Article Text

Abstract
Background T cell-based immunotherapies using chimeric antigen receptors (CAR) or bispecific antibodies (BsAb) have produced impressive responses in hematological malignancies. However, major hurdles remained, including cytokine release syndrome, neurotoxicity, on-target off-tumor effects, reliance on autologous T cells, and failure in most solid tumors. BsAb armed T cells offer a safe alternative.
Methods We generated ex vivo armed T cells (EATs) using IgG-[L]-scFv-platformed BsAb, where the anti-CD3 (huOKT3) scFv was attached to the light chain of a tumor-binding IgG. BsAb density on EAT, in vitro cytotoxicity, cytokine release, in vivo trafficking into tumors, and their antitumor activities were evaluated in multiple cancer cell lines and patient-derived xenograft mouse models. The efficacy of EATs after cryopreservation was studied, and gamma delta (γδ) T cells were investigated as unrelated alternative effector T cells.
Results The antitumor potency of BsAb armed T cells was substantially improved using the IgG-[L]-scFv BsAb platform. When compared with separate BsAb and T cell injection, EATs released less TNF-α, and infiltrated tumors faster, while achieving robust antitumor responses. The in vivo potency of EAT therapy depended on BsAb dose for arming, EAT cell number per injection, total number of EAT doses, and treatment schedule intensity. The antitumor efficacy of EATs was preserved following cryopreservation, and EATs using γδ T cells were safe and as effective as αβ T cell-EATs.
Conclusions EATs exerted potent antitumor activities against a broad spectrum of human cancer targets with remarkable safety. The antitumor potency of EATs depended on BsAb dose, cell number and total dose, and schedule. EATs were equally effective after cryopreservation, and the feasibility of third-party γδ-EATs offered an alternative for autologous T cell sources.
- immunotherapy
- adoptive
- neuroblastoma
- sarcoma
- therapies
- investigational
- translational medical research
Data availability statement
All data relevant to the study are included in the article or uploaded as supplementary information.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
T cell-based immunotherapy using chimeric antigen receptor (CAR), immune checkpoint inhibitors, or T cell-engaging bispecific antibody (T-BsAb) has shown major responses in human cancers, including metastatic stage or relapsed disease.1–4 However, clinical success has been limited to hematological malignancies and a few solid cancers with high tumor mutational burden. The immune suppressive tumor microenvironment (TME), cytokine release syndrome (CRS), life-threatening neurotoxicity, and permanent on-target off-tumor side effects are persisting immunological hurdles.5–8
Beyond CAR T cells, BsAb armed T cells have been investigated clinically.9–11 Using chemically conjugated anti-GD2 × anti-CD3 (hu3F8 × mouse OKT3; NCT02173093), anti-HER2 × anti-CD3 (trastuzumab × mouse OKT3; NCT00027807), and anti-EGFR × anti-CD3 (cetuximab × mouse OKT3; NCT04137536), BsAb armed T cells have been administered safely with minimal neurotoxicities or cytokine storm. They were safe at cell doses as high as 4×1010/dose in more than 170 patients in breast, prostate, and pancreatic cancers, as well as in hematological malignancies.9 11–13 However, robust responses remained elusive due to insufficient T cell infiltration and persistence, and/or impaired T cell function in the TME.
We previously demonstrated that the interdomain distance and cis-configuration of BsAb are critical for in vivo antitumor response.14 We take advantage of our structural design strategy for BsAb armed T cell. We generate ex vivo IgG-[L]-scFv-platformed BsAb armed T cells (EATs) and test their efficacy. First, we show that altering BsAb structural format significantly affects BsAb armed T cell infiltration into tumors and in vivo antitumor response, as expected by the relationship between tumor infiltrating lymphocyte (TIL) density and clinical outcome.15 16 Second, using EATs, we investigate the optimal surface density of BsAb, cytokine release, and in vivo T cell trafficking into tumors and persistence. Third, we study the factors determining the antitumor efficacy of EAT therapy. Fourth, we test the stability and efficacy of EATs after cryopreservation. Finally, we evaluate gamma delta T cells (γδ T cells) for EAT therapy as an off-the-shelf unrelated T cells that should avoid or minimize graft-versus-host disease.
