Article Text

Abstract
Background NKTR-255 is a novel polyethylene glycol-conjugate of recombinant human interleukin-15 (rhIL-15), which was designed to retain all known receptor binding interactions of the IL-15 molecule. We explored the biologic and pharmacologic differences between endogenous IL-15 receptor α (IL-15Rα)-dependent (NKTR-255 and rhIL-15) and IL-15Rα-independent (precomplexed rhIL-15/IL-15Rα) cytokines.
Methods In vitro pharmacological properties of rhIL-15, NKTR-255 and precomplex cytokines (rhIL-15/IL-15Rα and rhIL-15 N72D/IL-15Rα Fc) were investigated in receptor binding, signaling and cell function. In vivo pharmacokinetic (PK) and pharmacodynamic profile of the cytokines were evaluated in normal mice. Finally, immunomodulatory effect and antitumor activity were assessed in a Daudi lymphoma model.
Results NKTR-255 and rhIL-15 exhibited similar in vitro properties in receptor affinity, signaling and leukocyte degranulation, which collectively differed from precomplexed cytokines. Notably, NKTR-255 and rhIL-15 stimulated greater granzyme B secretion in human peripheral blood mononuclear cells versus precomplexed cytokines. In vivo, NKTR-255 exhibited a PK profile with reduced clearance and a longer half-life relative to rhIL-15 and demonstrated prolonged IL-15R engagement in lymphocytes compared with only transient engagement observed for rhIL-15 and precomplexed rhIL-15 N72D/IL-15Rα Fc. As a consequent, NKTR-255 provided a durable and sustained proliferation and activation of natural killer (NK) and CD8+ T cells. Importantly, NKTR-255 is more effective than the precomplexed cytokine at inducing functionally competent, cytotoxic NK cells in the tumor microenvironment and the properties of NKTR-255 translated into superior antitumor activity in a B-cell lymphoma model versus the precomplexed cytokine.
Conclusions Our results show that the novel immunotherapeutic, NKTR-255, retains the full spectrum of IL-15 biology, but with improved PK properties, over rhIL-15. These findings support the ongoing phase 1 first-in-human trial (NCT04136756) of NKTR-255 in participants with relapsed or refractory hematologic malignancies, potentially advancing rhIL-15-based immunotherapies for the treatment of cancer.
- immunotherapy
- hematologic neoplasms
- natural killer T-cells
Data availability statement
All data relevant to the study are included in the article or uploaded as supplementary information.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Interleukin-15 (IL-15) is a common γ-chain cytokine that plays a crucial role in the development and function of natural killer (NK) and CD8+ memory T cells.1 2 It signals through the heterotrimeric IL-15 receptor (IL-15R) comprizing the IL-15Rα, IL-2Rβ and IL-2Rγ subunits.3 Once bound to membrane IL-15Rα, the IL-15/IL-15Rα complex is presented to IL-2Rβγ on the same (cis) or adjacent (trans) cells3 4 or cleaved from the cell surface allowing the soluble complex to bind to IL-2Rβγ.3 5 Engagement of IL-2Rβγ can activate Janus kinase (JAK)-signal transducer and activator of transcription (STAT5), phosphoinositide 3-kinase (PI3K) and mitogen-activated protein kinase (MAPK) signaling pathways.3 These responses have a fundamental impact on the development, function, and survival of NK and CD8+ T cells that is reflected in the loss of these cell populations in mice that are genetically deficient in IL-15 or IL-15Rα.6 7 Recombinant human IL-15 (rhIL-15)-based immunotherapeutics are of great interest for cancer treatment. In non-human primates, rhIL-15 induced the preferential activation and proliferation of NK and CD8+ T cells.8 Furthermore, in multiple experimental models, rhIL-15 demonstrated antitumor efficacy by enhancing cytotoxic function in host immune cells or adoptively transferred T cells.9 10 Despite a strong mechanistic rationale, the pharmacology of rhIL-15 has hindered its advance into clinical practice. In the first-in-human study of rhIL-15, the mean plasma half-life was approximately 2.5 hours following intravenous bolus administration.11 This relatively short half-life meant that daily dosing was required to achieve an optimal pharmacodynamic (PD) response, but was associated with unfeasible peak concentration-related toxicity.11 Alternative dosing strategies have been evaluated to reduce toxicity but the pharmacology of rhIL-15 continues to be a challenge.12
Alternative rhIL-15-based agonists have been developed to improve the pharmacokinetic (PK) profile of rhIL-15. Many are based on precomplexed rhIL-15/IL-15Rα molecules which combine rhIL-15 with a soluble version of IL-15Rα4 13 to act as an IL-2Rβ chain agonist independent of IL-15Rα cellular expression.14 Initially, precomplexed rhIL-15/IL-15Rα molecules were expected to mediate IL-15 responses with higher potencies, leading to preferential stimulation of IL-2Rβ expressing lymphocyte subsets.4 13 However, there is now a concern that greater stimulation of immune cells by precomplexed rhIL-15/IL-15Rα molecules may lead to lymphocyte exhaustion.4 13 One such molecule, N-803 (ALT-803), is a complex of an amino acid-substituted (N72D) rhIL-15 agonist and an IL-15R sushi domain fused to an IgG1 Fc.15 Despite having demonstrated some degree of therapeutic efficacy in clinical trials,16 17 the proliferative response to N-803 diminishes after repeat dosing, suggesting that prolonged treatment may lead to attenuated biological responsiveness (tachyphylaxis).15
NKTR-255 was engineered to improve on existing rhIL-15-based immunotherapeutics. This novel polyethylene glycol (PEG)-conjugate of rhIL-15 was designed to retain all known receptor binding interactions of the native IL-15 molecule, and to have an improved PK profile over rhIL-15. Here, we report the preclinical effects of NKTR-255 to characterize its potential as an anticancer therapeutic. We also explore the biologic and pharmacologic differences between endogenous IL-15Rα-dependent (NKTR-255 and rhIL-15) and IL-15Rα-independent (precomplexed rhIL-15/IL-15Rα) cytokines. Our results support the future development of NKTR-255 for the treatment of cancer.
Methods
Reagents
Reagents such as rhIL-15, rhIL-15/IL-15Rα (rhIL-15 and human IL-15Rα sushi complex), and rhIL-15 N72D/IL-15Rα Fc (rhIL-15 N72D and human IL-15Rα sushi/Fc fusion protein complex) were manufactured according to published literature (see online supplemental methods). NKTR-255 was manufactured by Nektar Therapeutics (San Francisco, California, USA).
Supplemental material
For in vivo experiments, NKTR-255 was dosed in protein equivalents, not including the mass of PEG. The concentration was adjusted to achieve the specified dose at an injection volume of approximately 5 mL/kg for mouse.
Cell lines
Daudi Burkitt lymphoma and FreeStyle 293-F cell lines from American Type Culture Collection and Thermo Fisher, respectively, were maintained in the recommended medium.
Animals
Female BALB/c mice aged 6–8 weeks and female Fox Chase severe combined immunodeficiency (SCID) mice (CB17/Icr-Prkdcscid/IcrIcoCrl) aged 6–7 weeks were from Charles River Laboratories (Wilmington, Massachusetts, USA). Female C57BL/6 wild-type and IL-15Rα knock out (KO) mice (B6;129×1-Il15ratm1Ama/J) aged 8–12 weeks were from Vivo Bio Tech (Hyderabad, India) and The Jackson Laboratory (Bar Harbor, Maine, USA), respectively. All animals were cohoused with dose-group members and provided standard irradiated diet and water ad libitum.
Flow cytometry
To characterize IL-15-mediated signal transduction, flow cytometry experiments were performed to evaluate the phosphorylation status of specific proteins in immune cell populations. Details on the antibodies used are provided in the online supplemental methods. Flow cytometry experiments were performed using a Fortessa X-20 cell analyzer (Becton Dickinson, Franklin Lakes, New Jersey, USA) and FACSCanto II (Becton Dickinson). Instrument settings (gain, compensation, and threshold) were set with machine software in conjunction with calibration beads, and a total of 100,000 events were collected. Data were analyzed using FlowJo software (FlowJo, Oregon, USA). After gating for lymphocytes, positive populations were identified based on fluorescence-minus-one controls. Immunophenotyping results were enumerated as percent relative (%) values for each phenotype.
