Article Text

Abstract
Background Merkel cell carcinoma (MCC) is an aggressive skin cancer associated with poor survival. Programmed cell death-1 (PD-1) pathway inhibitors have shown high rates of durable tumor regression compared with chemotherapy for MCC. The current study was undertaken to assess baseline and on-treatment factors associated with MCC regression and 3-year survival, and to explore the effects of salvage therapies in patients experiencing initial non-response or tumor progression after response or stable disease following first-line pembrolizumab therapy on Cancer Immunotherapy Trials Network-09/KEYNOTE-017.
Methods In this multicenter phase II trial, 50 patients with advanced unresectable MCC received pembrolizumab 2 mg/kg every 3 weeks for ≤2 years. Patients were followed for a median of 31.8 months.
Results Overall response rate to pembrolizumab was 58% (complete response 30%+partial response 28%; 95% CI 43.2 to 71.8). Among 29 responders, the median response duration was not reached (NR) at 3 years (range 1.0+ to 51.8+ months). Median progression-free survival (PFS) was 16.8 months (95% CI 4.6 to 43.4) and the 3-year PFS was 39.1%. Median OS was NR; the 3-year OS was 59.4% for all patients and 89.5% for responders. Baseline Eastern Cooperative Oncology Group performance status of 0, greater per cent tumor reduction, completion of 2 years of treatment and low neutrophil-to-lymphocyte ratio were associated with response and longer survival. Among patients with initial disease progression or those who developed progression after response or stable disease, some had extended survival with subsequent treatments including chemotherapies and immunotherapies.
Conclusions This study represents the longest available follow-up from any first-line anti-programmed death-(ligand) 1 (anti-PD-(L)1) therapy in MCC, confirming durable PFS and OS in a proportion of patients. After initial tumor progression or relapse following response, some patients receiving salvage therapies survived. Improving the management of anti-PD-(L)1-refractory MCC remains a challenge and a high priority.
Trial registration number NCT02267603.
- skin neoplasms
- immunotherapy
- programmed cell death 1 receptor
Data availability statement
Data are available on reasonable request.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Background
Merkel cell carcinoma (MCC) is an aggressive neuroendocrine skin cancer that frequently spreads to nodal and distant sites. Prior to the use of immunotherapies targeting programmed cell death-1 (PD-1) or its major ligand programmed death-ligand 1 (PD-L1), patients with advanced MCC (aMCC) had an expected 5-year overall survival (OS) of 14%–27%.1 The incidence of MCC is increasing mainly due to an aging population, with nearly 3000 cases in the USA this year.2 MCC is an immunogenic cancer, with a higher incidence and poorer prognosis in immunosuppressed individuals.3–6 Evidence of active immunity within and near the tumor has been described; notably, cell surface expression of PD-L1 by tumor cells and by tumor infiltrating lymphocytes is present in 49% and 55% of specimens, respectively.7 Approximately 80% of MCCs are caused by the Merkel cell polyomavirus (MCPyV).8 Virus-positive tumors (VP-MCC) persistently express T-antigen oncoproteins required for tumor cell proliferation, which are recognizable by the immune system as indicated by detection of MCPyV-specific T cells in peripheral blood and tumors from most patients with VP-MCC.9 Furthermore, MCPyV-specific T cells often have high expression of PD-1 and Tim-3 on their surface indicating evidence of potentially reversable immune dysfunction.10 The remaining ~20% of MCCs are caused by ultraviolet light (UV) exposure (MCPyV-negative or VN-MCC). VN-MCCs contain abundant UV-induced mutations potentially generating neoantigens for immune recognition; their aggregate mutational burden is nearly 100-fold higher than that of VP-MCC tumors.11–14
Just a few years ago, standard-of-care treatment for aMCC was cytotoxic chemotherapy, which induced tumor regressions in ~60% of cases. However, responses to chemotherapy were not durable,15 with a median progression-free survival (PFS) of only ~90 days. More recently, several clinical trials of PD-1 pathway inhibitors in patients with aMCC demonstrated improved PFS and OS compared with historical data for conventional cytotoxic chemotherapy. Favorable outcomes from these trials supported US Food and Drug Administration approvals for avelumab (Bevencio, anti-PD-L1) in March 2017 and pembrolizumab (Keytruda, anti-PD-1) in December 2018. Response rates achieved in the first-line treatment setting were 50%–60%; unlike results from chemotherapy, these responses had greater durability.16–18 Across all anti-PD-(L)1 trials in aMCC, response rates appeared similar regardless of tumor viral status, suggesting that tumor antigens in both VP-MCC and VN-MCC can serve as effective targets for tumor elimination by the immune system. These outcomes led to rapid changes in the National Comprehensive Cancer Network guidelines for treating aMCC, and anti-PD-(L)1 agents are now included as preferred first-line systemic therapies.19
The current study was undertaken to further characterize long-term outcomes and explore factors associated with survival after first-line anti-PD-1 therapy in aMCC. Here, we report findings from the phase II Cancer Immunotherapy Trials Network (CITN)-09/KEYNOTE-017 trial of pembrolizumab. This report represents the longest available follow-up for any first-line anti-PD-(L)1 therapy in aMCC, with a median period of 31.8 months. Furthermore, we investigated survival in individuals who manifested primary or acquired resistance to first-line anti-PD-1 therapy and received subsequent treatments, in an effort to devise improved therapeutic strategies for these patients.
