Article Text

Abstract
The ongoing pandemic caused by the novel coronavirus SARS-CoV-2 has disrupted the global economy and strained healthcare systems to their limits. After the virus first emerged in late 2019, the first intervention that demonstrated significant reductions in mortality for severe COVID-19 in large-scale trials was corticosteroids. Additional options that may reduce the burden on the healthcare system by reducing the number of patients requiring intensive care unit support are desperately needed, yet no therapy has conclusively established benefit in randomized studies for the management of moderate or mild cases of disease. Severe COVID-19 disease is characterized by a respiratory distress syndrome accompanied by elevated levels of several systemic cytokines, in a profile that shares several features with known inflammatory pathologies such as hemophagocytic lymphohistiocytosis and cytokine release syndrome secondary to chimeric antigen receptor (CAR) T cell therapy. Based on these observations, modulation of inflammatory cytokines, particularly interleukin (IL)-6, was proposed as a strategy to mitigate severe disease. Despite encouraging recoveries with anti-IL-6 agents, especially tocilizumab from single-arm studies, early randomized trials returned mixed results in terms of clinical benefit with these interventions. Later, larger trials such as RECOVERY and REMAP-CAP, however, are establishing anti-IL-6 in combination with steroids as a potential option for hypoxic patients with evidence of hyperinflammation. We propose that a positive feedback loop primarily mediated by macrophages and monocytes initiates the inflammatory cascade in severe COVID-19, and thus optimal benefit with anti-IL-6 therapies may require intervention during a finite window of opportunity at the outset of hyperinflammation but before fulminant disease causes irreversible tissue damage—as defined clinically by C reactive protein levels higher than 75 mg/L.
- cytokines
- COVID-19
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
More than a year since the first case report of an atypical pneumonia caused by the novel coronavirus SARS-CoV-2 the COVID-19 pandemic continues to rage unabated, surpassing 1.5 million deaths worldwide in early December 2020.1 The case fatality rate varies by region, with the most deaths occurring in people over 70 years of age.2 Even though the mortality rate in the USA is less than 2%, almost 200,000 excess deaths were attributed to COVID-19 through October, 2020,3 and current trends indicate that the human toll will only continue to increase. Although vaccines showed promising results in topline results from phase III trials reported in late 2020, distribution infrastructure is lacking, and several months may still elapse before a sufficient proportion of the population is inoculated to achieve herd immunity. COVID-19 is an ongoing emergency for the global community, and it is imperative to identify treatments to alleviate the burden on the healthcare system. Furthermore, although COVID-19 has disrupted all aspects of modern life, the pandemic also has important consequences for cancer care—delaying or interrupting treatments, halting clinical trials, and dramatically reducing routine screening.4 Conflicting evidence has emerged whether cancer was associated with worse outcomes,5 6 or if increased mortality among patients with malignancies is due to covariant factors such as smoking.7 Regardless, effective therapies for COVID-19 that may be integrated into anticancer regimens are needed.
The primary driver of organ damage in COVID-19 is an ongoing area of investigation, but likely a combination of direct tissue necrosis due to uncontrolled viral replication as well as the fulminant systemic inflammatory pathology and an accompanying coagulopathy instigated by SARS-CoV-2 infection both contribute to severe disease. Across studies, the most consistent predictors of mortality in COVID-19 are advanced age, male sex, and the presence of comorbidities such as diabetes, hypertension, and obesity.8–16 A recent pair of papers showed that defective interferon (IFN) signaling is associated with severe disease—either through inborn variation at key loci or the presence of autoantibodies to type I IFNs.17 18 Additionally elevated neutrophil to lymphocyte (NLR) ratio, high serum C reactive protein (CRP), and increased interleukin (IL) 6 have consistently been linked to severe disease.19–23
COVID-19 pneumonia is characterized by an acute respiratory distress syndrome (ARDS), accompanied by a hypercytokinemia with similarities to cytokine release syndrome (CRS) secondary to chimeric antigen receptor (CAR) T cell therapy as well as hemophagocytic lymphohistiocytosis (HLH)/macrophage activation syndrome (MAS).24–29 Based on the observed inflammatory pathology, immune-modulatory strategies, especially targeting IL-6, were proposed as a potential intervention to break the inflammatory cycle.30 Although uncontrolled studies seemed to demonstrate miraculous recoveries with anti-IL-6 therapies, randomized trials returned more mixed results. It is becoming clear that IL-6 modulation in COVID-19 is not a “magic bullet.” However, some patients may benefit with anti-IL-6, emphasizing a need for readily available biomarkers to identify candidates for intervention. This review will contextualize the results of the published accounts of anti-IL-6 therapy for COVID-19 through the lens of the monocyte-driven and macrophage-driven hyperinflammatory pathology. Extrapolating from published trials and mechanism-based insights into the pathology of the hyperinflammatory state, a potential “window of opportunity” is defined, clinically identifiable as CRP levels above 75 mg/L, during which cytokine modulation may provide maximal benefit.
Clinical course and inflammatory pathology of SARS-CoV-2 infection
COVID-19 may present as a silent asymptomatic infection, a mild upper-respiratory tract illness, or severe disease characterized by fulminant inflammation, systemic coagulopathies, and severe damage to the lungs and cardiovascular system.2 8 16 31–34 Evidence is also emerging that SARS-CoV-2 infection may occasionally cause long-term and sometimes debilitating symptoms, including fatigue, dyspnea, joint pain, and chest pain.35 Additionally, a delayed-onset inflammatory vasculitis with features akin to Kawasaki shock syndrome has been reported in children whose primary disease was asymptomatic or minimally symptomatic.36 37 Much remains unknown about the mechanisms responsible for the dramatic variations in disease presentation and long-term sequalae. However, since the first reported cases emerged in 2019, the infectious disease and immunology communities have gained some insight into correlates of severe disease and mortality.