IL-15 receptor binding by surface plasmon resonance
Surface plasmon resonance was used to determine IL-15 receptor binding of each rhIL-15, NKTR-255 and rhIL-15/IL-15Rα (see online supplemental methods). The binding affinities were obtained by kinetic measurement: kon rates measured by flowing analytes for 3 min and koff rates measured during a 10-min wash step. The KD values were a calculated ratio between koff and kon . Each assay was done in duplicate, and the results summarized as individual numbers and as averages and SD. Data were analyzed using Biacore T200 Evaluation Software V.1.1.1 (GE Healthcare).
In vitro binding and signaling assay with IL-15Rα-overexpressing 293-F cells
To generate the IL-15Rα-overexpressing cell line, the sequence of human IL-15Rα was obtained from GenBank (Accession # Q13261). The amino acid sequence, including the signal sequence, was codon-optimized for expression in human cell lines. This gene was inserted into a pcDNA3.1+ plasmid (Thermo Fisher) and transfected into FreeStyle 293-F cells (Thermo Fisher). Stably transfected clones were selected using neomycin. For the cell binding and signaling assay, IL-15Rα-overexpressing 293-F cells were incubated with serial dilutions of either rhIL-15, NKTR-255, or rhIL-15 N72D/IL-15Rα Fc (3, 10 and 30 nM) for 30 min at 37°C. Cells were washed three times and then surface binding of the cytokines to IL-15Rα was assessed by flow cytometry. The 293-F cells pre-incubated with rhIL-15, NKTR-255, and rhIL-15 N72D/IL-15Rα Fc were co-cultured with human peripheral blood mononuclear cells (PBMCs; n=3) for 20 min at 37°C. The induction of STAT5 phosphorylation (pSTAT5) was assessed in the CD3+, CD8+ T cells by flow cytometry.
In vitro signaling assay in mouse whole blood
Fresh whole blood samples from C57BL/6 wild-type or IL-15Rα KO mice were stimulated in vitro for 30 min at 37°C with serial dilutions of rhIL-15, NKTR-255, or rhIL-15/mIL-15Rα Fc (0.00155–1550 nM). The induction of pSTAT5 was assessed in CD3+, CD8+ T cells by flow cytometry.
In vitro signaling characterization in human whole blood
Fresh whole blood samples from six healthy donors were stimulated in vitro for 20 min at 37°C with serial dilutions of either rhIL-15, NKTR-255, rhIL-15/IL-15Rα, or rhIL-15 N72D/IL-15Rα Fc (0.00008–775 nM). The induction of pSTAT5, phosphorylated AKT (pAKT) and phosphorylated ERK (pERK) were assessed in CD3−, CD56+ NK cells, CD3+, CD8+ T cells and CD3+, CD4+ T cells by flow cytometry. The EC50 (half maximal effective concentration) value for each donor was calculated with the percentage of pSTAT5, pAKT and pERK positivity for each cell population from 4-parameter non-linear regression curves using Prism V.8.0 (GraphPad Software, San Diego, California, USA).
In vitro functional characterization in human PBMCs
For the proliferation assay, carboxyfluorescein succinimidyl ester (CFSE)-labeled human PBMCs from three healthy donors were stimulated in vitro for 5 days with serial dilutions of either rhIL-15 (0.02–30 nM), NKTR-255 (0.2–300 nM), rhIL-15/IL-15Rα (0.02–30 nM), or rhIL-15 N72D/IL-15Rα Fc (0.02–30 nM). The proliferation in each CD3−, CD56+ NK cell, CD3+, CD8+ T cell and CD3+, CD4+ T-cell population was assessed with the percentage of the divided cell population as measured by CFSE dilution using flow cytometry. EC50 values for dose responses were determined from 4-parameter non-linear regression curves using Prism V.8.0. For the immunophenotyping assay, human PBMCs from three healthy donors were stimulated in vitro overnight with serial dilutions of either rhIL-15 (0.1–100 nM), NKTR-255 (1–1000 nM), rhIL-15/IL-15Rα (0.1–100 nM), or rhIL-15 N72D/IL-15Rα Fc (0.1–100 nM) to assess CD69 and CD107a surface expression on CD3−, CD56+ NK cells and CD3+, CD8+ T cells by flow cytometry. For the granzyme B secretion assay, human PBMCs from three healthy donors were stimulated in vitro overnight with serial dilutions of either rhIL-15 (0.3–30 nM), NKTR-255 (1–100 nM), rhIL-15/IL-15Rα (0.3–30 nM), or rhIL-15 N72D/IL-15RαFc (0.3–30 nM) and secreted granzyme B was assessed by Meso Scale Discovery.
In vivo PK evaluation in mice
Mice (n=3 per time point in each group) received a single intravenous dose of rhIL-15 (0.3 mg/kg) and NKTR-255 (0.3 mg/kg). Blood was collected from the terminal bleed 0.03, 0.08, 0.25, 0.5, 1, 2, 4, and 6 hours and 1, 2, 3, and 4 days post-dose for rhIL-15 or 0.03, 0.25, 1, 4, and 8 hours and 1, 2, 3, 4, 5, 6, 7, and 10 days post-dose for NKTR-255. For PK analysis, the plasma IL-15 concentrations were measured using a qualified ELISA method. PK parameters were determined by non-compartmental analysis using Phoenix WinNonlin software (V.6.4; Certara USA, Princeton, New Jersey, USA).
In vivo PD evaluation in mice
For in vivo pSTAT5 studies, mice (n=3 per time point in each group) were administered a single intraperitoneal (i.p.) dose of rhIL-15 (0.3 mg/kg). Blood was collected 0.03, 0.25, 1, 6, and 24 hours post-dose. In a separate pSTAT5 study, mice (n=3 per time point in each group) were administered a single intravenous dose of NKTR-255 (0.3 mg/kg). Blood was collected 0.25, 1, and 6 hours and 1, 2, 3, 4, and 5 days post-dose and measured by the percentage of positivity of intracellular pSTAT5 in CD3−, NKp46+ NK cells, CD3+, CD8+ T cells and CD3+, CD4+ T cells by flow cytometry. In an in vivo proliferation study, mice (n=3 per time point in each group) were administered a single or three consecutive daily i.p. doses of rhIL-15 (0.3 mg/kg). After the first dose, blood was collected 6 hours and 1, 2, 3, 4, and 6 days. In a separate proliferation study, mice (n=4 per time point in each group) were administered a single intravenous dose of NKTR-255 at 0.01, 0.03, 0.1, or 0.3 mg/kg. Blood was collected on days 1, 3, 6, 10, and 14 post-dose and assessed for absolute number and Ki67 staining (proliferation marker) in CD3−, NKp46+ NK cells, CD3+, CD8+ T cells and CD3+, CD4+ T cells. Lymphocyte subpopulations were defined as the following groups: immature NK cells (CD11b−, CD27−), early NK cells (CD11b−, CD27+), high effector NK cells (CD11b+, CD27+), terminal effector NK cells (CD11b+, CD27−), naïve CD8+ T cells (CD44low, CD62Lhigh), CD8+ central memory T cells (Tcm; CD44high, CD62Lhigh), CD8+ effector memory T cells (Tem; CD44high, CD62Llow), and CD4+ Tregs (CD25+, FoxP3+). In a PD study with repeated doses of NKTR-255, mice (n=4 per time point in each group) received a single or three times weekly intravenous dose of NKTR-255 (0.3 mg/kg). After the final dose, blood was collected on Days 3 and 6 to assess Ki67+ proliferating populations in NKp46+ NK cells and CD3+, CD8+ T cells. In a comparison PD study between NKTR-255 and the rhIL-15/IL-15Rα complex molecule, mice (n=4 per time point in each group) were administered a single 0.1 mg/kg intravenous dose of NKTR-255 or rhIL-15 N72D/IL-15Rα Fc. Blood was collected on days 1, 3, 6, and 10 post-dose and assessed for absolute number, pSTAT5, pAKT, proliferation (Ki67), granzyme B and IL-2Rβ expression in CD3−, NKp46+ NK cells and CD3+, CD8+ T cells. Fold changes of absolute number were calculated relative to vehicle.