Methods
Patients
Patients with aMCC (distant metastatic or locoregional disease) not amenable to definitive surgery or radiation therapy, and measurable per Response Evaluation Criteria in Solid Tumors RECIST v1.1, were enrolled. Patients who had prior systemic therapy for MCC were excluded, with the exception of adjuvant chemotherapy if completed >6 months prior to initiating study treatment. More detailed patient eligibility criteria have been reported previously.20 An initial cohort of 26 patients was enrolled between January and December 2015, with results reported in 2016.20 The protocol was then amended to include 24 additional patients enrolled between March 2016 and May 2017, and preliminary results were reported with a median follow-up of 14.9 months.16 Potential financial conflicts of the investigators were reported and managed according to institutional policies at each center.
Study design
The CITN-09/KEYNOTE-017 trial is a phase II, open-label, non-randomized Simon two-stage multicenter study. Per the Simon two-stage design for efficacy estimation, at least one response among the first group of nine treated patients was required in order to enroll additional patients. Patients received pembrolizumab 2 mg/kg intravenously every 3 weeks. Treatment continued for up to 2 years, or until the development of unacceptable adverse event(s) (AEs), progressive disease (PD), a complete response (CR) with at least 24 weeks of therapy and at least two treatments beyond the date of confirmed CR, consent withdrawal or physician discretion. Patients were followed for AEs, PFS, OS and treatments received after discontinuing the study drug.
Study objectives
The primary objective of the CITN-09/KEYNOTE-017 trial was to determine the clinical efficacy of systemic first-line therapy for aMCC with pembrolizumab (Keytruda/MK-3475). The primary end point was overall response rate (ORR) measured by RECIST V.1.1, defined as CR+partial response (PR). Secondary end points included PFS, duration of response (DOR) and OS. The study also collected data on subsequent treatments received by patients who had primary or acquired resistance to pembrolizumab. Exploratory objectives were to determine associations between clinical outcomes and baseline and on-treatment patient and tumor characteristics, including tumor viral status and PD-L1 expression.
Disease assessment
CT scans were performed at screening, 12 weeks after treatment initiation and at 9-week intervals thereafter as previously described.20 Patients who appeared to have PD were allowed to continue to the next cycle of therapy if they were asymptomatic, had Eastern Cooperative Oncology Group performance status (ECOG PS) ≤1 and had no evidence of rapid tumor progression; patients were evaluated 4 weeks later to assess possible further progression. After 1 year of treatment, the CT scan frequency was decreased to 12-week intervals. RECIST V.1.1 evaluations of scans were initially conducted at the investigator/institutional level, followed by central radiological review.
Specimen acquisition
Pretreatment fresh or archival tumor biopsy samples (formalin-fixed paraffin-embedded) were obtained from all patients. Blood samples were collected at the time of radiographic studies.
Laboratory assessments
Patients were determined to have MCPyV-positive tumors if they produced small T-antigen-specific serum antibodies21 or manifested large T-antigen expression in tumor biopsies via immunohistochemistry.22 PD-L1 staining (anti-PD-L1 clone 22C3, Merck & Co, Kenilworth, New Jersey, USA) was performed at QualTek Molecular Laboratories on pretreatment tumor specimens as previously described.17 Specimens were considered PD-L1 positive if ≥1% of tumor cells expressed PD-L1 at the cell surface.16 20
Neutrophil-to-lymphocyte ratio (NLR) was calculated using absolute neutrophil counts and absolute lymphocyte counts (ALC), determined from automated complete blood counts in peripheral blood specimens obtained at study visits. NLR was calculated at baseline (before initial pembrolizumab infusion) and after each of the first four treatment cycles.
Statistical analysis
All statistical analyses were based on a database cut-off date of October 23, 2019. Responses were evaluated with point estimates and 95% CIs based on the exact binomial method. Median DOR (for patients who had a CR or PR), PFS and OS with 95% CIs were estimated by the Kaplan-Meier (KM) method for censored data. For DOR, subjects who had not progressed by the last disease assessment were censored at the date of last disease assessment. For PFS, subjects without documented PD/death were censored at the last disease assessment date. Any subject who was lost to follow-up was included in the analysis, and their PFS time was censored on the last date the subject was known to be progression-free, defined as the date of the last tumor assessment not indicating progression. For OS, subjects without documented death at the time of data cut-off were censored at the date last known to be alive. Post hoc analyses of the relationships between baseline patient and tumor characteristics and survival time were also conducted using KM methods. HRs and corresponding 95% CIs were estimated. NLR and ALC analyses were performed with a mixed model approach. Briefly, each time point used a t-test allowing for unequal variance. P values for trends across all time points were based on mixed model, with treatment cycle and response status (CR/PR vs SD/PD) or survival status (alive vs dead) as fixed effects, with a random intercept.
Results
Patient and treatment characteristics
Fifty patients with aMCC were enrolled between January 2015 and May 2017. Data were analyzed as of October 23, 2019, representing ≥30 months since treatment initiation for all patients. For those who received pembrolizumab continuously for the maximum treatment period of 2 years, the follow-up period included ≥6 months after completing treatment. Median follow-up at the time of analysis was 31.8 months (range 0.4–56.9). Baseline patient and tumor characteristics have been detailed previously.16 Briefly, 43 (86%) patients had stage IV MCC and 7 (14%) had stage IIIB MCC23 at the time of enrollment, and all subjects had an ECOG PS of 0 or 1.24 Their median age was 70.5 years (range 46–91), similar to other studies of aMCC. Patients received a median of 10.5 doses of pembrolizumab (SD 12.7 doses; range 1–35). Twelve patients (24%) completed 2 years of treatment. Thirty-seven patients did not complete 2 years of therapy due to PD (n=19), AE (n=13), death (n=2), physician decision (n=2) or consent withdrawal (n=1). One patient was lost to follow-up; see online supplemental table S1.