Pulmonary failure secondary to ARDS is the primary cause of death in severe COVID-19.8 14 33 38 Almost all cases of severe COVID-19 present with bilateral lung involvement, with characteristic ground glass opacities visible on CT imaging.34 39 Multiple phenotypes on CT imaging have been described,40 41 and the different patterns correlate with disease severity. The signature findings in severe COVID-19 pneumonia, namely, intra-alveolar edema, fibrin, and variable cellular infiltrates with a hyaline membrane, have been shown to associate with systemic inflammation, including elevated creatine kinase levels, increased neutrophil percentage, and high serum CRP.42 Autopsies have revealed pronounced mononuclear inflammatory infiltrates in the lungs,8 43 44 microthrombi present in alveolar microvasculature as well as vessels of the heart, liver and kidneys,45 46 as well as evidence of hemophagocytosis in the bone marrow.47 48 T cell counts are dramatically reduced in severe cases, and high lymphocyte expression of PD-1 is seen, which increases along with TIM-3 as disease severity progresses.49
SARS-CoV-2 tissue tropism and pathological features of infection
Angiotensin-converting enzyme 2 (ACE2) is the major entry receptor for SARS-CoV-2.50 51 Emerging evidence suggests the virus may be able use additional targets, such as transferrin,52 however, the biological relevance of alternate entry receptors remains unknown. Cell surface engagement occurs through the receptor binding domain of the SARS-CoV-2 spike protein, and cleavage by host proteases, including TMPRSS2, lysosomal cathepsins, and furin, is essential for viral entry.50 51 53 ACE2 mRNA is found in almost all cell types, with especially high levels in renal, cardiovascular, and gastrointestinal system tissues,54 whereas protein expression is predominant on lung alveolar epithelial cells, enterocytes of the small intestine, as well as arterial and venous endothelial cells and arterial smooth muscle cells.55 ACE2 is a key component of the renin-angiotensin-aldosterone system (RAAS) as the catalytic enzyme that cleaves angiotensin II (Ang II) into angiotensins 1–7 (Ang1–7).56 In addition to the well-established role of the RAAS in maintaining blood vessel tone, Ang II modulates numerous responses in local tissues, such that disruptions in ACE2 are associated with a number of inflammatory conditions, including atherosclerosis, heart failure, and chronic kidney disease.57
The potential tissue tropism of SARS-CoV-2 is vast given the widespread distribution of its entry receptors throughout the body, and possibly even further expanded through spike protein pre-activation by furin to overcome a requirement for cell-surface TMPRSS2.51 SARS-CoV-2 viral particles have been observed in renal, intestinal, cardiac, and lymphoid tissues.58 SARS-CoV-2 has been demonstrated to directly infect type 2 pneumocytes and ciliated airway epithelial cells,48 59 60 and viral RNA persists in lung endothelium and pneumocytes for several weeks after diagnosis.61 Cytopathic effects underlie some of the diffuse alveolar damage found in the lungs of patients with severe COVID-19. However, mononuclear inflammatory infiltrates dominated by monocytes and macrophages and lacking eosinophils are also characteristic of severe disease.8 43 44 62–64 Although it is difficult to untangle the relative contributions of uncontrolled viral replication and aberrant immune reactions on COVID-19 mortality, it is becoming apparent that a damaging inflammatory pathology initiated in the lower respiratory tract eventually causes systemic tissue damage, disseminated coagulation, and multi-organ failure.
Cytokine storm in COVID-19
“Cytokine storm” is a catch-all term that has used to describe a range of dysregulated immune reactions with a variety of instigating events ranging from sepsis to stem cell transplant.24 In recent years, the term has come to most commonly be associated with CRS after CAR T cell therapy—defined clinically by fever, hypoxia, and hypovolemia65 and characterized by profoundly elevated serum levels of CRP, IL-6, IL-10, and IFNγ.66–69 CAR T cell-associated CRS shares several features with secondary HLH/MAS, in which serum levels of IFN‐γ, tumor necrosis factor (TNF)‐α, IL‐1β, IL‐2, IL‐6, IL‐12, IL‐16, and IL‐18 are elevated.70 71 Even though the levels of IL-6 measured in patients infected with SARS-CoV-2 are on the order of 10-fold lower than those seen in patients treated with CAR T cells, the hypercytokinemia seen with severe COVID-19, CRS, and HLH/MAS substantially overlap.8 25
The incidence of primary HLH, an autosomal recessive disorder, is roughly 1 in 100,000 live births. Secondary, or reactive, HLH/MAS may be triggered by infections (especially Epstein-Barr virus, CMV and HIV), autoimmune diseases, and some cancers. As many as 1% of patient with hematological cancers develop HLH, and mortality rates associated with this presentation approach 80%.72 73 Viral HLH occurs when persistence of infected cells causes cytotoxic T lymphocytes and NK cells to continuously secrete inflammatory cytokines thus activating macrophages and resulting in a vicious cycle of IL-6 secretion, activation and an out-of-control positive feedback loop.70 Although IL-6 is essential for both innate and adaptive immunity, uncontrolled secretion leads to inflammatory damage to host cells.74 In severe COVID-19, IL-6 has been shown to be central among a network of 12 cytokines that are elevated in cases with poor prognoses.75
Although the presence of a cytokine storm in COVID-19 has been demonstrated many times over, the definition of “hyperinflammation” has varied substantially. Multiple studies have set out to define the threshold levels of various laboratory parameters that are associated with severe outcomes or mortality, and some meta-analyses have attempted to define threshold levels with prognostic power.76 Because IL-6 measurements are not readily available in many centers, however, cytokine measurements have limited utility for decision-making. Hyperinflammation at admission defined as CRP >150 mg/L or doubling within 24 hours from greater than 50 mg/L, or a ferritin concentration greater than 1500 μg/L was shown significantly increase the risk of next-day escalation of respiratory support or death (hazard ratio (HR) 2.24; 95% CI 1.62 to 287) in one cohort.77 Additionally, a six-criteria clinically based scoring criteria for the COVID-19 hyperinflammatory state (cHIS) has been proposed and validated,78 which shares several parameters with the scoring system for secondary HLH.79 The cHIS and HLH scores are summarized in table 1.