Daudi Burkitt lymphoma xenograft model
CB17 SCID mice (n=6/treatment group) were inoculated intravenously with 10 million exponentially growing Daudi cells. Four days later, mice were intravenously treated with a single dose of NKTR-255 (0.3 mg/kg), rhIL-15 N72D/IL-15Rα Fc (0.3 mg/kg), or vehicle. Animals were monitored daily for meeting survival endpoint criteria (hindlimb paralysis and morbidity). Survival rate differences between treatments were analyzed using the Kaplan-Meier log-rank test (GraphPad Prism, San Diego, California, USA). NK cell number and cytotoxic function analysis in Daudi B-cell lymphoma-bearing mice was conducted by flow cytometry. Cytotoxic activation of NK cells within the tumor environment in the femoral bone marrow was assessed by intracellular granzyme B staining and degranulation ability was measured by cell surface CD107a staining. Tumors were induced and treatments administered as for efficacy studies. Details on bone marrow collection can be found in the online supplemental methods. NK cells were identified as mouse CD45+, CD3–, NKp46+ cells.
Results
NKTR-255 maintains a similar receptor binding profile to that of rhIL-15, but with an improved PK profile
NKTR-255 comprises rhIL-15 covalently attached to a PEG moiety that is designed to retain binding affinity to IL-15Rα. Binding affinities of NKTR-255, rhIL-15, and rhIL-15/IL-15Rα for human IL-15Rα and IL-2Rβ are summarized in table 1.
Receptor affinity of rhIL-15, NKTR-255 and rhIL-15/IL-15Rα
As previously reported,18 19 rhIL-15 showed high affinity for human IL-15Rα versus modest affinity for human IL-2Rβ. The rhIL-15 had a unique slow dissociation rate when bound to IL-15Rα, which was consistent with the physiological function of membrane-bound IL-15/IL-15Rα to present to IL-2Rβγ. Relative to rhIL-15, NKTR-255 showed 4.5-fold and 4.4-fold reduced binding affinity for IL-15Rα and IL-2Rβ, respectively, due to its slower kon value, while the koff value was unchanged. The rhIL-15/IL-15Rα complex predictably showed no appreciable binding to IL-15Rα, and showed an approximately 10-fold higher affinity for IL-2Rβ than rhIL-15 (table 1). Equivalent data for mouse IL-15Rα is included in online supplemental table 1).
We next investigated cell-surface binding of NKTR-255 using an IL-15Rα-overexpressing 293-F cell line. NKTR-255 bound to surface IL-15Rα in 293-F cells with a similar magnitude to rhIL-15 (figure 1A), while rhIL-15 N72D/IL-15Rα Fc showed negligible binding. Importantly, surface-bound NKTR-255 retained the ability to induce IL-15R-mediated signaling to the same extent as surface-bound rhIL-15 in a co-culture assay with human PBMCs (figure 1B). We investigated IL-15Rα dependency in NKTR-255-induced signal transduction using whole blood from IL-15Rα KO mice. The dose-response curves of rhIL-15 and NKTR-255 were right-shifted in IL-15Rα KO mice while the dose-response curve of rhIL-15/mIL-15Rα Fc was unchanged (figure 1C). These data indicate that rhIL-15 and NKTR-255 use endogenous IL-15Rα, when available, for optimal IL-2Rβγ engagement and biological activity. The rhIL-15 and NKTR-255 retain some capacity for IL-15 signaling in the absence of IL-15Rα. In contrast, the rhIL-15/IL-15Rα complex directly engages IL-2Rβγ.

NKTR-255 is an IL-15Rα-dependent polymer-conjugated cytokine with an improved pharmacokinetic profile versus rhIL-15. (A) Representative flow cytometry histogram plots of surface IL-15 binding on IL-15Rα-overexpressing 293-F cells after incubation with rhIL-15, NKTR-255 or rhIL-15 N72D/IL-15Rα Fc (10 nM) for 30 min from two independent experiments. Solid line: cytokine treatment; dotted line: vehicle control. (B) pSTAT5 expression in CD8+ T cells derived from human peripheral blood mononuclear cells (n=3 donors indicated by a different color dot) in co-culture with cytokine pre-incubated IL-15Rα-overexpressing 293-F cells. ***p≤0.001, ****p≤0.0001 (Dunnett’s multiple comparisons test vs vehicle). (C) Cytokine-induced pSTAT5 expression in CD8+ T cells derived from C57BL/6 wild-type or IL-15Rα KO mice. Results are shown from two independent experiments. (D) Log-linear plasma concentration versus time profiles for rhIL-15 and NKTR-255 in mice (n=3). IL, interleukin; IL-15Rα, IL-15 receptor α; KO, knock out; pSTAT5, STAT5 phosphorylation; rhIL-15, recombinant human IL-15; STAT5, signal transducer and activator of transcription.
We confirmed that NKTR-255 displayed an improved mouse PK profile compared with rhIL-15. A single dose of 0.3 mg/kg intravenous NKTR-255 in mice exhibited a PK profile with reduced clearance and a longer half-life (clearance: 2.31 mL/hour/kg; effective half-life: 15.2 hours) relative to rhIL-15 (clearance: 507 mL/hour/kg; effective half-life: 0.168 hours) (figure 1D).
NKTR-255 has different signaling and leukocyte degranulation properties in vitro compared with precomplexed cytokines
We assessed the in vitro activity of rhIL-15, NKTR-255, and precomplexed cytokines (rhIL-15/IL-15Rα and rhIL-15 N72D/IL-15Rα Fc) by evaluating signaling pathways (JAK/STAT, PI3K and MAPK) downstream of IL-15R. Human whole blood was utilized to simultaneously evaluate the effects of the cytokines on different immune cell subsets since surface expression levels of IL-15Rα and IL-2Rβ differed on NK, CD8+ and CD4+ T cells (online supplemental figure 1).
Supplemental material
The pSTAT5, pAKT and pERK were dose-dependently activated in NK cells, while pSTAT5 was dominantly induced in CD8+ T and CD4+ T cells. The maximal response of each pathway in each cell type was equivalent between cytokines (figure 2A). The pSTAT5 EC50 values for NKTR-255 were approximately sixfold higher in NK cells and fourfold higher in CD8+ T and CD4+ T cells versus rhIL-15. The pSTAT5 EC50 values for rhIL-15/IL-15Rα and rhIL-15 N72D/IL-15Rα Fc were approximately fivefold lower in NK cells compared with rhIL-15, and were similar in CD8+ and CD4+ T cells (online supplemental table 2).

The rhIL-15 and NKTR-255 have different signaling and leukocyte degranulation properties in vitro compared with precomplexed rhIL-15/IL-15Rα cytokines. (A) Dose-response curves for pSTAT5, pAKT and pERK in NK, CD8+ T and CD4+ T cells following stimulation with rhIL-15, NKTR-255, rhIL-15/IL-15Rα or rhIL-15 N72D/IL-15Rα in human whole blood (n=6). (B) pSTAT5 EC50 ratios for CD8+ T/NK cells and CD4+ T/NK cells and (C) EC50 ratios for pAKT/pSTAT5 or pERK/pSTAT5 in NK cells (*p<0.05, **p<0.01; Dunnett’s multiple comparisons test vs rhIL-15). Human peripheral blood mononuclear cells (n=3) were stimulated in vitro with rhIL-15, NKTR-255, rhIL-15/IL-15Rα and rhIL-15 N72D/L-15Rα Fc overnight to assess (D) CD107a surface expression by flow cytometry (n=3) and (E) secreted granzyme B by Meso Scale Discovery (n=3; *p≤0.05, **p≤0.01, ***p≤0.001, ****p≤0.0001; Dunnett’s multiple comparisons test vs vehicle). EC50, half maximal effective concentration; IL, interleukin; IL-15Rα, IL-15 receptor α; NK, natural killer; pAKT, phosphorylated AKT; pERK, phosphorylated ERK; pSTAT5, STAT5 phosphorylation; rhIL-15, recombinant human IL-15; STAT5, signal transducer and activator of transcription.