Methods
T cell expansion ex vivo
Serially expanded T cells from a single donor were used for each individual experiment. Peripheral blood mononuclear cells (PBMCs) were separated from buffy coats (New York Blood Center) by Ficoll. Naïve T cells were purified using Pan T Cell Isolation Kit (Miltenyi Biotec, Cat#130096535) and expanded by CD3/CD28 Dynabeads (Gibco, Cat#11 132D) for 7–14 days in the presence of 30 IU/mL of interleukin-2 (IL-2). Expanded T cells were analyzed for their proportion of CD3(+), CD4(+), and CD8(+) T cells, and the fraction of CD4 or CD8 T cells was allowed between 40% and 60% to maintain consistency. Unless stated otherwise, these activated T cells were used for all T cell experiments.
Autologous T cell activation
Naïve T cells were separated from cryopreserved peripheral blood stem cell collections with institutional review board (IRB) approval. These cells were purified using Dynabeads untouched human T cell kit (Invitrogen, Cat# 11 344D) and expanded with CD3/CD28 Dynabeads (Gibco, Cat#11 132D) and 30 IU/mL of IL-2 for 10–14 days.
Gamma delta T cell activation
γδ T cells were expanded in one of two ways: (1) fresh PBMCs separated from buffy coats were cultured with 2 µM of zoledronic acid (Sigma, #Cat 165800066) and 800 IU/mL of IL-2 for 12–14 days according to protocols; and (2) fresh PBMCs were cultured with 2 µM of zoledronic acid and 30 ng/mL of IL15Rα-IL15 for 12–14 days. Cultured PBMCs were tested for their antigen expression using antibodies against human CD3 (BioLegend, Cat# 300308, RRID:AB_314044), CD4 (BioLegend, Cat# 357410, RRID:AB_2565662), CD8 (BioLegend, Cat# 300912, RRID:AB_314116), γδ T cell receptor (TCR) (BioLegend, Cat# 331207, RRID:AB_1575111), and αβ TCR (BioLegend, Cat# 306723, RRID:AB_2563001). The IL15Rα-IL15 complex was prepared as described.17
Tumor cell lines
Representative neuroblastoma cell line IMR-32 (ATCC-CCL-127), osteosarcoma cell line 143B (ATCC-CRL-8303) and U-2 OS (ATCC-HTB-96), primitive neuroectodermal tumor cell line TC-71 (ATCC-CRL-1598), prostate cancer cell line LNCaP-AR (ATCC-CRL-1740), and melanoma cell line M14 (UCLA-SO-M14) were used. All cancer cells were authenticated by short tandem repeats profiling using PowerPlex V.2.1 System (Promega, Cat# DC8942) and periodically tested for mycoplasma infection using a commercial kit (Lonza, Cat# LT07-318). The luciferase-labeled melanoma cell line M14Luc, osteosarcoma cell line 143BLuc, and neuroblastoma cell line IMR32Luc were generated by retroviral infection with an SFG-GF Luc vector.
GD2-BsAb or HER2-BsAb was mainly used for arming T cells. Hu3F8-BsAb specific for GD2 was built on the IgG-[L]-scFv format, in which the anti-CD3 huOKT3 single-chain variable fragment (ScFv) was linked to the carboxyl end of the anti-GD2 hu3F8 IgG1 light chain, where the N297A mutation was introduced to remove glycosylation and the K322A to remove complement activation—a combination to reduce spontaneous cytokine release.18 HER2-BsAb built on the IgG-[L]-scFv format carried a heavy chain variable (VH) identical to that of trastuzumab IgG1, again with both N297A and K322A mutations to silence fragment crystallisable (Fc) functions.19 Hu3F8 × mOKT3 and Herceptin × mOKT3 chemical conjugates were made as previously described by Sen et al.10 20 The other BsAbs were synthesized as previously described (US Patent #62/896415).18 21–23 Anti-CD33/anti-CD3 BsAb or anti-GPA33/anti-CD3 BsAb was used as a control BsAb.22 Biochemistry data of these BsAbs are summarized in online supplemental table 1.
Supplemental material
Antibody-dependent T cell-mediated cytotoxicity
T cell-mediated cytotoxicity was performed using 51Cr release,18 and EC50 was calculated using SigmaPlot software. Target cells were labeled with sodium 51Cr chromate (51CrNa2CrO4; Amersham, Arlington Heights, Illinois) at 100 µCi/106 cells at 37°C for 1 hour. After washing twice, these radiolabeled target cells were plated in 96-well plates. BsAb armed T cells were added to target cells at decreasing effector to target cell (E:T) ratios, at twofold dilutions from 50:1. After incubation at 37°C for 4 hours, the released 51Cr was measured by a gamma counter (Packed Instrument, Downers Grove, Illinois). The percentage of specific lysis was calculated using the formula where cpm represented counts per minute of 51Cr released.