Comparison of scoring systems for COVID-19 hyperinflammation and HLH
Viral HLH/MAS represents an overexuberant failsafe mechanism when primary protection measures do not clear infection. A similar process may be occurring in the COVID-19 cytokine storm. Recent evidence for defective type I IFN signaling as risk factor for severe disease in COVID-1917 18 further underscores the notion that a failure of fist-line antiviral defenses leads to poor outcomes. Coronaviruses, like many other viruses have been shown to directly promote IL-6 production in immune and epithelial cells.80 81 Although the hyperinflammation in COVID-19 ARDS eventually affects the entire body, accumulating evidence implicates the myeloid lineage, specifically monocytes and macrophages, in driving severe disease.63 82 83
Contribution of monocytes and macrophages to COVID-19 inflammatory pathology
Multiple mechanisms have been proposed to account for the hyperinflammatory state seen in severe COVID-19. Pyroptotic death of infected cells, leading to release of IL-1β along with danger-associated molecular patterns could contribute to inciting an inflammatory cascade.84 Additionally, stem cell-derived type 2 alveolar cells infected with SARS-CoV-2 have been shown in vitro to upregulate expression of NF-κB target genes, including IL6, CXCL8, CXCL2, CXCL3, CXCL10, and CXCL11.85 The viral spike protein itself has been proposed to possess superantigenicity, potentially inciting systemic cytokine release through major histocompatibility complex (MHC)-independent T cell activation.86 87
Emerging evidence, however, suggests a prominent role for monocytes and macrophages in the hyperinflammatory pathology that causes COVID-19 ARDS. Patients with severe COVID-19 have dramatically reduced numbers of CD4+ and CD8+ T cells, especially those requiring intensive care unit (ICU) care, and lymphocyte counts negatively correlate with levels of serum IL-6, IL-10, and TNF-α.49 Monocytes have been shown upregulate expression of IL-6 in response to spike protein from 2003 pandemic SARS-CoV.81 Monocyte-derived IL-6 has been also shown to drive CRS after CAR T cell therapy.88 Alveolar macrophages potently produce IL-6, and epithelial cells upregulate its expression in response to interferon gamma and danger-associated molecular patterns.89 Additionally, macrophages and dendritic cells express ACE2,63 90 and interactions with the virus may contribute to the generation of an inflammatory milieu.
SARS-CoV-2 binding to ACE2 on monocytes and macrophages leads to receptor internalization,91 thus enhancing local Ang II and vascular permeability. In the lungs, CD14+CD16+ monocytes produce high levels of Ang1–7 and are thought to be vasoprotective.74 Loss of surface ACE2 on infection could therefore promote local endothelial dysfunction. Activation of the RAAS in monocytes plays a well-established role in the inflammatory and coagulation pathology in acute coronary syndromes.92 Ang II also promotes reactive oxygen species production and the expression of proinflammatory chemokines, leading to local accumulation of immune cells.93 The monocyte compartment is extensively remodeled in COVID-19, with phenotypic shifts in both circulating and tissue-resident populations. Although the original insult of SARS-CoV-2 infection in the lungs may initially target the epithelium, a resulting increased cytokine production by macrophages and perturbations to the RAAS initiate the cascade that leads to ARDS. A model for a monocytic and macrophage-driven inflammatory cascade originating in the alveoli is illustrated in figure 1.
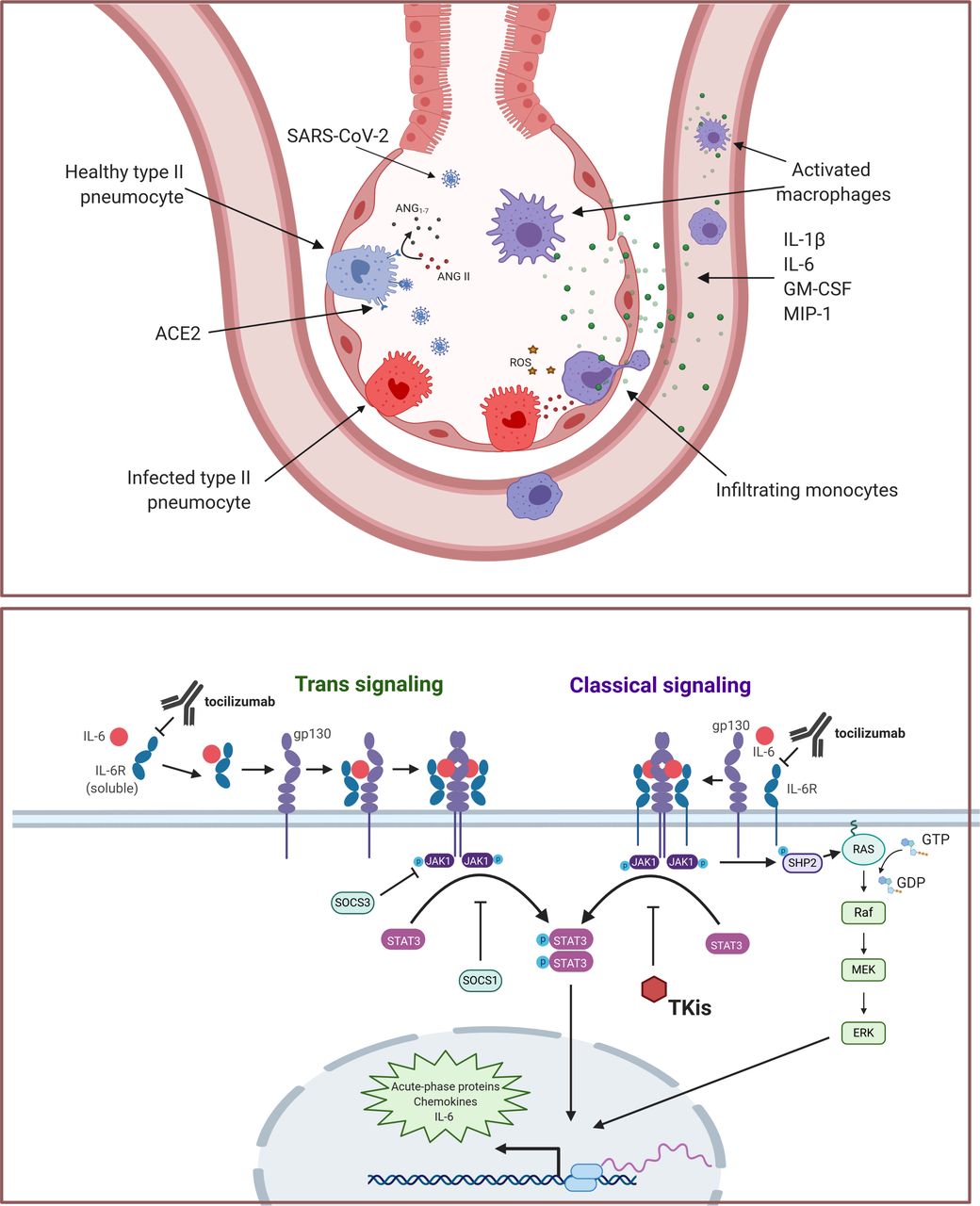
{kind=link}
(A) Monocytic and macrophage contribution to SARS-CoV-2 hyperinflammation. SARS-CoV-2 directly infects pulmonary pneumocytes, causing cell death and the release of danger-associated molecular patterns, activating macrophages. Viral spike protein also triggers release of inflammatory cytokines, including interleukin (IL)-1β, IL-6, granulocyte-macrophage colony-stimulating factor (GM-CSF) and macrophage inflammatory protine (MIP)-1 by alveolar-resident macrophages, promoting monocytic infiltration. Additionally, internalization of ACE2 on viral entry leads to increased angiotensin II, facilitating reactive oxygen species production, tissue damage, and an NF-κB-driven inflammatory gene expression program that includes production of chemokines, cytokines, and IL-6. (B) Contribution of IL-6 signaling to COVID-19 inflammatory cascade. Trimeric complex formation with IL-6, IL-6R and gp130 leads to dimerization and autophosphorylation of JAK1. Phosphorylated JAK1 triggers STAT3 phosphorylation and translocation to the nucleus, initiating acute-phase protein production, including chemokines, cytokines and IL-6. Phospho-JAK1 may also contribute to acute-phase transcription through an extracellular signal-regulated kinase (ERK)-dependent pathway that relies on SHP2 and Ras. Notably, IL-6 also induces expression of SOCS1 and SOC3, which negatively regulate JAK/STAT signaling. The classical signaling pathway is largely restricted to the lymphocyte compartment, where it contributes to adaptive immunity, whereas trans signaling may occur in any tissue type and is generally pro-inflammatory. Interventions that may alleviate inflammatory IL-6 signaling (ie, tocilizumab or tyrosine kinase inhibitors) are shown.