Supplemental material
Response and duration of response
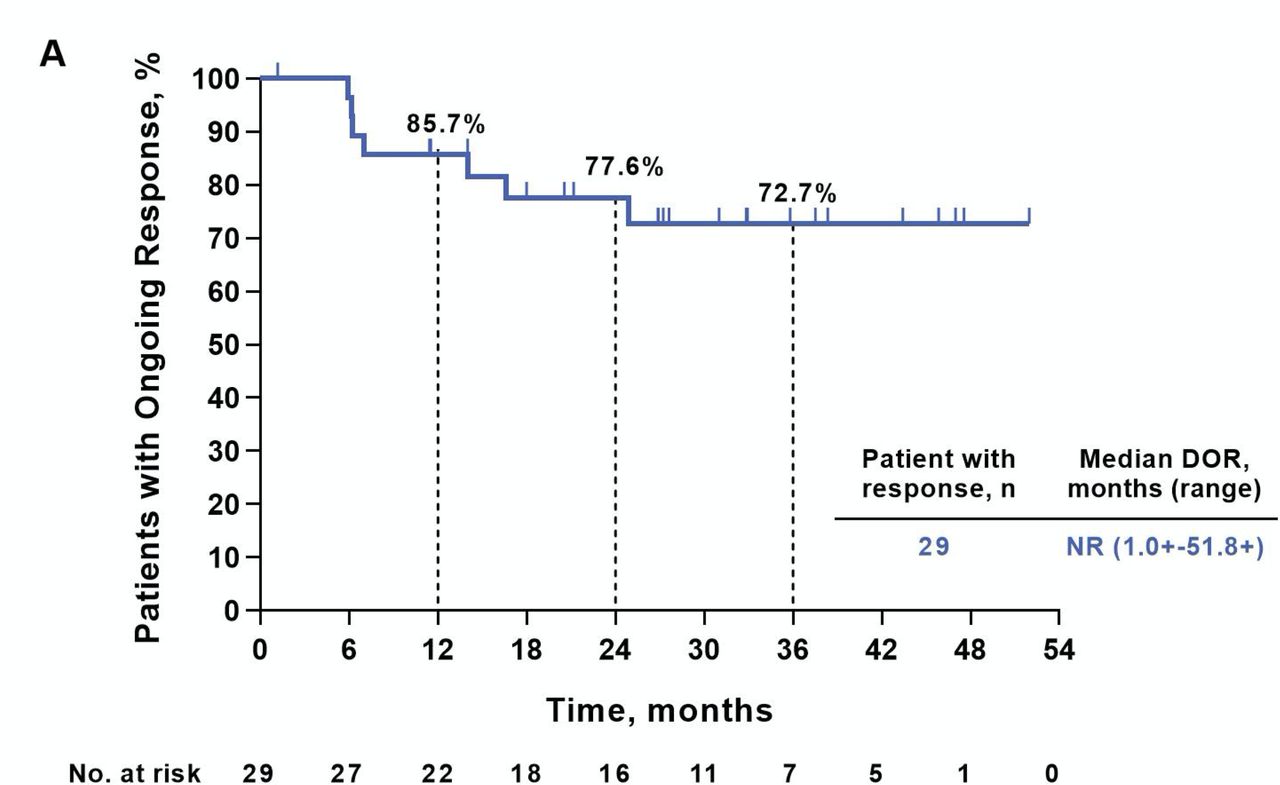
Similar to the ORR of 56% reported earlier in this study,16 20 with longer treatment and follow-up the ORR to pembrolizumab was 58% (95% CI 43.2 to 71.8); this included 15 patients with CR and 14 with PR (table 1). Among a total of 29 responders, the median response duration was not reached (NR, range 1.0+ to 51.8+ months; figure 1). At 3 years after treatment initiation, 72.7% of responders remained in response. Most objective tumor regressions occurred soon after treatment initiation, with 90% (26/29) of CRs and PRs documented at the initial ~12-week assessment (figure 2A,B).

Duration of response (DOR). Kaplan-Meier curve showing duration of response among 29 patients having a complete or partial tumor regression by Response Evaluation Criteria in Solid Tumors V.1.1. Patients without an event were censored (tick mark) at the last disease assessment date. Rates of ongoing response at 12, 24 and 36 months are indicated. NR, not reached.