Total release of 51Cr was assessed by lysis with 10% sodium dodecyl sulfate (SDS; Sigma, St Louis, Missouri, Cat# 71736) and background release was measured in the absence of effector cells and antibodies.
Cytokine release assays
Human T helper 1 (Th1) cell-released cytokines including IL-2, IL-6, IL-10, interferon (IFN)-γ and tumor necrosis factor (TNF)-α were measured using LEGENDplex Human Th1 Panel (BioLegend, Cat# 741035) in vitro and in vivo and analyzed by flow cytometry.
T cell arming
Activated T cells were armed with each BsAb for 20 min at room temperature and washed twice with phosphate-buffered saline (PBS). T cell surface BsAb density was measured by mean fluorescence intensity (MFI) using anti-idiotype antibody or anti-human IgG Fc antibody (BioLegend, Cat# 409303, RRID:AB_10900424). MFIs were referenced to antibody binding capacity using Quantum Simply Cellular microspheres (Bio-Rad, Cat# FCSC815A, RRID:AB_10061915).
Freeze-thaw of EATs
After arming, EATs were centrifuged at 500 g for 5 min at 4°C, and the cell pellet was suspended in T cell freezing medium (90% of fetal bovine serum (FBS) and 10% dimethyl sulfoxide (DMSO)) to achieve a cell concentration of 5×107 cells/1 mL, chilled to 4°C and aliquoted into 2 mL cryovials. Vials were immediately transferred to freeze at −80°C for 24 hours before transferring to liquid nitrogen. After storage cryovials were thawed in a 37°C water bath with gentle swirling for 1 min. The thawed cells were transferred to F10 media and centrifuged at 500 g for 5 min. They were analyzed for viability, phenotype, antibody binding, and cytotoxicity.
T cell transduction with tdTomato and click beetle red luciferase
T cells isolated from PBMCs were stimulated with CD3/CD28 Dynabeads (Gibco, Cat#11 132D) for 24 hours. T cells were transduced with retroviral constructs containing tdTomato and click beetle red luciferase in RetroNectin (Takara Bio, #Cat T100A/B)-coated six-well plates in the presence of IL-2 (100 IU/mL) and protamine sulfate (4 µg/mL). Transduced T cells were cultured for 8 days before use in animal experiments.
In vivo antitumor effects
Tumor cells suspended in Matrigel (Corning, Tewksbury, Massachusetts) were implanted in the flank of male BALB-Rag2−/−IL-2R-γc-KO (BRG) mice aged 6–10 weeks old (Taconic Biosciences).24 The following tumor cell lines were used: 1×106 of 143BLuc, 5×106 of IMR32Luc, 5×106 of M14Luc, 5×106 of HCC1954, 5×106 of LnCaP-AR, and 5×106 TC-71. Three osteosarcoma, two neuroblastoma, one Ewing sarcoma, and one breast cancer patient-derived tumor xenografts (PDXs) established from fresh surgical specimens with Memorial Sloan Kettering Cancer Center (MSKCC) IRB approval were used for in vivo experiments (online supplemental table 2). Treatment was initiated after tumors were established, with an average tumor volume of 100 mm3 when measured using TM900 scanner (Piera, Brussels, Belgium). Before treatment, mice were randomly assigned to each group. The T cell number administered per dose was 2×107 cells based on previous reports.25 When tumor growth reached 2000mm3or greater, mice were euthanized. CBC analyses, body weight, general activity, physical appearance and graft-versus-host-disease (GVHD) scoring were monitored. All animal experiments were repeated twice more with different donor’s T cells to ensure that our results were reliable.
Bioluminescence imaging
Luc(+) T cell engraftment and trafficking were quantified after intravenous injection of 3 mg D-luciferin (Gold Biotechnology, Cat# LUCK-100) on different days following T cell injection. Bioluminescence images were acquired using IVIS Spectrum CT In Vivo Imaging System (Caliper Life Sciences) and overlaid onto visible light images, to allow Living Image V.2.60 (Xenogen) to quantify bioluminescence in the tumor regions of interest (ROI). The total flux (photon/s) in the ROI was monitored.