NKTR-255 and rhIL-15 induced pSTAT5 signaling with the same potency in NK and T cells, while precomplexed cytokines preferentially induced signaling in NK cells (figure 2B). NKTR-255 and rhIL-15 showed higher sensitivity for the JAK/STAT5 pathway in NK cells relative to the PI3K and MAPK pathways versus precomplexed cytokines (figure 2C).
We next compared the functional effect of these agents in human PBMCs to determine their relative effects on immune cell populations and markers. In a proliferation assay, rhIL-15, NKTR-255, and precomplexed rhIL-15/IL-15Rα cytokines similarly induced proliferation of NK, CD8+ and CD4+ T cells in a dose-dependent manner and the profile of maximal response (the response of CD4+ T cells was lower than that of NK and CD8+ T cells) was also similar (online supplemental figure 2A). We further assessed the effect of each cytokine on the surface expression of CD69 (early activation marker) and CD107a (degranulation marker) in NK and CD8+ T cells. The maximal response of CD107a induction by rhIL-15 and NKTR-255 in both NK and CD8+ T cells was higher than that observed for rhIL-15/IL-15Rα and rhIL-15 N72D/IL-15Rα Fc (figure 2D), while the maximal responses for CD69 induction were similar across cytokines (online supplemental figure 2B). In agreement with observed differences in lymphocyte degranulation profiles, the maximal response of granzyme B secretion was also higher in human PBMCs stimulated with rhIL-15 and NKTR-255 compared with cells stimulated with rhIL-15/IL-15Rα and rhIL-15 N72D/IL-15Rα Fc (figure 2E). These data demonstrate that while all rhIL-15 cytokines induced granzyme B secretion in human PBMCs due to enhanced leukocyte degranulation, the maximal response was greater with NKTR-255 and rhIL-15 over precomplexed cytokines.
Supplemental material
NKTR-255 relative to rhIL-15 results in sustained IL-15R engagement and proliferation of NK and CD8+ T cells in vivo
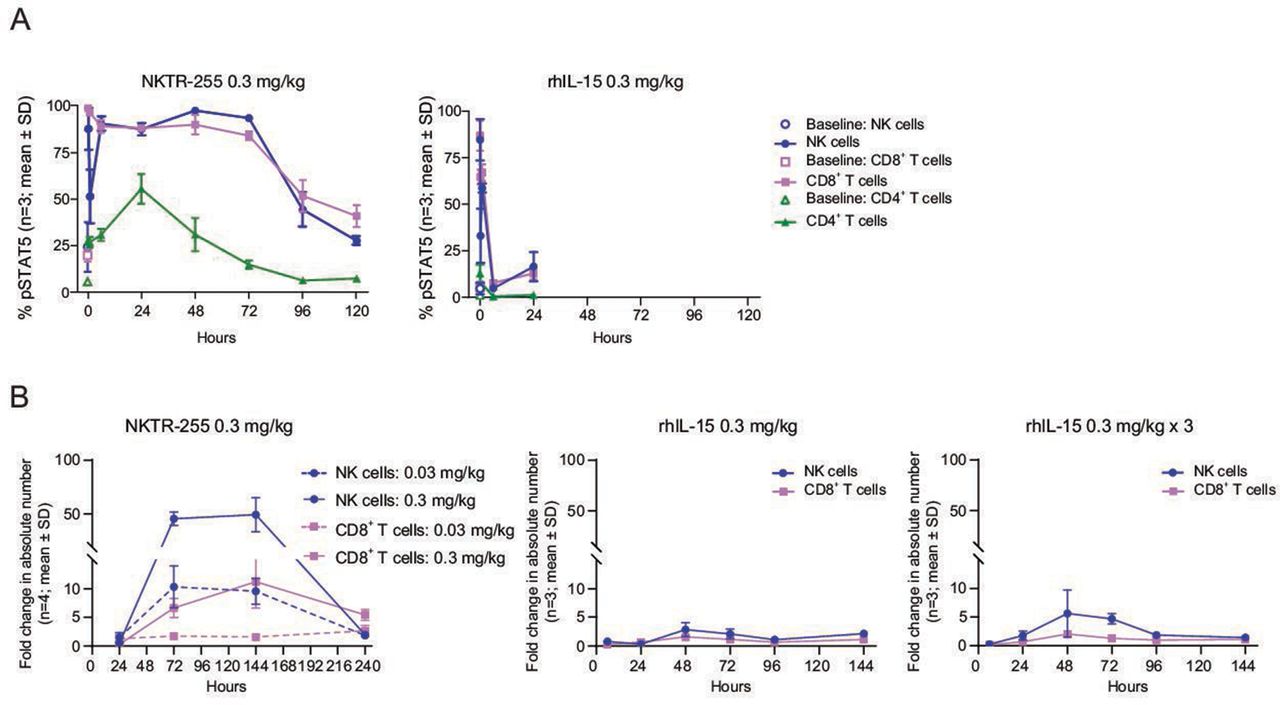
We next compared the in vivo PD profiles of rhIL-15 and NKTR-255 in normal mice. In concordance with the PK results, NKTR-255 showed a sustained pSTAT5 induction in all examined populations, maintaining the sensitivity order of rhIL-15 across the populations (NK cells>CD8+ T cells>CD4+ T cells). The pSTAT5 induction in NK and CD8+ T cells was rapid for NKTR-255 (returning to baseline by 120 hours) and was transient for rhIL-15 (returning to baseline by 6 hours post-treatment); there was a minimal effect in CD4+ T cells for both cytokines (figure 3A). In terms of NK and CD8+ T cell numbers, a single dose of NKTR-255 induced a strong and sustained response, whereas a single dose or three consecutive daily doses of rhIL-15 (0.3 mg/kg i.p.) only induced a mild increase (figure 3B). Increases in the number of NK cells relative to vehicle for NKTR-255 were 10-fold with 0.03 mg/kg and 46-fold with 0.3 mg/kg at 72 hours, and maintained until 144 hours, followed by a return to baseline at 240 hours. Whereas, for rhIL-15 the NK cell elevations were 2.9-fold with a single dose and 5.6-fold with three doses at 48 hours, followed by a return to baseline at 96 hours. A 6.7-fold increase in CD8+ T cells after treatment with NKTR-255 0.3 mg/kg (relative to vehicle) was observed at 72 hours, whereas CD8+ T cells only increased 1.6-fold for single-dose rhIL-15 and twofold for three doses of rhIL-15 at 48 hours.