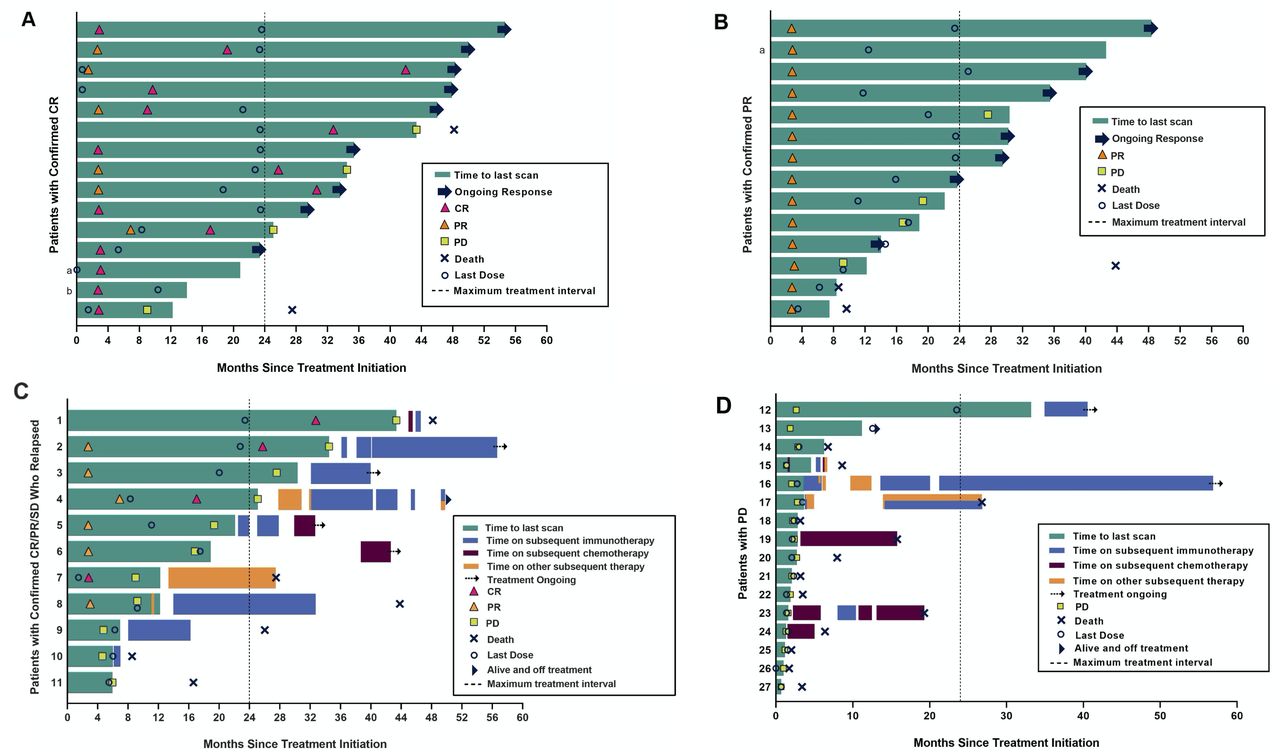
Kinetics of response to pembrolizumab, and subsequent treatments received by patients with tumor relapse or with no response. Each lane in these swimmer plots depicts an individual patient. Dotted vertical lines indicate the maximum on-study pembrolizumab treatment interval (24 months). (A) Patients with a confirmed complete response (CR) to pembrolizumab (n=15). a,bTwo patients were censored for progression/response because they started a new anticancer therapy without documented disease progression. (B) Patients with a confirmed partial response (PR) to pembrolizumab therapy (n=14). aThis patient was censored for progression/response because they started a new anticancer therapy without documented disease progression. (C) Patients with CR (red triangle), PR (yellow triangle) or stable disease (SD) (patients #9, 10, 11) after receiving pembrolizumab on-study, who later experienced disease progression (n=11). Subsequent treatments are shown. Patients with CR or PR are also depicted in panels (A) and (B), respectively. Details of subsequent treatments are presented in online supplemental table S6. (D) Patients with initial progressive disease (PD) (no CR, PR or SD) on pembrolizumab (n=16), showing subsequent treatments received. Details of subsequent treatments are presented in online supplemental table S6.
Summary of best response by blinded independent central review per Response Evaluation Criteria in Solid Tumors V.1.1
Progression-free survival and overall survival
PFS and OS estimates for first-line pembrolizumab therapy in aMCC are shown in figure 3. The median PFS was 16.8 months (95% CI 4.6 to 43.4), and the KM estimate of PFS at 3 years was 39.1% (figure 3A). The median OS was not reached at the time of analysis (95% CI 26 months, not estimable). Notably, while the KM estimate of OS at 3 years was 59.4% for all patients, it was 89.5% for responders (CR+PR; figure 3B).

Survival among patients with advanced Merkel cell carcinoma (aMCC) receiving pembrolizumab. (A) Progression-free survival (PFS). Kaplan-Meier curve depicting PFS measured from the time of treatment initiation until either disease progression (Response Evaluation Criteria in Solid Tumors V.1.1) or death, whichever occurred first. At 36 months, the estimated PFS was 39.1%. Median PFS was 16.8 months (95% CI 4.6 to 43.4). (B) Overall survival (OS). Kaplan-Meier curves depicting OS among all 50 patients in green, or among those with objective tumor regression (complete response (CR)+partial response (PR)) in blue. At 36 months, the estimated OS was 59.4% for all patients, and 89.5% for those with objective response. Median OS was not reached in either group at the time of analysis. NR, not reached.