Despite originating in the lungs, the inflammatory cascade set off by SARS-CoV-2 affects immune and stromal cells throughout the body. Elevated fractions of CD14+CD16+ inflammatory monocytes with the capability to secrete granulocyte-macrophage colony-stimulating factor (GM-CSF) and high IL-6 expression have been measured in the peripheral blood of patients with COVID-19.83 Also seen is an increase in circulating Th17 cells in patients with severe disease.44 Eosinopenia, which has now been linked with worse outcomes and poor responses to antivirals as well as to anti-IL-6 for COVID-19,62 94–96 is a well-established signature of inflammatory respiratory pathology.97 98 Eosinophils also play a role in the resolution of inflammation through their secretion of IL-4 and IL-14 along with a variety of lipid-based mediators of macrophage activity,99 suggesting that a failure of type 2 immunity to resolve inflammatory monocyte and macrophage activity may also contribute to the uncontrolled inflammation in COVID-19.
Rationale for targeting IL-6
Elevated IL-6 has consistently predicted mortality and severe outcomes in COVID-19.8 19 20 22 100 IL-6 signaling primarily occurs through the Janus kinase (JAK)/signal transducer and activator of transcription (STAT) pathway, predominantly via STAT3, which induces acute phase proteins, including CRP, amyloid A, and fibrinogen, and promotes differentiation, recruitment, survival and transformation programs in T and B cells and myeloid cells.101–103 JAK also modulates cellular survival through a phosphorylation cascade involving phosphorylates phosphatidylinositol-4,5-bisphosphate (PIP2), PI3K, phosphati-dylinositol-3,4,5-trisphosphate (PIP3), and PkB/Akt serine/threonine kinase. IL-6 signaling may also occur through RAS, RAF, and mitogen-activated protein kinases, culminating in acute phase protein generation.70 IL-6 is positively regulated by NF-kB, CAAT/enhancer-binding protein beta (C/EBPβ), specificity protein 1 (Sp1), cyclic AMP (cAMP) response element-binding protein (CREB), IFN regulatory factor 1 (IRF-1), activator protein 1 (AP-1), and TNF-α.101 A negative feedback loop modulates IL-6 signaling through JAK/STAT, as STAT3 upregulates SOCS1 and SOCS3, which directly inhibit the catalytic activity of JAK.69 Members of the Coronaviridae family may be especially likely to trigger dysregulated IL-6 production, as the 2003 pandemic SARS-CoV not only causes increased IL-6 production in epithelium compared with several influenza strains, but also induces lower levels of expression of the negative regulator SOCS3.104
IL-6 is a highly pleiotropic cytokine with the potential to perturb almost all tissues in the body. Signaling through IL-6 in the central nervous system leading to prostaglandin production has been shown to underly fever under conditions of inflammation.105 Although the membrane-bound form of the IL-6 receptor (IL-6R) is expressed primarily on hepatocytes, megakaryocytes, and leukocytes,103 both alternative splicing and cleavage by the membrane metalloproteases ADAM10 and ADAM17 may generate a soluble form.103 106 107 Signaling requires assembly trimeric complex of IL-6, IL-6R, and the ubiquitously expressed 130-kilodalton signal-transducing b-receptor glycoprotein (gp130, also called CD130).101 102 IL-6 activity through the membrane-bound form of the receptor (termed classical signaling) is generally involved in wound healing and the resolution of inflammation, whereas the interaction between the soluble IL-6R/IL-6 complex and gp130 (trans signaling) is generally considered to be pro-inflammatory.101–103 107
Cytokine modulation for the treatment of hyperinflammation has become an established paradigm for several autoimmune disorders such as rheumatoid arthritis and colitis, and immuno-oncologists have readily adopted some anti-inflammatory agents originally developed for the rheumatology field for the treatment of immune-related adverse events. For example, the TNF-α inhibitor infliximab is commonly used for the management of colitis secondary to checkpoint inhibitor therapy,108 and the IL-6R antagonist tocilizumab was approved by the Food and Drug Administration (FDA) for the treatment of CRS secondary to CAR T cell therapy in 2017 in a companion regulatory decision with tisagenlecleucel.109 Tocilizumab has also been used to treat some cases of checkpoint inhibitor-mediated pneumonitis.110 Administration of tocilizumab not only has anti-inflammatory properties, but the agent improves endothelial function leading to an increase of effective myocardial work through a reduction of inflammatory burden and oxidative stress.111
Based on the central role for IL-6 in the inflammatory pathology of COVID-1975 and a consistent association between elevated levels of the cytokine and severe disease outcomes,10 22 23 25 112 113 cytokine-modulatory therapies were proposed as a strategy to break the cycle of hyperinflammation. Reports from China28 114 and Italy,96 115 regions that were among the hardest-hit during the early months of 2020, seemed to demonstrate miraculous recoveries with anti-IL-6 therapies. Results of subsequent large-scale, randomized trials, however, were mixed, indicating that although IL-6 modulation for the management of COVID-19 may be useful in some circumstances, not all patients will benefit.