Flow cytometry of blood, spleen and tumor
Peripheral blood and tumors were collected and analyzed by flow cytometry. Tumor tissue was dissociated into single cell suspensions as previously described.26 Antibodies against human CD3 (BioLegend, Cat# 300308, RRID:AB_314044), CD4 (BioLegend, Cat# 357410, RRID:AB_2565662), CD8 (BioLegend, Cat# 300912, RRID:AB_314116), and CD45 (BioLegend, Cat# 304012, RRID:AB_314400) were used to quantify T cell engraftment and subpopulations. Fluorescence of stained cells was acquired using either a BD FACSCalibur or a BD LSRFortessa (BD Biosciences, Heidelberg, Germany) and analyzed using FlowJo V.10.6.0 software (FlowJo, Ashland, Oregon).
Immunohistochemical staining
Fresh tumors were embedded in Tissue-Tek OCT (Miles Laboratories, Elkhart, Indiana) and snap-frozen in liquid nitrogen for storage at −80°C. The tumor sections were stained with mouse IgG3 mAb 3F8, as previously described.27 Paraffin-embedded tumor sections were tested for T cell infiltration using immunohistochemical (IHC) staining of human CD3, CD4, and CD8 T cells performed by the Molecular Cytology Core Facility of MSKCC using Discovery XT processor (Ventana Medical Systems), as previously described.26 Histological images were acquired with NIS-Elements V.4.0 imaging software and Nikon ECLIPSE Ni-U microscope.
Statistics
Statistical analysis of tumor growth or cytokine release was conducted using area under the curve (AUC). Two-tailed Student’s t-test was used to determine statistical difference between two sets of data, while one-way analysis of variance with Tukey’s post-hoc test was used to determine statistical differences among three or more sets of data. All statistical analyses were performed using GraphPad Prism V.8.0 for Windows (GraphPad Software, La Jolla, California; www.graphpad.com). P<0.05 was considered statistically significant. Asterisks indicate that the experimental p value is statistically different from the associated controls at *p<0.05, **p<0.01, ***p<0.001, and ****p<0.0001.
Results
T cells armed with GD2-BsAb built on the IgG-[L]-scFv platform had superior antitumor potency
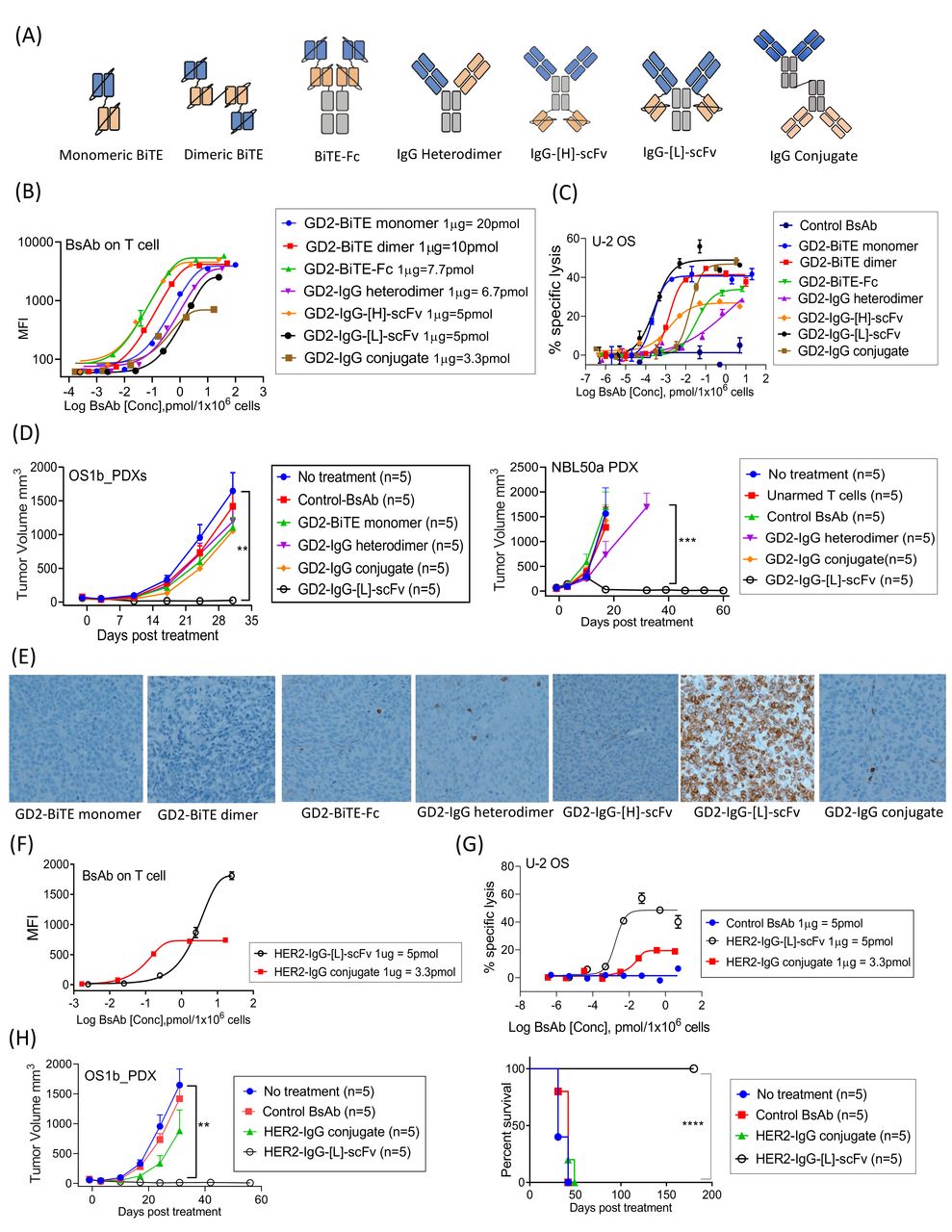