Factors associated with response and overall survival
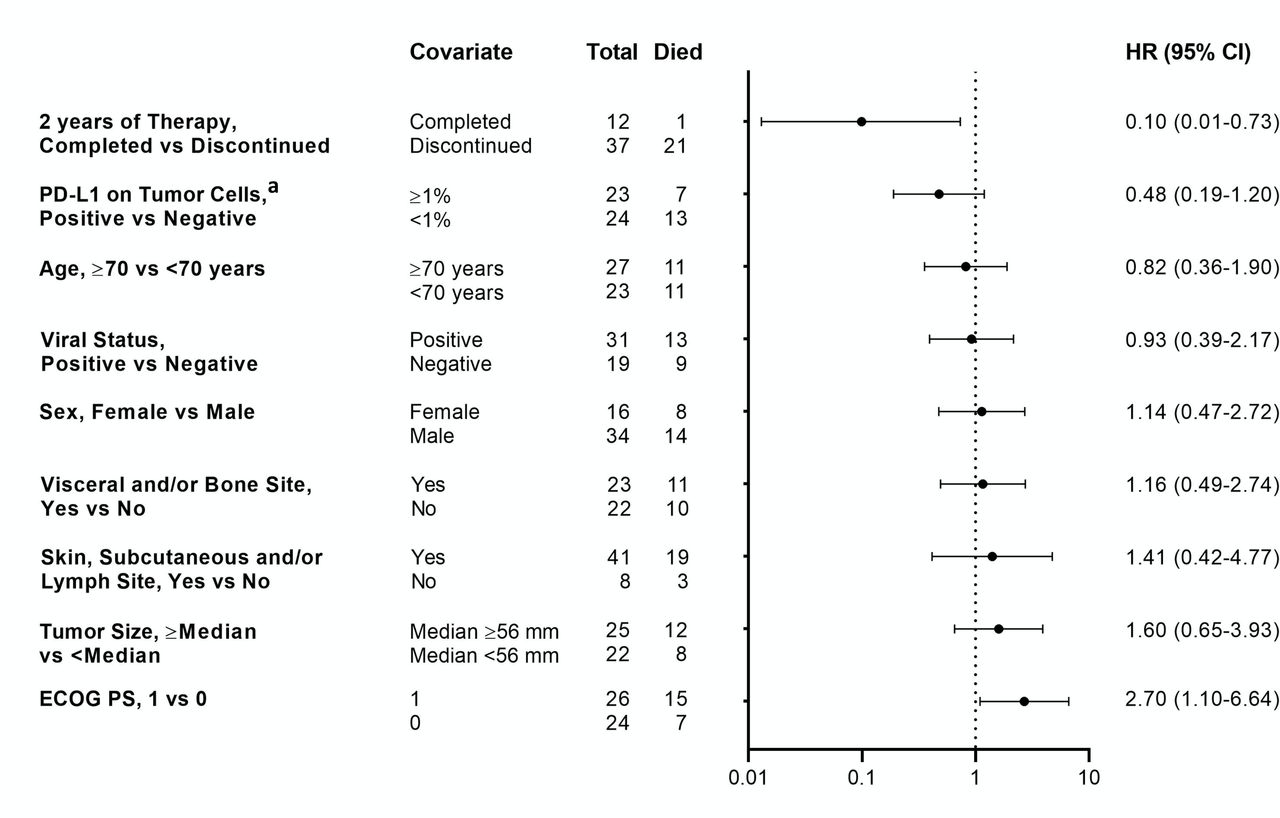
Based on outcomes reported for anti-PD-1 therapies in some other cancer types,25 26 we first asked if the degree of tumor burden reduction in patients with aMCC receiving pembrolizumab was associated with OS. Forty-five patients with evaluable tumor target lesions per RECIST V.1.1 were included in this analysis (figure 4). An increasing degree of tumor target lesion reduction was associated with prolonged OS, such that the majority of patients with 100% reductions survived for 30 months and beyond. These findings are consistent with the OS results shown in figure 3B, in which patients experiencing an objective response (CR+PR) to pembrolizumab therapy survived longer than the overall treatment population. Associations of several baseline patient and tumor features with OS were also assessed (figure 5 and online supplemental figure S1A). Patients who were able to complete 2 years of continuous pembrolizumab therapy were more likely to be alive with 30 months’ follow-up (HR 0.1; 95% CI 0.01 to 0.73), while a baseline ECOG PS of 1 vs 0 was associated with a decreased likelihood of survival (HR 2.7; 95% CI 1.10 to 6.64). Interestingly, the magnitude of baseline tumor burden (above or below the median, figure 5; or absolute dimensions, online supplemental figure S1A) was not associated with OS, nor were age (< vs ≥70 years), gender, anatomic sites of metastases, or tumor viral or PD-L1 status (figure 5). Analysis of the same factors with objective response did not yield any significant associations (online supplemental table S2 and figure S1B).
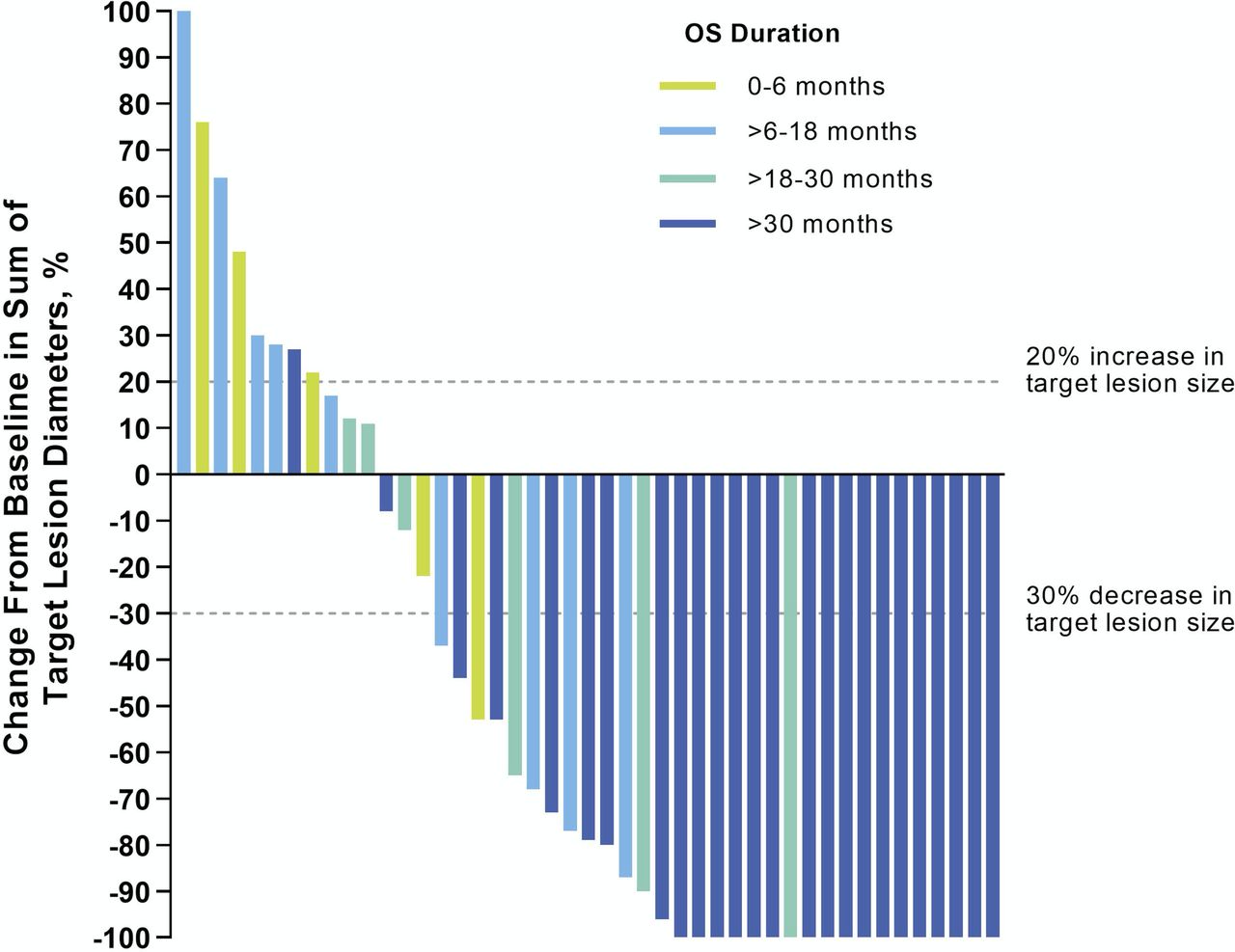
Association between magnitude of tumor burden reduction and overall survival (OS). Waterfall plot showing the maximum change in tumor burden (sum of target lesion diameters) compared with baseline, for radiographically evaluable patients (n=45). Horizontal dashed lines indicate Response Evaluation Criteria in Solid Tumors V.1.1 criteria for partial response (≥30% decrease in sum of target lesion diameters from baseline, in the absence of new lesions) and progressive disease (≥20% increase in sum of target lesion diameters). Vertical bars are color-coded to indicate OS duration in individual patients.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Association of overall survival with 30 months’ follow-up, with baseline demographics and tumor and treatment characteristics. Forest plot showing overall survival HRs (with 95% CI) for characteristics which are listed from top to bottom in increasing order of HR magnitude. Total numbers of evaluable patients in each category are shown. Patients with baseline Eastern Cooperative Oncology Group performance status (ECOG PS) of 1 vs 0 had significantly reduced survival, while those who completed 2 years of pembrolizumab therapy experienced significantly longer survival. programmed death-ligand 1 (PD-L1) positive, ≥1% of tumor cells expressed cell surface PD-L1, assessed by immunohistochemistry.