Outcomes of anti-IL-6 interventions on COVID-19
During the initial months of the pandemic, a case series that included 21 severe and critical patients from The First Affiliated Hospital of University of Science and Technology of China (Anhui Provincial Hospital) and Anhui Fuyang Second People’s Hospital treated with tocilizumab in early February, 2020, demonstrated fever reduction within the first day after administration, and improvements of other symptoms within 5 days, with 15 of 20 patients (75.0%) lowering O2 intake, radiographic improvement in 19 patients (90.5%), a return to normal ranges of lymphocyte percentages in peripheral blood in 10 of 19 patients (52.6%), and significantly decreased CRP in 16 of 19 patients (84.2%).28 The single-arm TOCIVID-19 study enrolled 300 patients in Italy within 24 hours its launch, and subsequently concluded that tocilizumab reduced 14-day and 30-day mortality compared with a priori expected rates.116 Spurred on by these encouraging findings, numerous subsequent reports emerged of rapid and sustained recoveries in patients with severe COVID-19 after the administration of anti-IL-6 therapies, most commonly the IL-6R antagonist tocilizumab.114 115 117 118
Case studies evaluating sarilumab, another anti-IL-6R therapy also surfaced, describing improvements in respiratory parameters96 and shortened time to clinical improvement in patients with lung consolidation119 with treatment. Interpretation of individual non-randomized studies is challenging, and cross-trial comparisons are difficult, because anti-IL-6 was initiated at different stages during the course of disease and a rapidly evolving standard of care meant that patients may or may not have received a heterogenous assortment of concomitant medications, including steroids and/or antivirals.
Multiple randomized trials have now evaluated IL-6-directed therapies in the management of COVID-19. The phase III study investigating sarilumab was halted early for futility because it did not meet its primary and key secondary endpoints.120 However, topline results have become available from eight randomized trials evaluating tocilizumab: RCT-TCZ-COVID-19, CORIMUNO-TOCI-1, COVACTA, EMPACTA, BACC Bay TCZ, REMAP-CAP, RECOVERY, and TOCIBRAS. Characteristics and key outcomes from randomized trials are summarized in table 2.
Key outcomes from randomized trials evaluating tocilizumab for the treatment of COVID-19
Of the major randomized trials with available results, four met at least one primary endpoint for efficacy. The CORIMMUNO-TOCI-1 study,121 showed significant improvement for reducing the need for ventilation with tocilizumab treatment, despite the fact that the threshold for efficacy for improvement to a score of greater than 5 on the World Health Organization (WHO) Clinical Progression Scale122 was not met and there was no difference in 28-day mortality between the groups. In the EMPACTA trial, which investigated the safety and efficacy of tocilizumab in hospitalized, non-ventilated patients, with an emphasis on enrolling high-risk and racial and ethnic minority populations, tocilizumab was shown to reduce the rate of mechanical ventilation or death by day 28.123
Importantly, the two largest trials with data available both met primary and secondary efficacy endpoints. REMAP-CAP, which randomized patients to tocilizumab, sarilumab, or standard of care within 24 hours of receiving organ support found that tocilizumab and sarilumab were effective not only for the primary outcome of organ support-free days, but also all secondary outcomes, including 90-day survival, time to ICU and hospital discharge, and improvement according to WHO ordinal scale at day 14.124 The RECOVERY trial similarly saw significant reductions in mortality rates as well as met key secondary endpoints such as reduced intubation rate and reduced requirement for dialysis with tocilizumab.125
Although not randomized, the observational retrospective STOP-COVID study is also of note due to its large population size, as well as its stringent consistency with the timing of intervention—all enrolled patients received tocilizumab within 2 days of ICU admission. In the study, early tocilizumab treatment led to reduced risk of death across multiple sensitivity analyzes.126 In the study, the time to death HR with tocilizumab was 0.71 (95% CI 0.56 to 0.92) and the 30-day mortality risk difference (RD) was 9.6% (95% CI 3.1% to 16.0%).
