus therapy can promote immune responses within the TME that may overcome resistance to anti-PD-1 therapy.7 29 30 Preclinical studies in murine melanoma models indicate that PVSRIPO induces activation of tumor antigen specific cytotoxic T lymphocytes.15 Intratumoral delivery of PVSRIPO could ultimately be an effective strategy to support sustained antitumor responses alone or in combination with systemic anti-PD-1 therapy by converting ‘cold’ tumors to ‘hot’ tumors characterized by active engagement of antitumor immune responses.15 16
While multiple strategies to overcome anti-PD-1 resistance are ongoing, defining and characterizing resistance to anti-PD-1 can be complicated in some patients.31 Given the complexity of defining response and resistance in these patients, recent SITC guidelines have been established to provide a standard definition for anti-PD-1 resistance.4 These guidelines, developed and published during the current study time period and therefore not available during trial design, include recommendation of confirmatory imaging of anti-PD-1 progression 4–12 weeks after the first observation of progression to account for pseuodoprogression. In our study, failure of anti-PD-1 therapy as needed for study entry was determined by clinical judgment of treating providers, and confirmation of progression on anti-PD-1 therapy 4–12 weeks after the first observation was not required.
Five patients in this study received PVSRIPO within 30 days of their last dose of anti-PD-1 therapy and therefore did not have confirmatory observation of anti-PD-1 progression prior to trial entry; this includes three of the four patients (patients 8, 9, and 12) who had an objective response after PVSRIPO. Thus, prior anti-PD-1 therapy (including possible pseudoprogression) could have contributed to response noted in patients 8, 9, and 12. While this potentially limits interpretation of the effects of PVSRIPO, pseudoprogression is only thought to occur in about 10% of patients; thus, it would be unlikely that pseudoprogression occurred in 3 of 12 patients (25%) in this study.
All four patients who achieved an objective response in our study received three injections of PVSRIPO. Two received three injections into a single lesion only, while the other two responders had three separate lesions injected (each lesion injected once). Interestingly, all four of these patients began to have clinical evidence of tumor regression approximately 10 days after the first PVSRIPO injection. The optimized dosing regimen for PVSRIPO in unresectable mucosal or cutaneous melanoma is being examined in an ongoing multicenter phase II study; this includes injecting multiple lesions per dosing cycle and repeated dosing with PVSRIPO every 3–4 weeks. Patients with confirmed anti-PD-1 refractory unresectable melanoma will be randomized to receive PVSRIPO alone (to determine contribution of effect/optimized dosing) or in combination with anti-PD-1 therapy (see clinicaltrials.gov). This trial requires confirmation of disease progression on anti-PD-1 therapy in concordance with SITC guidelines prior to randomization.4
While patients with stage IV disease were included in the current study, only two patients had M1b disease, both of whom had progression of visceral (non-injected) lesions and died at 9 months and 23 months after PVSRIPO therapy. This phase I trial is therefore limited in evaluating activity in non-injected visceral lesions, although response was observed in non-injected subcutaneous and nodal lesions (eg, patient 9; see figure 3). Other limitations include the small sample size, as this was primarily a safety and feasibility study. Thus, while our results suggest that PVSRIPO may have benefit for patients with refractory melanoma, the role of injectable therapies is still being defined, particularly in the setting of anti-PD1 and CTLA-4 checkpoint inhibitors.
Conclusion
In this phase I trial, intratumoral PVSRIPO was well tolerated; encouraging antitumor responses were observed. The favorable safety profile demonstrated in this study, along with the antitumor responses, suggests that PVSRIPO, either alone or in combination with anti-PD-1, may be an effective treatment in anti-PD-1 refractory melanoma.
Data availability statement
All data relevant to the study are included in the article or uploaded as supplemental information.
Ethics statements
Ethics approval
Because of conflict of interest and intellectual property considerations, an external data and safety monitoring board oversaw the conduct of the study. The study was approved by the institutional review board at Duke University and the Western institutional review board. The study was conducted in accordance with the provisions of the Declaration of Helsinki and the Good Clinical Practice guidelines of the International Conference on Harmonization. All patients provided written informed consent.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Presented at Presented in part at Society for Immunotherapy of Cancer 35th Annual Meeting, November 9–14, 2020.
Contributors GMB, SKN, NF, KL, ATK, MG, DDB, and AKS made substantial contributions to the conception and design of the work. GMB, SKN, NF, KL, SHJ, AS, CAW, DDB, MG, ATK, and AKS made significant contributions to data acquisition, analysis, and interpretation of data for the work. GMB, SKN, NF, ATK, MG, and AKS made significant contributions to drafting the manuscript and critical revision for important intellectual content. All authors gave the final approval of the version to be published. All authors were accountable to ensure that questions related to the accuracy or integrity of any part of the work were appropriately investigated and resolved.
Funding The clinical trial was funded by Istari Oncology. GB is supported by National Institutes of Health (NIH) (NIHK08 CA237726-01A1) and the Duke Cancer Institute’s Pilot Grant (P30 Cancer Center Grant NIH CA014236). SKN was supported by the Department of Defense Breast Cancer Research Program award (W81XWH-16-1-0354). NF was supported by an NIH T-32 grant (T32-CA093245) for translational research in surgical oncology and received support from a Polka Dot Mama Melanoma Foundation research grant.
Disclaimer GMB has clinical trial funding from Istari Oncology, Delcath, Oncosec Medical, Replimune, and Checkmate Pharmaceuticals paid to Duke University. AS has research funding paid to the institution from Bristol Myers Squibb, Immunocore, Merck, Pfizer Advisory roles: GB served on an advisory board for Regeneron and Cardinal Health. AKS has served on advisory boards for Array, Novartis, Iovance, and Regeneron.
Competing interests SKN owns intellectual property related to this research, which has been licensed to Istari Oncology, Inc. DDB and MG have financial interest in Istari Oncology, Inc. Duke University (Licensor of PVSRIPO) has a financial interest in Istari Oncology, Inc.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.