Cell counts in the peripheral blood at baseline and during pembrolizumab treatment were also assessed for potential correlations with objective response and OS. When trends were assessed across the first 3 months of therapy, the NLR but not the ALC was associated with objective response (CR+PR, p=0.043) and OS (p=0.028) at 30 months (online supplemental figure S2). Specifically, a lower NLR across all time points was associated with improved outcomes. However, the results of similar assessments conducted at baseline only, or at any individual time point during therapy, were not statistically significant.
Adverse events
AEs experienced by patients in this study are summarized in online supplemental table S3. Treatment-related adverse events (TRAEs) of any grade occurred in 49 of 50 patients (98%), and 15 patients (30%) had grade ≥3 TRAEs, similar to earlier reported results from this trial.16 In the setting of longer treatment duration, eight patients (16%) discontinued treatment due to TRAEs, similar to seven patients (14%) reported earlier. A single treatment-related death occurred and was detailed previously.16 These results suggest that TRAEs were not cumulative with prolonged anti-PD-1 therapy for aMCC, as previously shown for patients with other cancer types receiving anti-PD-1 continuously for up to 2 years.27 Immune-mediated TRAEs and infusion reactions occurred in 16 patients (32%) (summarized in online supplemental table S4).
Salvage therapies for anti-PD-1-resistant aMCC
Currently, available data describing effective subsequent therapies for patients with cancer who experience primary or acquired resistance to anti-PD-(L)1 therapy are limited. To gain insights into potentially effective therapeutic options for patients with anti-PD-1-refractory aMCC, we collected subsequent treatment data from those who received pembrolizumab on the CITN-09/KEYNOTE-017 trial. In total, there were 22 patients who received other therapies for MCC after discontinuing on-study pembrolizumab, including a variety of chemotherapies, immunotherapies and experimental treatments (listed in online supplemental table S5).
Eleven patients depicted in figure 2C developed resistance to pembrolizumab after an initial response (CR, n=4; PR, n=4) or SD (n=3; patients #9–11 as shown); for the eight patients with CR or PR, the time interval between first response and disease progression varied widely. Among these 11 patients, 10 received additional therapies; 8 received subsequent immunotherapies, including pembrolizumab, nivolumab, avelumab, ipilimumab and combination nivolmab+ipiliumumab. Five of the 10 (50%) patients with subsequent therapies were alive at the time of data analysis, 4 of whom had received immunotherapies. Eight of 10 (80%) patients with initial CR/PR/SD who relapsed and received subsequent therapies survived for >12 months after disease progression was documented on-study.