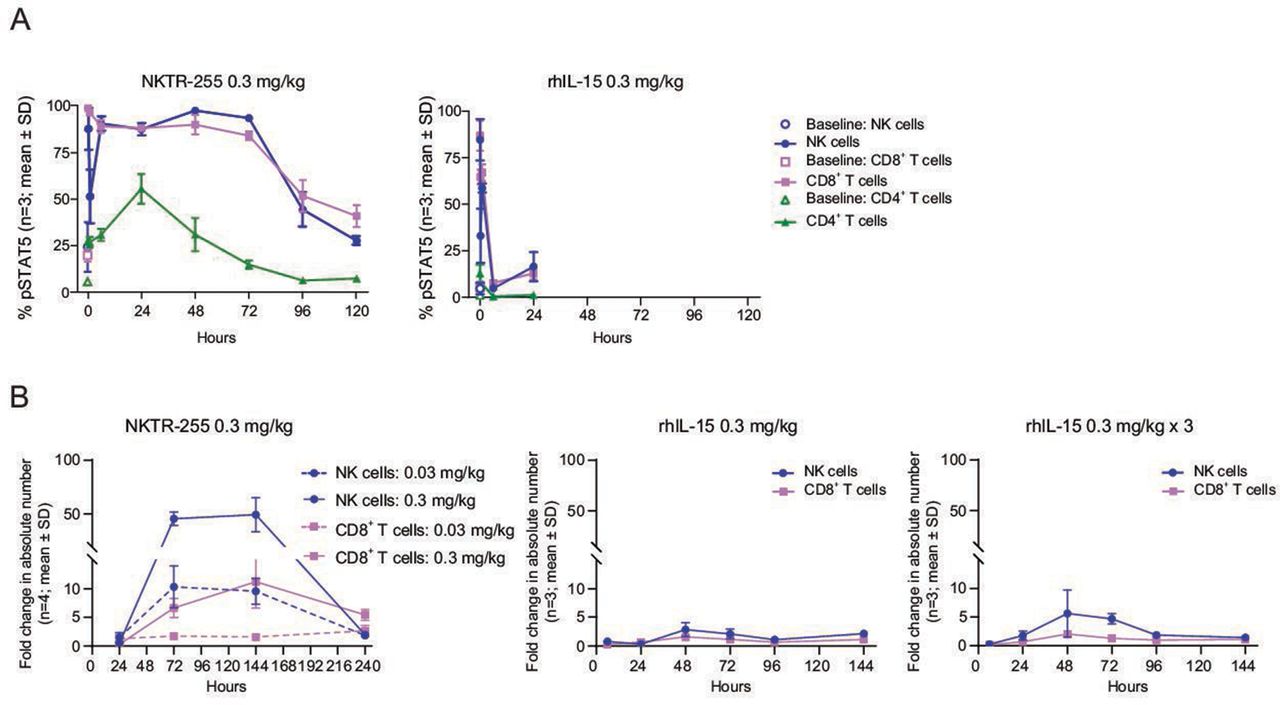
Improved pharmacokinetic profile of NKTR-255 relative to rhIL-15 results in sustained IL-15R engagement and proliferation of NK and CD8+ T cells. (A) Time-course of pSTAT5 induction in peripheral blood NK, CD8+ T and CD4+ T cells in mice after a single dose of rhIL-15 (0.3 mg/kg intraperitoneally) or NKTR-255 (0.3 mg/kg intravenously; n=3 per time point in each group). (B) Time-course of fold change (relative to vehicle) in absolute number of NK and CD8+ T cells after a single dose or three consecutive daily doses of rhIL-15 (0.3 mg/kg intraperitoneally) or a single dose of NKTR-255 (0.03 or 0.3 mg/kg intravenously) in mice. IL, interleukin; NK, natural killer; pSTAT5, STAT5 phosphorylation; rhIL-15, recombinant human IL-15; STAT5, signal transducer and activator of transcription.
We further investigated the proliferation effects of NKTR-255 on NK, CD8+ T and CD4+ T cell subpopulations. NKTR-255 dose-dependently induced an expansion of all NK cell subpopulations, in which immature, early, high effector and terminal effector NK cell numbers increased 32-fold, 94-fold, 160-fold and 26-fold at 0.3 mg/kg, respectively, compared with vehicle (online supplemental figure 3A). In concordance with cell number observations, NKTR-255 also induced Ki67 expression (proliferation marker) in nearly 100% of cells in each NK subpopulation at all dose levels (online supplemental figure 3A). Substantial and dose-dependent expansion of CD8+ T-cell subpopulations was also observed after a single dose of NKTR-255. Preferential expansion was observed in memory subpopulations, Tcm and Tem) compared with naïve CD8+ T cells (130-fold, 18-fold and 5.6-fold peak increase, respectively) at 0.3 mg/kg (online supplemental figure 3B). The enhanced responsiveness of CD8+ Tcm and CD8+ Tem populations was also evident by saturation of Ki67 expression at 0.03–0.3 mg/kg NKTR-255, which was not observed in naïve CD8+ T cells (online supplemental figure 3B). In contrast to NK and CD8+ T cells, NKTR-255 exhibited minimal increases in total CD4+ T cells or CD4+ regulatory T cells (online supplemental figure 3C).
Supplemental material
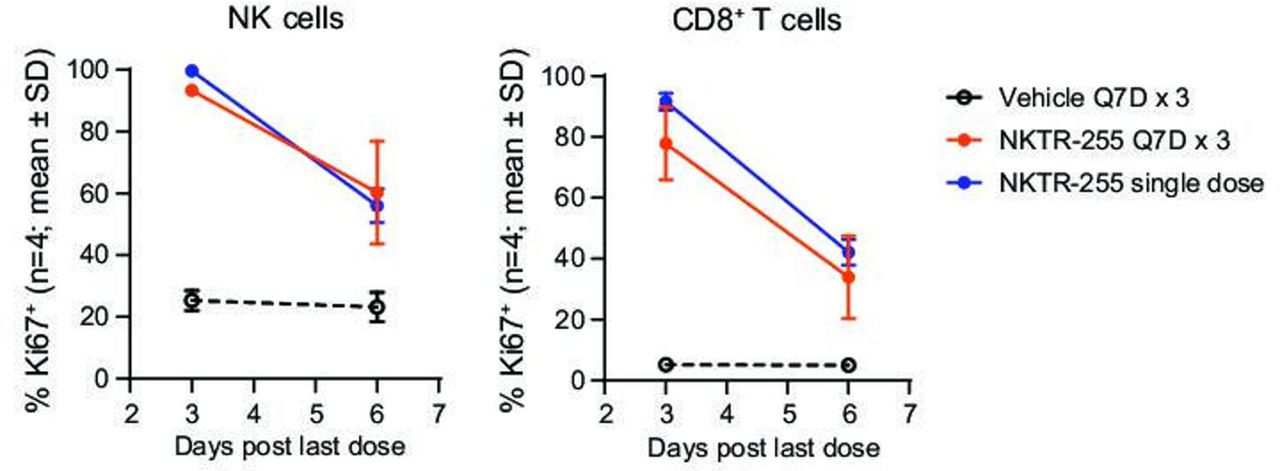
To investigate the potential for tolerance or tachyphylaxis in the PD response on repeated administration of NKTR-255, we assessed the proliferation response after a single administration or three consecutive weekly doses. NKTR-255 increased the Ki67+ population in NK and CD8+ T cells in agreement with the single-dose PD study described above. The magnitude of the proliferation response was maintained with a similar time course for single and repeated doses, without signs of tachyphylaxis (figure 4). The in vivo PD studies showed a substantial and sustained induction of receptor engagement, proliferation, and activation of NK and CD8+ T cells by NKTR-255.

NKTR-255 induces similar proliferation responses after single and repeated doses. Time-course of Ki67 positivity in NK and CD8+ T cells after a single or a third repeat intravenous dose of NKTR-255 (treatment repeated once every 7 days for three doses (Q7D×3) in mice). NK, natural killer; Q7D, once every 7 days.