We armed T cells with the major formats of GD2-BsAb (figure 1A) and compared the efficacy. The biochemical properties of GD2-BsAbs are summarized in online supplemental table 3. Surface BsAb densities (measured by MFIs) on T cells were analyzed in figure 1B. While chemical conjugate (IgG conjugate) armed T cells had lower MFIs, other GD2-BsAb armed T cells showed comparable MFIs. In vitro cytotoxicity assay showed that IgG-[L]-scFv or monomeric bi-specific T-cell engagers (BiTE) armed T cells had the strongest in vitro potency, while dimeric BiTE or IgG conjugate armed T cells had comparable cytotoxicity (figure 1C). To compare in vivo antitumor potency, a fixed dose of T cells armed with different GD2-BsAb formats (2 µg of each BsAb/2×107 T cells) was intravenously administered into PDX-bearing mice (figure 1D and online supplemental figure 1). All EATs were well tolerated irrespective of BsAb formats, but only IgG-[L]-scFv GD2-BsAb armed T cells (GD2-EATs) exerted significant and durable antitumor responses prolonging survival. The in vivo efficacy of GD2-BsAb armed T cells strongly correlated with the density of tumor infiltrating CD3(+) T cells (TILs) by IHC staining of PDXs (figure 1E); GD2-EATs (T cells armed with GD2-IgG-[L]-scFv) showed substantially more abundant TILs compared with those armed with other BsAb formats.

Bispecific antibody platform has profound effects on the antitumor activity of bispecific antibody armed T cell immunotherapy. (A) BsAb structural platform.10 14 62 (B) BsAb armed T cells were stained with anti-idiotype antibody, and surface BsAb densities were measured as MFI by flow cytometry. (C) Antibody-dependent T cell-mediated cytotoxicity assay of GD2-BsAb armed T cells at increasing BsAb arming doses. The effector to target cell ratio (E:T ratio) was 10:1. (D) 2×107 of T cells were armed with 2 µg of each BsAb and administered intravenously two doses per week for 2–3 weeks in GD2(+) osteosarcoma PDX and GD2(+) neuroblastoma PDX mouse models. In vivo antitumor responses were compared among groups. (E) Immunohistochemical staining of CD3(+) tumor infiltrating lymphocytes in neuroblastoma PDX tumors treated with a variety of GD2-BsAb armed T cells (on day 10 after the beginning of treatment). (F) Surface BsAb density (MFI) of HER2-BsAb armed T cells. (G) HER2-BsAb armed T cell induced cytotoxicity against osteosarcoma cell line U-2 OS at an E:T ratio of 10:1. (H) HER2-BsAb armed T cells (2 µg of HER2-BsAb/2×107 of T cells) were administered intravenously twice per week for 3 weeks in HER2(+) osteosarcoma PDX-bearing mice. In vivo antitumor effect was compared. **P<0.01, ***P<0.001, ****P<0.0001. BiTE, Bi-specific T-cell engagers; BsAb, bispecific antibodies; GD2, disialoganglioside; HER2, human epidermal growth factor receptor 2; MFI, mean fluorescence intensity; PDX, patient-derived tumor xenograft; scFv, single-chain variable fragment.