There were 16 patients with primary resistance to pembrolizumab (figure 2D). Among them, seven received subsequent therapies, while others expired soon after developing PD. Five of 16 (38%) patients survived >12 months after disease progression on-study, all having received subsequent treatment(s). Three patients were alive at the time of data analysis. Details of treatments received on a per-patient basis are shown in online supplemental table 6).
Discussion
This multi-institutional study provides the longest available follow-up for first-line anti-PD-(L)1 therapy in advanced unresectable MCC. All 50 patients were assessed ≥30 months following treatment initiation, with a median follow-up of 31.8 months. After a potential maximum continuous treatment period of 2 years, the ORR of 58% was very similar to earlier reports from this trial (56%). This likely reflects the rapid kinetics of anti-PD-1 response in MCC, with most responses occurring at the first radiographic evaluation (12 weeks).16 20 With prolonged follow-up, the majority of responses were durable: 73% persisted at 3 years, and the median DOR was not reached. Furthermore, the median OS for all patients in this study was not reached. Importantly, objective responders had a substantially improved OS (89.5%) compared with the total study population (59.4%) at 3 years, suggesting that objective response is an early predictor of long-term survival in patients with aMCC receiving first-line anti-PD-1 therapy. Similarly, in studies of anti-PD-1 therapy in patients with advanced melanoma, non-small-cell lung cancer or renal cell carcinoma, objective responses correlated with long-term OS.25 26 In the current MCC study in which 86% of patients had stage IV disease,16 regardless of response status, the median OS far exceeded the 9.6-month median survival anticipated for patients with a new diagnosis of distant metastatic MCC before the advent of anti-PD-(L)1 therapies.28 These findings of high response rate and durability, associated with extended survival, supported regulatory approval of pembrolizumab for aMCC based on non-randomized data and underline the enormous impact that anti-PD-(L)1 therapy has had on the outlook for patients with aMCC.
Here, we identify several baseline and on-treatment factors associated with survival assessed 30 months after initiating first-line pembrolizumab therapy for aMCC: ECOG PS 0, greater magnitude of reduction in tumor burden, and successful completion of 2 years of continuous therapy. Conversely, we also identified factors not associated with OS, including age, gender, baseline tumor burden, anatomic sites of metastasis and tumor PD-L1 expression and viral status. While baseline ECOG PS, magnitude of tumor burden reduction and duration of continuous anti-PD-1 administration29 have been associated with response and survival in studies of anti-PD-1 therapy for various cancer types, the lack of association of several other factors as reported here for aMCC diverges from prior experience.25 30 31 This may reflect an extremely robust response to anti-PD-1 therapy in highly immunogenic MCCs that can override the influence of other demographic or on-treatment factors. Although the current study permitted a maximum continuous treatment period of 2 years, it is unknown if this is sufficient or optimal for aMCC, or if treatment duration should be individualized depending on anti-PD-(L)1 response status. This important issue has been examined in a randomized trial in non-small-cell lung cancer,29 which demonstrated survival benefit from continuous anti-PD-1 vs discontinuing at 1 year; this remains to be explored in MCC and other cancers.
Interestingly, our study also associated low peripheral blood NLR over the treatment course with objective tumor response and survival. A prognostic association between high baseline blood NLR and decreased OS has been reported for several different cancer types,32 33 and specifically for MCC.34 Furthermore, in the context of anti-PD-(L)1 therapy, high baseline and/or on-treatment NLRs have been reported to predict OR and OS in melanoma, non-small-cell lung cancer, renal cell carcinoma and other cancers.35–38 In the current study of first-line pembrolizumab for aMCC in immunocompetent patients, most ALCs were within the normal range and ALC as a single factor was not associated with response or survival, suggesting the importance of blood neutrophils as potentialy reflecting immune-suppressive inflammation, which might be driven by tumor-secreted IL-8 or other neutrophil-stimulating factors.39
The immunotherapy field is currently challenged with managing primary anti-PD-(L)1 resistance or relapse after an initial response (acquired resistance).40 Improving the management of anti-PD-(L)1-refractory MCC remains a high priority. Our study describes salvage treatments received by these patients. Several initial responders with subsequent relapse had sustained survival after retreatment with immune checkpoint blockade, similar to published experience in other cancers.41–44 However, among those with primary anti-PD-1 resistance, many expired soon after disease progression, although a few patients derived sustained survival from subsequent immunotherapies or chemotherapies. Beyond available immunotherapies and chemotherapies for advanced MCC, innovative clinical trial development is needed to address or prevent anti-PD-(L)1-refractory disease. Diverse approaches to address this problem include the addition of anti-CTLA-4 to anti-PD-(L)1,45 toll-like receptor agonists,46 histone deacetylase inhibitors47 and oncolytic virotherapy.48 In particular, infusion of MCPyV-specific T cells combined with immune checkpoint inhibitors may reduce the chance of tumor escape by boosting T cell numbers, increasing diversity of T cell responses and augmenting terminally exhausted T cells (NCT03747484). A therapeutic vaccine targeting MCPyV antigens is also an appealing approach to prevent recurrent disease as well as potentially overcome PD-(L)1 pathway resistance.49 Both adjuvant and neoadjuvant anti-PD-(L)1 immunotherapies hold promise for preventing high-risk early stage resectable MCC from advancing to stage IV.50
Data availability statement
Data are available on reasonable request.