Four randomized studies did not meet their primary outcome endpoints; however, even in these trials, exploratory analyses suggest that anti-IL-6 may provide meaningful benefit for some patients. For example, although the COVACTA trial found no significant difference in clinical status at day 28 between study groups, the incidence of ICU transfer was 23.6% (30/127) in the tocilizumab arm and 40.6% (26/64) in the placebo arm (weighted difference, –17.2%; 95% CI –31.3% to –3.0%; Cochran-Mantel-Haenszel nominal p=0.01). In post hoc analysis of the patients not ventilated at randomization, more patients in the placebo group died, withdrew during hospitalization, were transferred to an ICU, or required invasive mechanical ventilation within 28 days of baseline (29.0% (53 of 183 patients) in the tocilizumab arm and 42.2% (38 of 90) in the placebo) (HR 0.614; 95% CI 0.40 to 0.94; nominal p=0.03).127 Additionally, although not statistically significant, numerically reduced duration of mechanical ventilation was observed with tocilizumab in the 19 patients who were intubated during the BACC Bay trial (median duration, 15.0 days in the tocilizumab group and 27.9 days in the placebo group).128 A significant interaction was also found between treatment and required respiratory support in the TOCIVID-19 trial, such that the effect of tocilizumab on lethality was larger among patients not requiring mechanical ventilation (OR 0.37 (95% CI 0.18 to 0.74) vs 0.50 (95% CI 0.27 to 0.92)).116 TOCIBRAS, an open-label randomized trial conducted in Brazil was terminated early due to increased mortality at 15 days in the tocilizumab arm (8% vs 20%; odds ratio (OR) 1.54; 95% CI 0.66 to 3.66; p=0.32), though the difference was not statistically significant. However, despite not meeting mortality endpoints, patients treated with tocilizumab were found to have significantly reduced duration of hospital stay.129
Several meta-analyses have now evaluated the available cohort studies and randomized trials.130–132 Meta-analyses of randomized controlled trials found no effect on mortality (pooled risk ratio (RR) 1.09; 95% CI 0.80 to 1.49, I2=0%), tocilizumab was associated reduced risk of mechanical ventilation (pooled RR 0.71; 95% CI 0.52 to 0.96; I2=0%) and decreased risk of composite poor outcome (pooled RR 0.71; 95% CI 0.56 to 0.89; I2=0%).132 By contrast, another analysis, which included both controlled trials and single-arm studies, found that mortality was slightly lower with tocilizumab compared with standard of care (RD −0.06; 95% CI −0.12 to −0.01; p = 0.03; I2=80.9%), and the difference was more pronounced among the studies that included on only patients with severe disease (RD −0.12; 95% CI −0.18 to −0.06; p < 0.01; I2=53.7%). The subgroup analysis for high quality studies was consistent, that is, lower mortality in TOC group (RD: −0.07; 95% CI −0.13 to −0.01; p=0.02; I2=82.6%). In that analysis, lower rates of mechanical ventilation with tocilizumab were only found in patients with severe COVID-19 (RD −0.11; 95% CI −0.19 to −0.02; p=0.01; I2=74.0% for severe disease vs RD: −0.04; 95% CI −0.15 to 0.06; p=0.44; I2=89.5%; for the combined analysis).130 Reduced mortality with tocilizumab treatment was also demonstrated in a meta-analysis including nine high-quality small studies, with an RR of 0.27 (95% CI 0.12 to 0.59) and RD of 12% (95% CI 4.6% to 20%) in favor of IL-6 blockade.
Effect modulators: steroids, severity and timing?
Cross-trial comparisons are complicated because the major studies evaluated tocilizumab in disparate patient populations at varying points in the COVID-19 disease course. Because hyperinflammation is a positive-feedback regulated response, intervention with anti-IL-6 either too early or too late in the disease course may negate any potential benefit. Furthermore, most patients admitted to the hospital for COVID-19 received multiple interventions—especially at the beginning of the pandemic when options were limited, and efficacy data was sparse. Although remdesivir was initially shown to reduce the duration of hospitalization,133 updated WHO guidelines recommend against its use.134 Other organizations, however, including the Infectious Disease Society of America (IDSA) recommend remdesivir over no antiviral treatment for hospitalized patients with severe COVID-19.135 Currently, the only intervention shown to significantly reduce mortality for severe COVID-19 is dexamethasone.136 Reflecting this ever-shifting landscape, patient in published trials may have received concomitant antivirals, corticosteroids, or other investigational agents along with IL-6-directed therapies. In addition to the potential confounding effects of comedications, the timing of administration and the patient selection criteria has varied between studies. The eligibility criteria for major studies of tocilizumab as well as the timing of administration of therapy and allowed concomitant medications is summarized in table 3.
Potential effect modulators for anti-interleukin 6 in large-scale studies of tocilizumab for COVID-19
Disease severity may be an effect modulator. In one meta-analysis, the mortality benefits for tocilizumab was more pronounced among patients with severe disease (RD 12%; 95% CI 18% to 6%; p<0.01) compared with the overall population (RD 6%; 95% CI 12% to 1%; p=0.03),130 though the effects of steroids were not investigated. Another systematic review found that tocilizumab was associated with reduced mortality across disease severities (pooled adjusted RR 0.58; 95% CI 0.51 to 0.66, I2=2.5%), although the effect only held for cohort studies and not randomized trials.132 Supporting this, the REMAP-CAP trial recruited patients with severe disease, requiring organ support at the time of tocilizumab administration. In RECOVERY, 562 patients (14%) were receiving invasive mechanical ventilation at the time of hospitalization, while 1686 (41%) required non-invasive respiratory support, and 1868 (45%) were on supplemental oxygen. While more data are needed on tociliumab for moderate or mild COVID-19 (not requiring oxygen support), no benefit was seen in the BACC Bay trial that enrolled patients with minimal symptoms and only mildly elevated inflammatory parameters.128 The RCT-TCZ-COVID-19 trial also showed that early administration of tocilizumab in patients with PaO2/FiO2 between 200 and 300 mm Hg, and a mild inflammatory phenotype defined by fever or CRP ≥10 mg/dL did not reduce the risk of clinical worsening.137