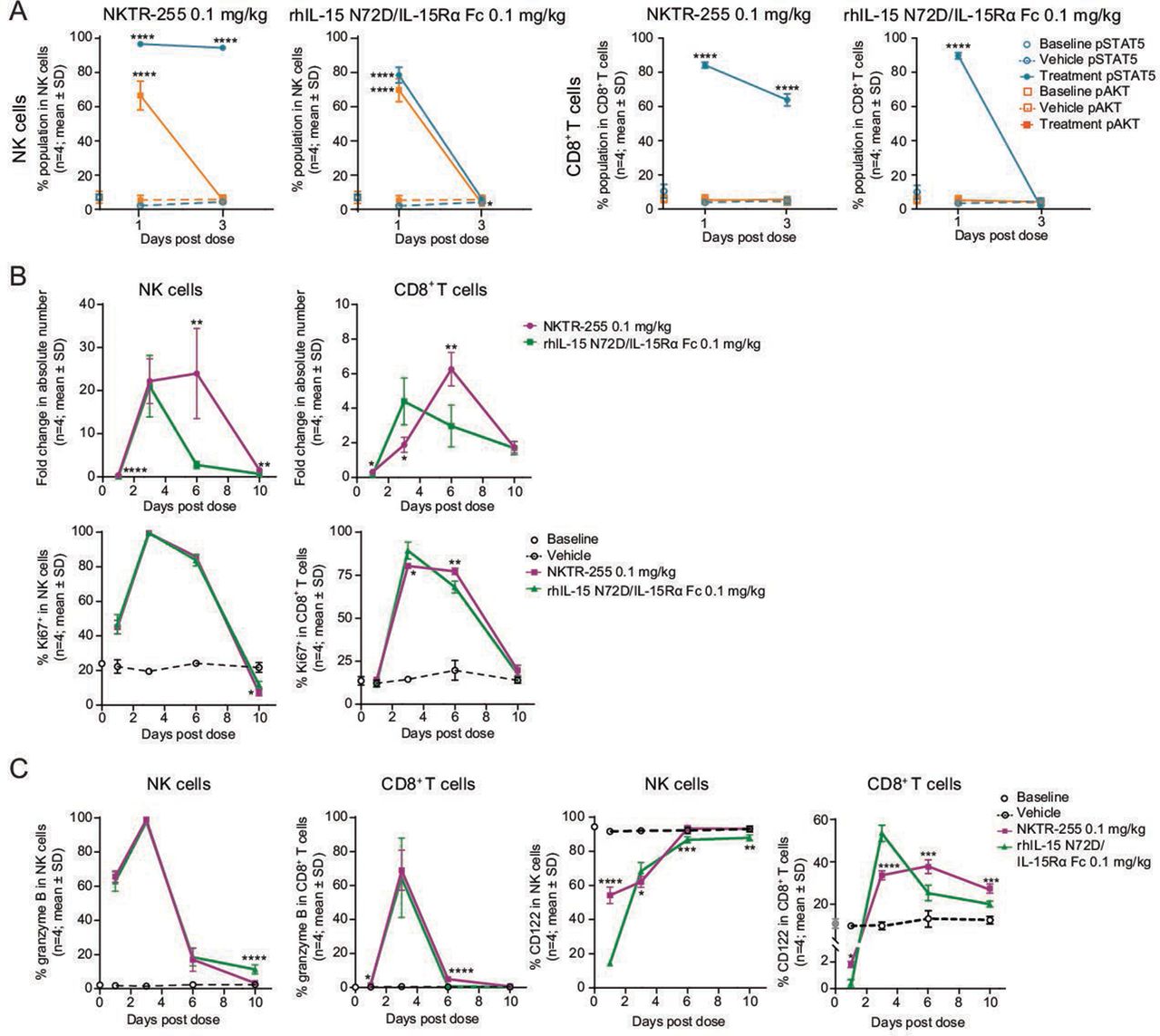
NKTR-255 has more sustained IL-15R engagement and provides a prolonged increase in NK cell numbers compared with precomplexed rhIL-15 N72D/IL-15Rα Fc cytokine
To compare in vivo PD properties between NKTR-255 and rhIL-15 N72D/IL-15Rα Fc, we investigated IL-15R engagement and cell expansion, proliferation, and activation after single doses of each agent in normal mice. NKTR-255 showed substantial pSTAT5 induction in both NK and CD8+ T cells that was maintained for at least 3 days after administration. rhIL-15 N72D/IL-15Rα Fc also activated pSTAT5 in the same cell populations and returned to baseline by day 3. The pAKT induction was activated and returned to baseline levels by day 3 for NKTR-255, and rhIL-15 N72D/IL-15Rα Fc (only observed in NK cells (figure 5A). In concordance with the kinetics of pSTAT5 induction, both cytokines induced robust NK cell expansion on day 3 (NKTR-255: 22-fold; rhIL-15 N72D/IL-15Rα Fc: 21-fold). On day 6, NKTR-255 showed sustained NK cell increases (24-fold over baseline) while NK cell increases induced by rhIL-15 N72D/IL-15Rα Fc had returned to baseline (figure 5B). For CD8+ T cells, the peak response for NKTR-255 was observed on day 6 (6.3-fold increase), while for rhIL-15 N72D/L-15Rα Fc was observed on day 3 (4.4-fold increase; figure 5B). In contrast, there was no difference in Ki67 induction kinetics between NKTR-255 and rhIL-15 N72D/IL-15Rα Fc in both NK and CD8+ T cells (figure 5B). Furthermore, both cytokines upregulated intracellular granzyme B expression in NK and CD8+ T cells with a similar magnitude and trajectory (figure 5C).

NKTR-255 has more sustained IL-15R engagement and prolonged NK cell increase compared with precomplexed rhIL-15 N72D/IL-15Rα Fc NK and CD8+ T cell time-course of (A) pSTAT5 and pAKT induction, (B) cell number increase (fold change relative to vehicle) and Ki67 positivity, and (C) intracellular granzyme B and cell surface IL-2Rβ (CD122) expression after a single dose of NKTR-255 (0.1 mg/kg intravenously) or rhIL-15 N72D/IL-15Rα Fc (0.1 mg/kg intravenously) in mice. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001, one-way analysis of variance, Sidak’s post-test treatment versus matching vehicle (A) or NKTR-255 versus rhIL-15 N72D/IL-15Rα Fc (Ki67 positivity) (C), and unpaired t-test (B). IL, interleukin; IL-15Rα, IL-15 receptor α; NK, natural killer; pAKT, phosphorylated AKT; pSTAT5, STAT5 phosphorylation; rhIL-15; recombinant human IL-15; STAT5, signal transducer and activator of transcription.
Finally, we measured IL-2Rβ (CD122) expression on the surface of NK and CD8+ T cells to examine ligand-induced receptor down-modulation as a possible cause of the difference in signaling kinetics between NKTR-255 and rhIL-15 N72D/IL-15Rα Fc. On day 1 post-treatment, IL-2Rβ expression by NK cells was substantially reduced versus baseline in IL-15 N72D/IL-15Rα Fc treated mice, whereas IL-2Rβ expression was only marginally reduced in NKTR-255 treated mice. IL-2Rβ surface expression recovered to baseline levels by day 6 in NK cells with both cytokines. In CD8+ T cells, IL-2Rβexpression remained above baseline after day 3 (figure 5C).