Ethics statements
Ethics approval
This study was conducted in accordance with the Declaration of Helsinki and the International Conference on Harmonisation Guidelines for Good Clinical Practice. All patients provided written informed consent before study entry. The protocol was approved by the Institutional Review Board at each participating center.
Acknowledgments
The authors would like to thank the patients and their families and caregivers for participating in this study. The authors acknowledge the efforts of the Cancer Immunotherapy Trials Network (CITN) across the participating clinical sites in conducting this trial, and Judith Kaiser for budgetary and regulatory oversight of CITN; Sumia Dakhil, University of Washington/Fred Hutchinson Cancer Research Center, Seattle, USA, for clinical trial support; Trish Brothers, Uchechi Nwaba and Katrina Purtell, Johns Hopkins Kimmel Cancer Center and Bloomberg~Kimmel Institute for Cancer Immunotherapy, Baltimore, USA for clinical trial support; and Elliot K. Chartash, Steven M. Townson, Scot Ebbinghaus, Scott Diede, Rachel Lewis, Jenia Booth, Diana Yurewicz, Steven R. Bird and Nageatte Ibrahim, Merck & Co., Inc., Kenilworth, New Jersey, USA, for collaboration. The authors would also like to thank Doyel Mitra, ApotheCom, Yardley, Pennsylvania, USA, for editorial assistance; this assistance was funded by Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, New Jersey, USA in accordance with Good Publications Practice guidelines.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Twitter @PaulNghiem, @evanlipson, @ragikudchadkar, @ad1600, @michi_shinohara, @EladSharonMD
MAC and SLT contributed equally.
Contributors PN, SB, RK, HK, JT, ES, MC, SLT were involved in the conception, design and planning of the study. PN, SB, EL, WHS, RK, ASB, PF, AD, HK, SR, BCB, AR, MB, BAH, TO, KK, CC, NR, MMS and JT collected data. PN, CC, TA, BS, JT, EJ, MK, SPF, BHM, ES, MC and SLT analyzed and interpreted data. PN, CC, TA and SLT drafted the manuscript and critically reviewed and revised the manuscript for important intellectual content with contributions from all authors. All authors reviewed the manuscript and agreed with its content and submission.
Funding Supported by the National Cancer Institute (NCI) Grants No. 1U01CA154967 (MC), P01 CA225517 (PN), R01 CA142779 (SLT and JT) and the National Institutes of Health/NCI Cancer Center Support Grant in Seattle Grant No. P30 CA015704. This study was also funded by the Merkel cell carcinoma (MCC) patient gift fund at the University of Washington, the Kelsey Dickson MCC Challenge Grant from the Prostate Cancer Foundation and Merck & Co., Inc., Kenilworth, New Jersey, USA, which provided pembrolizumab and partial funding for this study.
Competing interests PN reports grants from Bristol-Myers Squibb and EMD-Serono; advisory fees from EMD-Serono, Pfizer and Merck & Co.; travel expenses from Sanofi/Regeneron and Merck & Co. and has a pending patent related to high-affinity T-cell receptors that target the Merkel polyomavirus. SB reports personal fees from Bristol-Myers Squibb, EMD-Serono and personal fees and other from Sanofi-Genzyme; grants from Bristol-Myers Squibb, EMD-Serono, Merck & Co., NantKwest, Novartis, Immune Design, Oncosec, Exicure, Nektar; and personal fees from Castle Biosciences. EJ reports grants from Merck & Co., Bristol-Myers Squibb and Sanofi/Regeneron; personal fees from Bristol-Myers Squibb, Novartis, Array BioPharma, Macrogenics, Sanofi/Regeneron and Genentech. RK reports grants from Merck & Co., Bristol-Myers Squibb and Regeneron; advisory fees from Merck & Co., Bristol-Myers Squibb, Regeneron, Novartis and Array. ASB reports personal fees from Bayer, Deciphera and EMD Serono. BAH reports grants from Merck & Co., Tempest Therapeutics, Olatec Therapeutics, A*STAR Singapore, Sanofi, Leap Therapeutics, GSK and AstraZeneca; personal fees from Merck & Co., Novartis, G1 Therapeutics and CE Concepts; travel fees from ASCO and ASCI and patents related to dendritic cell vaccines, immunotherapy biomarkers and methods for augmenting anti-PD-1 therapy. CC reports a pending patent related to high-affinity T-cell receptors that target the Merkel polyomavirus. JT reports consulting/advisory fees from Merck & Co, Bristol-Myers Squibb, AstraZeneca and Compugen; and a grant from Bristol-Myers Squibb. EJ, MK and BHM are employees of Merck & Co. SPF reports research funding from Merck. MAC reports research funding from Merck & Co. SLT reports that she or an immediate family member has stock and other ownership interests in Aduro Biotech, DNAtrix, Dracen Pharmaceuticals, Dragonfly Therapeutics, Ervaxx, Five Prime Therapeutics, Potenza Therapeutics, RAPT, Tizona Therapeutics, Trieza Therapeutics and WindMIL; a consulting or advisory role in Amgen, DNAtrix, Dragonfly Therapeutics, Dynavax, Ervaxx, Five Prime Therapeutics, Immunocore, Immunomic Therapeutics, Janssen Pharmaceuticals, MedImmune/AstraZeneca, Merck & Co., RAPT and WindMIL; research grants from Bristol-Myers Squibb and Compugen; patents, royalties and/or other intellectual property with Aduro Biotech, Arbor Pharmaceuticals, Bristol-Myers Squibb, Immunomic Therapeutics, NexImmune and WindMIL and travel, accommodations, and expenses from Bristol-Myers Squibb, Dragonfly and Five Prime Therapeutics.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.