Steroids are now recognized as an important component of effective interventions for COVID-19, although early reports paint conflicting pictures of the interplay between glucocorticoids and IL-6 antagonism. In the retrospective STOP-COVID trial, corticosteroid use was not shown to significantly affect mortality for tocilizumab treatment (HRs 0.71; 95% CI 0.53 to 0.96 with steroids vs 0.68; 95% CI 0.46 to 0.99 without; p=0.83), although the proportion of patients who received steroids in that trial was low.126 A subsequent meta-analysis found that glucocorticoid use was associated with smaller effects of tocilizumab on mortality with an RD of 9.1% (95% CI 2.8% to 15%) compared with 31% (95% CI 15% to 47%). Importantly, however, no difference in mortality was seen between the tocilizumab and control groups for studies that used steroids without antivirals.131 Corticosteroids use was also associated with a lower OR for death both at 14 and 30 days (OR 0.36 (95% CI 0.21 to 0.62) and OR 0.62 (95% CI 0.40 to 0.95)) in the TOCIVID-19 study.116 In the TOCIBRAS trial, where corticosteroids use was evenly balanced between treatment groups, yet remdesivir was not available in Brazil, increased mortality was seen in the tocilizumab arm. The interpretation of these data is challenging because the sample size is small and the majority of patients were treated at a single center.129 By contrast, results from REMAP-CAP and RECOVERY support the benefits of combining IL-6 modulation with corticosteroids for COVID-19. At the time of randomization, 82% of patients in RECOVERY and 93% of patients in REMAP-CAP were receiving steroids. In both studies, the combination of anti-IL-6 and corticosteroids led to greater magnitude of benefit than any single intervention alone.124 125
The historically controlled prospective CHIC trial found that a 5-day course of high-dose methylprednisolone followed by dose-reduction for the steroid and tocilizumab administration led to a 79% higher likelihood of reaching ≥2 stages of improvement on the 7-item WHO-endorsed disease severity scale (HR 1.8; 95% CI 1.2 to 2.7) as well as 65% less mortality (HR 0.35; 95% CI 0.19 to 0.65), and 71% less need for invasive mechanical ventilation (HR 0.29; 95% CI 0.14 to 0.65).138 The CHIC study was notable because it incorporated a lab parameter-based selection to enroll only patients experiencing a COVID-19-related cytokine storm (defined as rapid respiratory deterioration plus elevation in at least two out of three biomarkers: CRP >100 mg/L, ferritin >900 µg/L or d-dimer >1500 µg/L).
Another retrospective study found that timing of administration of anti-IL-6 had important effects on outcomes among patients with severe COVID-19 (defined as a SpO2/FiO2<325 with bilateral pneumonia and a clinical diagnosis of infection with SARS-CoV-2) who were treated with both tocilizumab and steroids. In the early therapy subgroup who received tocilizumab after 24 hours of admission and before SpO2/FiO2 decreased to ≤250, 6.25% required ICU admission or died in hospital. By contrast, mortality or ICU admission occurred in 45.3% of patients who received tocilizumab and steroids when SpO2/FiO2 was <250 (6.25% vs 34.9%, p<0.01).139
Other effect modulators that may underlie inter- and intra-trial heterogeneity include geography, patient ethnicity, comorbidities, and the microbiota. Marked regional differences in mortality due to COVID-19 have been observed,140 with proposed mechanisms involving differences in vitamin D levels by latitude, geographic and dietary influences on the gut microbiome, and others. Altered composition of the bacterial and fungal microbiota have been correlated with disease severity in COVID-19.141–143 Bacterially produced butyrate and other short-chain fatty acids have been shown to modulate CD8+ T cell effector functions as well as IL-6 production by monocytes,144 145 a potential mechanism linking dysbiosis to COVID-19 disease severity. Although not currently actionable, interactions between the gut microbiota and SARS-CoV-2 pathogenesis may contribute to the clinical course of COVID-19. Future studies may identify microbiota-targeted perturbations or patient selection strategies to enhance the efficacy of anti-IL-6.
It has also become apparent that racial and ethnic minorities experience disproportionately high mortality rates from COVID-19 in the USA and the UK.3 146 The confounding effects of differential access to healthcare, comorbidities, and demographics complicates the interpretation of these results, however.146 Despite being multi-center, international trials, patients in REMAP-CAP and RECOVERY were majority white (roughly 70%), whereas TOCIBRAS was conducted in Brazil. Of note, EMPACTA trial, which recruited patients with an emphasis on inclusion of racial minorities met its primary efficacy endpoint of reduced 28-day mortality with tocililzumab in a population that was 56.0% Hispanic or Latino, 14.9% Black, 12.7% American Indian or Alaska Native, 12.7% non-Hispanic White, and 3.7% were of other or unknown race or ethnic group.123 Recruiting diverse patient populations should remain a priority for ongoing and future trials, both to identify any potential biological differences in response to therapy among different groups, as well as to improve access to care.
Defining the window of opportunity
Taken together, the results of randomized trials and meta-analyses clearly demonstrate that anti-IL-6 therapies are not universally effective for COVID-19, however, cytokine modulation offers definitive benefit for some patients, if administered at the appropriate time after symptom onset. Additionally, the optimal use of tocilizumab is likely in combination with glucocorticoids. Updated guidelines from IDSA reflect this evolving understanding of the standard of care, now suggesting the addition of anti-IL-6 to standard care (ie, steroids) in hospitalized patients with severe disease and elevated levels of inflammatory markers.147 In patients where the inflammatory cascade has advanced beyond a “point of no return” modulation of IL-6 is unlikely to provide benefit—exemplified by the futility of tocilizumab administration in the COVACTA trial,127 which included patients with critical disease. Conversely, IL-6 blockade too early in the disease course is also ill-advised. Because IL-6 a master regulator of inflammation and immunity, anti-cytokine therapy during the initial stages of infection may hinder the development of robust anti-viral T cell responses.101 148
Optimal incorporation of anti-IL-6 into the anti-COVID-19 arsenal will require defined criteria for the patients who may benefit. Parameters for patient selection should be readily available, and biologically informed. Trials to date have used CRP as marker for patient selection, and prespecified analysis in REMAP-CAP demonstrated the greatest magnitude of benefit with anti-IL-6 in the highest tercile for pretreatment CRP. Based on the validated definitions of COVID-19 hyperinflammation,77 78 and evidence from the large anti-IL-6 trials, a threshold of CRP >75 mg/L likely may be used to identify patients for anti-IL-6 therapy. This threshold is the same value used for eligibility in the RECOVERY trial.125 Notably, this value for CRP is higher than the cut-off for inflammatory markers used in the BACC Bay trial, which showed no benefit for tocilizumab administration in moderate disease.128
Future directions and other anti-inflammatory strategies
Anti-IL-6 therapies remain an ongoing area of investigation, both alone and in combination with other interventions such as steroids and antivirals. The phase III REMDACTA trial (NCT04409262), evaluating tocilizumab with remdesevir for severe COVID-19 pneumonia, is ongoing. Additional trials are studying the effects of varying the dose or route of administration of anti-IL-6, and at least one case report has shown benefit with subcutaneous tocilizumab.149 Future studies of anti-IL-6 therapies should include detailed biomarker analyses at baseline and on-treatment to definitively establish indicators of response to therapy as well as tools for patient selection.