Together, these findings demonstrated the ability of NKTR-255 to induce more sustained IL-15R engagement and prolonged expansion of NK cells compared with precomplexed rhIL-15 N72D/IL-15Rα Fc.
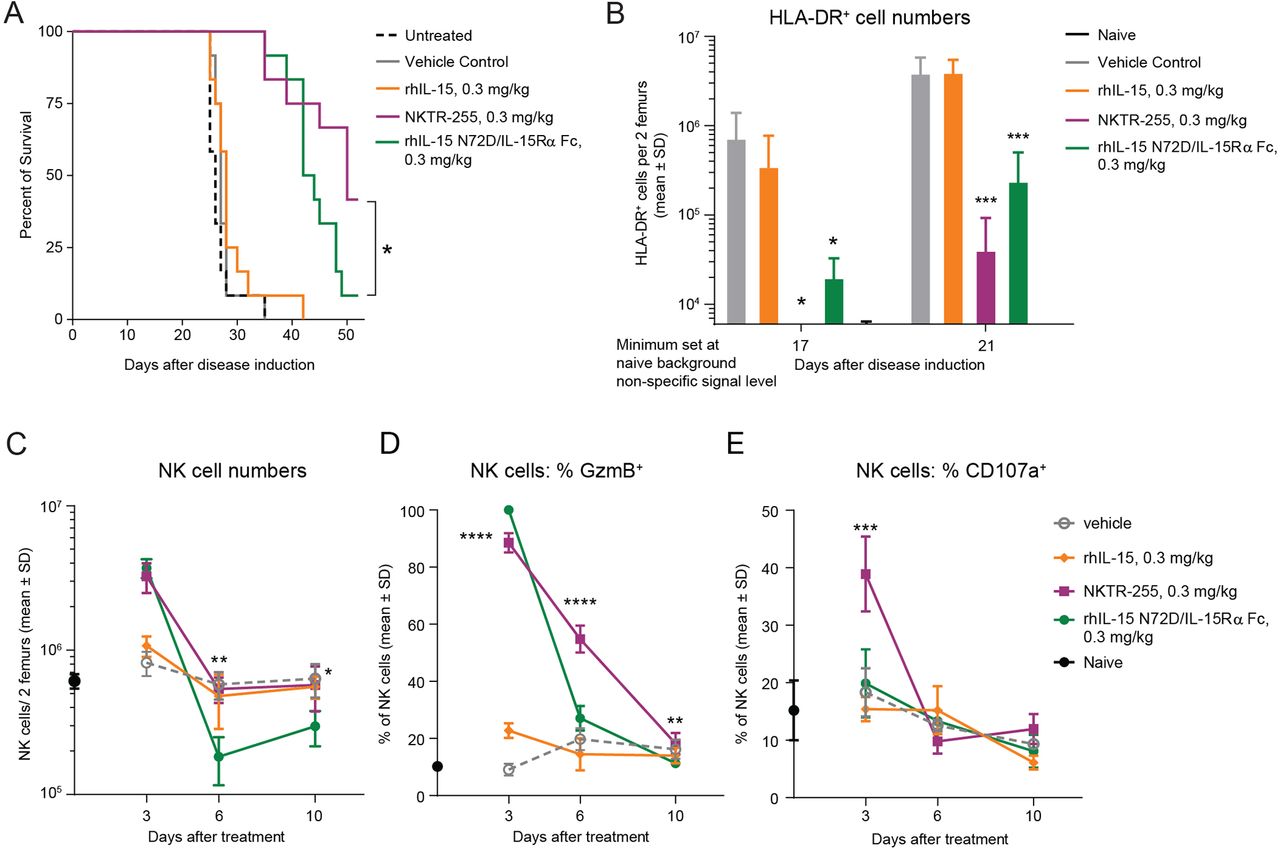
NKTR-255 has superior antitumor activity compared with precomplexed rhIL-15 N72D/IL-15Rα Fc in a Daudi lymphoma model
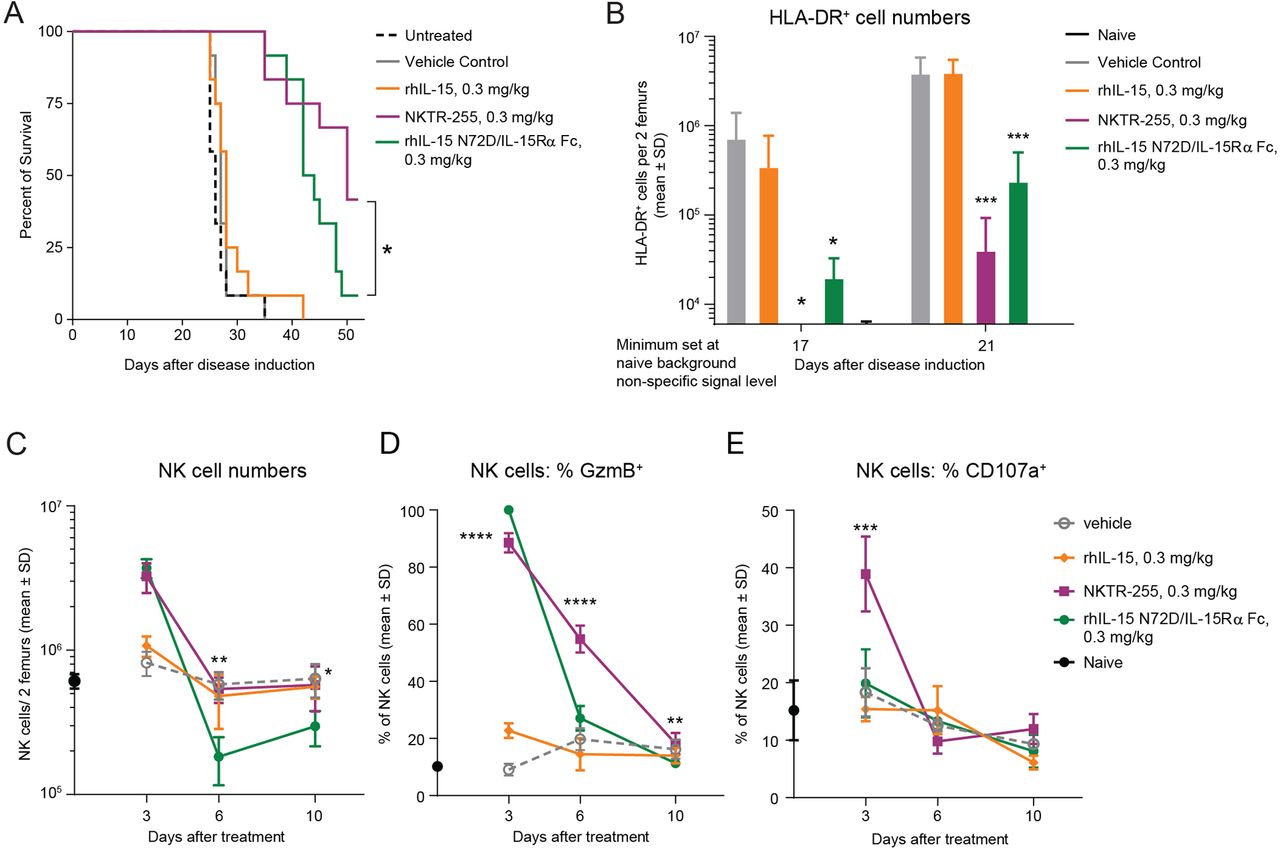
To compare the antitumor efficacy of NKTR-255 versus rhIL-15 N72D/IL-15Rα Fc, we assessed the survival rate of SCID mice bearing Daudi lymphoma xenografts. Mice in two independent efficacy studies treated with NKTR-255 reproducibly showed significantly increased survival rates compared with rhIL-15 N72D/IL-15Rα Fc-treated mice (42%–84% versus 8%–33%, respectively; p=0.05 log-rank test) on day 53 after tumor inoculation, indicating superior tumor control by NKTR-255 (figure 6A, online supplemental figure 4). As a control group analogous rhIL-15 treatment provided no significant survival benefit compared with vehicle or untreated animals. None of the untreated and vehicle control group animals survived past day 35. These results demonstrated that NKTR-255 elicited greater antitumor efficacy compared with rhIL-15 N72D/IL-15Rα Fc in a mouse lymphoma model. Superior survival efficacy of NKTR-255 was also reflected in tumor burden measurements in the femoral bone marrow of tumor bearing mice on days 17 and 21 after inoculation (figure 6B). While both NKTR-255 and rhIL-15 N72D/IL-15Rα Fc treatments resulted in significantly lower overall tumor cell counts in the bone marrow compared with vehicle or rhIL-15 treatment, on day 17 after tumor inoculation, NKTR-255 animals had undetectable tumor burden. Meanwhile, the rhIL-15 N72D/IL-15Rα Fc-treated mice showed observable tumor growth in the femoral bone marrow. These findings thus indicate superior Daudi cell depletion by NKTR-255 treatment. As tumor growth proceeded in the bone marrow, by day 21 after inoculation, lower tumor burden was observed in NKTR-255-treated animals compared with rhIL-15 N72D/IL-15Rα Fc treatment. These data indicate that NKTR-255 treatment was significantly more effective in delaying tumor outgrowth in the bone marrow compared with rhIL-15 N72D/IL-15Rα Fc, potentially explaining superior survival rates observed for NKTR-255-treated mice.
Supplemental material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
NKTR-255 has superior antitumor activity and NK cell cytotoxic function activation compared with precomplexed rhIL-15 N72D/IL-15Rα Fc and rhIL-15 in a Daudi lymphoma model. (A) Survival plot of Daudi Burkitt lymphoma xenograft model. *p<0.05, log-rank test. (B) Femoral bone marrow tumor load (human leukocyte antigen-DR+ [HLA-DR+] Daudi lymphoma cell count) on days 17 and 21 after tumor inoculation. (C) NK cell expansion, (D) granzyme B induction and (E) CD107a induction in the femoral bone marrow of tumor-bearing mice at 3, 6 and 10 days after treatment with vehicle, NKTR-255 (0.3 mg/kg, intravenously), rhIL-15 N72D/IL-15Rα Fc (0.3 mg/kg, intravenously) or rhIL-15 (0.3 mg/kg, intravenously). **p<0.01, ***p<0.001, ****p<0.0001, one-way analysis of variance, Sidak’s post-test; NKTR-255 versus IL-15 N72D/IL-15Rα Fc,. IL, interleukin; IL-15Rα, IL-15 receptor α; NK, natural killer; rhIL-15, recombinant human IL-15.