Alongside GD2-BsAb armed T cells, we also compared the efficacy between HER2-EATs (armed with HER2-IgG-[L]-scFv) and HER2-IgG conjugate (Herceptin × OKT3) armed T cells proven safe in clinical trials.9 28 HER2-EATs showed higher BsAb densities (MFIs) (figure 1F) and more potent tumor cell killing than HER2-IgG conjugate armed T cells (figure 1G). HER2-EATs were much more effective in vivo for tumor response (p<0.01) and for survival (p=0.0020) (figure 1H).
EATs acquired target antigen-specific cytotoxicity
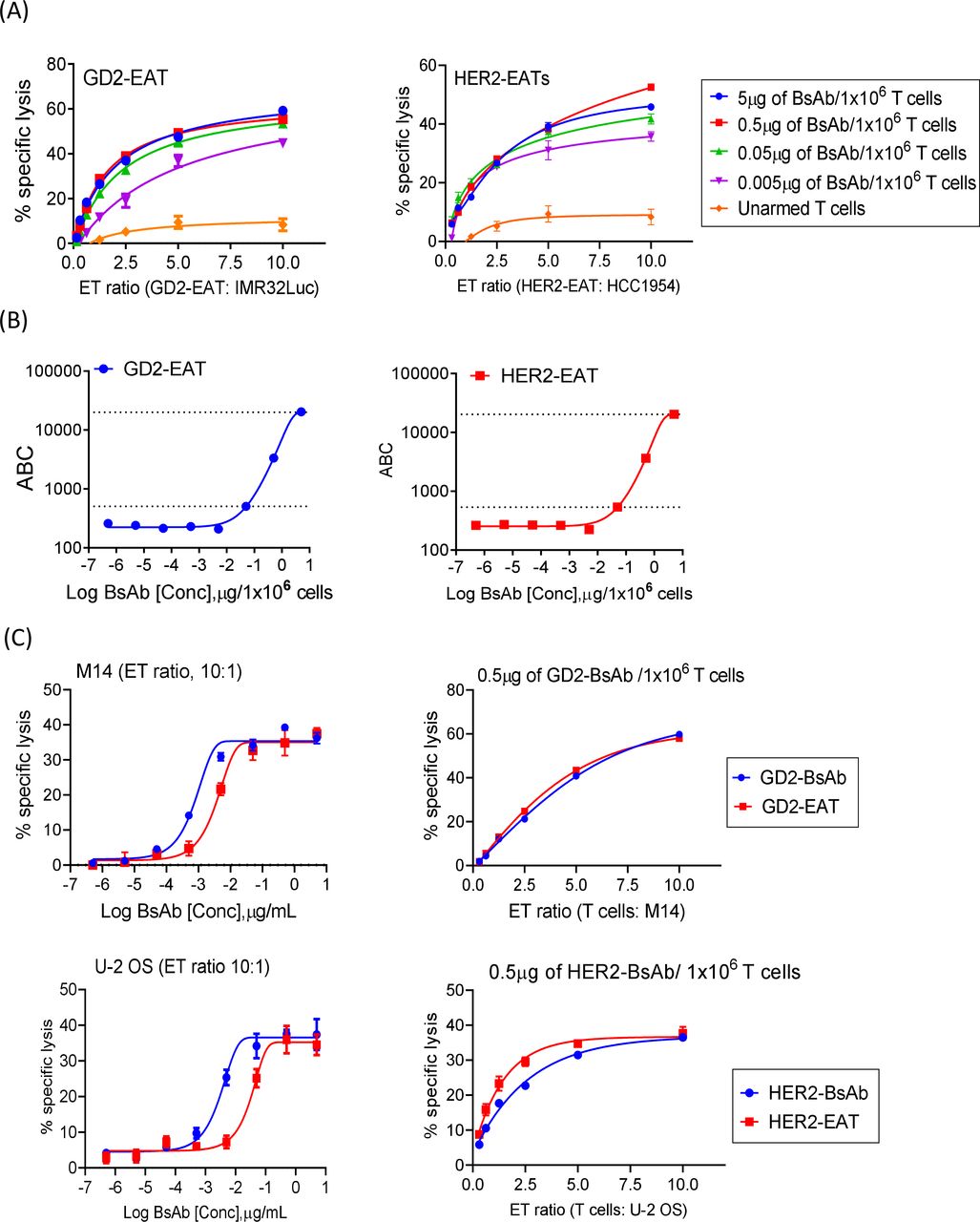
With this IgG-[L]-scFv BsAb platform we conducted the rest of our experiments. Given the finite CD3 TCR density on human T cell,29 we set out to identify the optimal BsAb density on T cell. The in vitro cytotoxicity of EAT over a range of E:T ratios and BsAb arming dosages was studied (figure 2A). GD2-EATs and HER2-EATs both showed maximal cytotoxicity between 0.05 µg and 5 µg of BsAb/1×106 T cells where quantitated between 500 and 20,000 BsAb molecules per T cell (figure 2B). When we compared the in vitro cytotoxicity of EATs with soluble BsAb under identical E:T ratios (figure 2C), the potencies of GD2-EATs and HER2-EATs were ~10-fold lower (EC50) than soluble BsAbs; however, the maximal killing efficacy was comparable.

Ex vivo arming of T cells with IgG-[L]-scFv-platformed BsAb. (A) Antibody-dependent T cell-mediated cytotoxicity assay of GD2-EATs and HER2-EATs at increasing effector to target ratios (E:T ratios) and at increasing BsAb doses. (B) Surface BsAb density on EAT was analyzed using anti-human IgG Fc-specific antibody and anti-mouse quantum beads. Geometric MFIs of GD2-EATs and HER2-EATs were measured with increasing arming dose of either GD2-BsAb or HER2-BsAb, and BsAb density (MFI) on EAT was referenced to antibody binding capacity (ABC). (C) Comparison of in vitro cytotoxicity between EATs and soluble BsAb with identical E:T ratios, for both anti-GD2 and anti-HER2 systems. BsAb, bispecific antibodies; EATs, ex vivo armed T cells; GD2, disialoganglioside; HER2, human epidermal growth factor receptor 2; MFI, mean fluorescence intensities.