Additionally, future studies will be needed to rule out the potential for harm with tocilizumab treatment, specifically the possibility of further immunosuppression leading to secondary infection. The FDA label for tocilizumab includes a black boxed warning that serious infections leading to hospitalization or death including tuberculosis, bacterial, invasive fungal, viral, and other opportunistic infections have occurred in patients on treatment for rheumatoid arthritis. Some retrospective studies have reported increased secondary infections in patients with COVID-19 treated with tocilizumab.150–152 No such increased risk was seen in several single-center randomized controlled trials, however.116 121 128 137 Similarly, the RECOVERY trial reported no significant increase in the rate of secondary infections with IL-6 modulation,125 and only one secondary bacterial infection was observed in the tocilizumab group in the REMAP-CAP study.124 Notably, experience from CAR T cell therapy indicates that tocilizumab administration for CRS is not associated with increased infection risk. Because the “single shot” tocilizumab dosing used for COVID-19 is more akin to CRS management than the long-term immunosuppression required to treat rheumatoid arthritis, the risk for secondary infections may be reduced. Future meta-analyses, however, will be necessary to determine if the single or double doses of tocilizumab given in the context of COVID-19 is associated with a significant risk signal for secondary infections.
Several other anti-inflammatory and immune-modulatory strategies have been proposed to calm the cytokine storm in COVID-19.153 Targeting the monocyte-driven inflammatory pathology through the administration of anti-GM-CSF antibodies is an active area of investigation.63 154 Modulation of IL-1β with anakinra has demonstrated improved survival in cohort-controlled studies,155 156 although it remains to be seen if the results are recapitulated in randomized trials. Targeting downstream components of the IL-6 signaling cascade is also being studied, with several janus kinase inhibitors being repurposed for COVID-19.153 Of note, baricitinib, which was identified through artificial intelligence as a potential treatment, reportedly led to prompt resolution of respiratory function and improvement radiological findings when used as a rescue therapy for a patient with disease that did not respond to sarilumab or antivirals.157 Baricitinib has been evaluated in the phase III ACTT-2 study (NCT04401579), which enrolled 1034 hospitalized patients and found that JAK inhibition plus remdesivir was superior to the antiviral alone in reducing recovery time and accelerating improvement in clinical status.158 Several small-scale studies are ongoing, and active phase III trials evaluating baricitinib and anakinra for COVID-19 are summarized in table 4.
Phase III trials evaluating immune-modulatory therapies for COVID-19 as of December, 2020*
Conclusion
The current standard of care for severe COVID-19 is dexamethasone, which has conclusively been shown to reduce mortality in patients requiring supplemental oxygen.136 There is currently no generally accepted recommended therapy for patients with mild or moderate disease, yet increasing evidence suggests that adding anti-IL-6 to corticosteroids for hypoxic patients may reduce mortality and shorten times in hospital. Although vaccine rollout is ramping up, the healthcare system has been strained beyond capacity, and interventions that reduce the burden on hospital resources are still desperately needed. In future trials, it will be important to evaluate endpoints beyond mortality, such as need for ICU care, duration of hospital stay, or need for ventilation, so that therapies that alleviate the demands on the healthcare system may be identified. Additionally, trials that specifically recruit participants from historically under-represented populations, such as was done in the EMPACTA study,123 will be important to not only provide equitable access to therapy but also potentially identify discrepancies in response to treatment among demographics.
In all likelihood, the optimal treatment approach for COVID-19 will be context-dependent and involve a combination of several different therapies. Establishing clinical benefit in such a complex landscape—a heterogeneous disease treated with a combination of therapies—may be challenging. For this reason, the immuno-oncology community may be uniquely poised to support our colleagues in infectious disease and intensive care. Cancer clinical trialists have extensive experience in the design and execution of adaptive, biomarker-selected studies that involve complex combination regimens. Additionally, immunotherapy practitioners may contribute expertize in assay development and sample handling considerations for the delicate and difficult immunological profiling experiments that will be necessary to gain additional insight into the pathology of COVID-19. As the world responds to COVID-19, interdisciplinary collaboration will be essential, and the lessons learned from this pandemic may provide crucial insight, not only in the next infectious outbreak, but in understanding the basic mechanisms of inflammation and immunity.
References
Footnotes
Twitter @PAscierto
Contributors All the authors contributed to the conception, content, writing and revisions, and have read and approved the submitted manuscript.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests PAA has/had a consultant/advisory role for Bristol Myers Squibb, Roche-Genentech, Merck Sharp & Dohme, Novartis, Array, Merck Serono, Pierre-Fabre, Incyte, Medimmune, AstraZeneca, Syndax, Sun Pharma, Sanofi, Idera, Ultimovacs, Sandoz, Immunocore, 4SC, Alkermes, Italfarmaco, Nektar, Boehringer-Ingelheim, Eisai, Regeneron, Daiichi Sankyo, Pfizer, Oncosec, Nouscom, Takis, Lunaphore. He also received research funding from Bristol Myers Squibb, Roche-Genentech, Array, Sanofi and travel support from MSD. All the other authors have no conflict of interest to declare.
Provenance and peer review Commissioned; externally peer reviewed.