We next evaluated intratumoral NK cell PD responses in the femoral bone marrow of tumor-bearing mice (figure 6C–E). The increase in NK cell number on day 3 was comparable for NKTR-255 and rhIL-15 N72D/IL-15Rα Fc at fourfold and 4.5-fold, respectively (figure 6C). By day 6, NK cell numbers had returned to vehicle-treatment levels for NKTR-255 treated mice but was significantly reduced in the rhIL-15 N72D/IL-15Rα Fc treatment group (p<0.01). Induction and durability of NK cell cytotoxic function was evaluated in the femoral bone marrow by measuring intracellular granzyme B as a cytotoxic activation marker and cell surface CD107a expression as a degranulation marker on days 3, 6, and 10. Although NKTR-255 and rhIL-15 N72D/IL-15Rα Fc induced a comparable increase in granzyme B expressing NK cells (88.5% and 100% of total NK cells, respectively) (figure 6D), significant differences were observed in the durability of the granzyme B induction. With NKTR-255, granzyme B expression was maintained in 54.8% of NK cells at day 6, whereas the granzyme B-positive NK-cell fraction had diminished to 27.1% (p<0.0001 one way analysis of variance, Sidak’s post-test) for rhIL-15 N72D/IL-15Rα Fc, which was comparable with 19.8% for vehicle-treated mice (figure 6D). NK cell degranulation marker CD107a was efficiently induced by NKTR-255 on day 3 in 38.9% of NK cells compared with 18.3% in vehicle-treated animals. The rhIL-15 N72D/IL-15Rα Fc treatment led to only a small increase (19.9%) in CD107a expressing NK cells that was not significantly different from vehicle-treated animals (figure 6E). These PD data in the Daudi tumor model suggest that while NKTR-255 and precomplexed rhIL-15 N72D/IL-15Rα Fc induce comparable expansion of NK cell numbers in the tumor environment, NKTR-255 is more effective at inducing NK cells with cytotoxic function.
Discussion
NKTR-255 was designed to improve the PK profile of rhIL-15 while retaining binding affinity for endogenous IL-15Rα. Binding of IL-15 to IL-15Rα is crucial for its natural signaling profile, irrespective of the cis or trans mode of presentation to IL-2Rβγ.20 The slow dissociation rate of IL-15 from IL-15Rα aligns with its biological response in which the stabilized membrane-bound IL-15/IL-15Rα complex is presented to IL-2Rβγ.21 Binding selectivity between IL-15Rα and IL-2Rβ is also a key factor in determining the biological characteristics of IL-15, since the expression levels of IL-15Rα and IL-2Rβ are different on NK, CD8+ T, and CD4+ T cells.22 Our data indicate that NKTR-255 maintains the characteristic binding profile of rhIL-15 and behaves as an IL-15Rα-dependent cytokine in both cis-presentation and trans-presentation modes. In contrast, as previously reported,21 a precomplexed rhIL-15/IL-15Rα had a higher binding affinity for IL-2Rβ than rhIL-15 and behaved as an IL-15Rα-independent cytokine.
The IL-15R signaling characterization results show that the activation ratios between NK and T cells and between JAK/STAT5 and other signal transduction pathways within NK cells are different between rhIL-15 or NKTR-255 and the precomplexed cytokines. Precomplexed cytokines preferentially activated NK cells over T cells due to their higher IL-2Rβ expression (online supplemental figure 2). Activation of IL-15R signaling pathways in different proportions was recently reported in a study of the IL-15/IL-15Rα complex, RLI in a human T-lymphoma cell line.23 Importantly, results reported herein demonstrate that NKTR-255 fully engages IL-15R downstream signaling, maintaining the characteristic profile of native IL-15. Furthermore, in vitro data indicate that rhIL-15 and NKTR-255 can induce higher granzyme B secretion in human PBMCs due to enhanced leukocyte degranulation, compared with precomplexed cytokines. In vivo, NKTR-255 showed superior induction of NK cell cytotoxic function in the bone marrow tumor environment, including more efficient degranulation marker CD107a expression compared with precomplexed rhIL-15 N72D/IL-15Rα Fc during peak NK cell expansion after cytokine treatment.
The in vivo PD studies showed that NKTR-255 induces substantial and sustained receptor engagement, proliferation, and activation of NK and CD8+ T cells. Sustained receptor engagement is a key property that gives NKTR-255 an enhanced ability to increase NK and CD8+ T cell populations compared with rhIL-15, despite having lower in vitro potency. This concept was validated by comparing lymphocyte expansion levels in two Phase 1 studies of rhIL-15 as a bolus11 or continuous intravenous infusion.12 The NK cell expansion effect was substantially improved by continuous infusion. The effects on CD8+ T cell expansion were equivalent between studies. This suggests that different concentrations of rhIL-15 are required for the in vivo proliferation of NK and CD8+ T cells, as seen in our study and possibly explained by the higher IL-2Rβ expression on NK cells. In mice, increasing the NKTR-255 dose resulted in a larger increase of CD8+ T cells compared with three daily doses of rhIL-15. Importantly, with repeated administration, NKTR-255 did not show attenuation of Ki67 induction in both NK and T cells at least up to the third dose. This is in contrast to precomplexed rhIL-15/IL-15Rα cytokines, which cause attenuation of Ki67 induction after repeated doses.15 24
Our study revealed three different in vivo PD profiles between NKTR-255 and rhIL-15 N72D/IL-15Rα Fc: (1) the kinetics of the STAT5 pathway activation; (2) the kinetics of NK cell expansion; and (3) the magnitude of cell surface IL-2Rβ down-modulation. NKTR-255 showed more sustained pSTAT5 induction and NK cell number increase than rhIL-15 N72D/IL-15Rα Fc, while kinetics of the proliferation (Ki67) response were similar, suggesting that the difference lies in the survival of expanded NK cells. Indeed, in vitro studies have revealed that NK cell survival requires much lower IL-15 concentrations than proliferation, with the concentrations coinciding with pSTAT5 but not pAKT induction.25 Although rhIL-15 N72D/IL-15Rα Fc caused more substantial IL-2Rβ down-modulation compared with NKTR-255, especially in NK cells, this may explain the shorter duration of IL-15R engagement. IL-15Rα, the primary receptor of NKTR-255, is reported to be recycled after ligand-induced internalization26; however, IL-2Rβ, the primary receptor of precomplexed rhIL-15/IL-15Rα cytokines, is sorted to the degradation pathway.27 RLI, for example, has been reported to induce in vitro down-modulation of IL-2Rβ but not IL-15Rα, while rhIL-15 induces down-modulation of IL-15Rα but not IL-2Rβ.23 Collectively, these different pharmacologic properties of NKTR-255 translate into a greater antitumor efficacy in the Daudi lymphoma model compared with IL-15 N72D/IL-15Rα Fc.
In conclusion, our results indicate the critical importance of the mode of IL-15 receptor binding for achieving optimal lymphocyte responses and therapeutic use of an investigational IL-15R agonist. NKTR-255 is being investigated in a Phase 1 open-label, multicenter trial (NCT04136756) in relapsed or refractory multiple myeloma and non-Hodgkin's lymphoma. Together with its improved PK profile, NKTR-255 has the potential to advance IL-15-based cancer immunotherapies.
Supplemental material
Data availability statement
All data relevant to the study are included in the article or uploaded as supplementary information.
Ethics statements
Ethics approval
Animal studies were performed under protocols approved by the Institutional Animal Care and Use Committee of Nektar Therapeutics and the Canadian Council on Animal Care. All studies met the ethical and humane criteria for transportation, housing, and care established by the US NIH guidelines or Canadian animal care regulators.
Acknowledgments
Editorial assistance was provided by Sara Shaw PhD CMPP of BOLDSCIENCE Inc., funded by Nektar Therapeutics.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors TM and SK contributed to the planning of experiments, interpreting the data, and writing the manuscript. MM, TC, MA, SS, and JW planned and conducted in vitro studies. MH, PKu, RP, PQ, JC, and AM planned and conducted in vivo studies PO and WR planned and conducted pharmacokinetic studies. JH, DS, PZ, PKi, TC and YT planned and carried out the generation and characterization of proteins described in the manuscript. LM and JZ supervised the work. All authors reviewed and approved the final manuscript for publication.
Funding The study was funded by Nektar Therapeutics.
Competing interests All authors are current or former employees of Nektar Therapeutics and/or have Nektar Therapeutics stock ownership interests to disclose. PKu is currently employed by Gilead Sciences. MA is currently employed by HIBC Biopharma. PKi is currently employed by Immunocore. LM is currently employed by Invivoscribe.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.