BsAb delivered on T cells had reduced cytokine release
Next, we compared cytokine release between EATs and T cells in the continuous presence of BsAb (BsAb plus T cells). Cytokine release was evaluated throughout each step of T cell arming (figure 3A). First, Th1 cell cytokines (IL-2, IL-6, IL-10, IFN-γ, and TNF-α) were measured in the supernatants after 20 min incubation (prewash) and then after the washing step (postwash) (figure 3B). Although the released cytokine levels were generally low, IFN-γ and TNF-α did increase with the increment of GD2-BsAb (online supplemental figure 2A), which were removed after the washing steps (online supplemental figure 2B). Next, after co-culture with target cells, T cell cytokine levels were measured again and compared among groups. The cytokines surged after exposure to target cells. Both GD2-EATs and T cells coincubated with GD2-BsAb (GD2-BsAb plus T cells) released more cytokines in proportion to BsAb dose (online supplemental figure 2C,D). However, GD2-EATs produced significantly less amount of IL-2 (p=0.0071), IL-10 (p=0.0203), IFN-γ (p=0.0043), and TNF-α (p=0.0162) compared with GD2-BsAb plus T cells. Finally, we analyzed in vivo cytokine levels at different time points after GD2-EAT or GD2-BsAb plus T cell injection (figure 3D and online supplemental figure 3). GD2-EATs showed an early IL-2 and IFN-γ release, while GD2-BsAb plus T cells showed a surge of TNF-α at 4 hours and IL-6 at 48 hours. The AUC for TNF-α was significantly higher in GD2-BsAb plus T cells compared with GD2-EATs (p=0.0033).

Ex vivo arming of T cells could significantly reduce cytokine release. (A) Th1 cell cytokines (IL-2, IL-6, IL-10, IFN-γ, and TNF-α) released by T cells were measured in each step of arming: (a) in the supernatants after 20 min of incubation with GD2-BsAb (prewash), (b) after the second washing step (postwash), and (c and d) after incubation with target cells (GD2(+) M14 melanoma cell line) at 37℃ for 4 hours. (B) Cytokine release was compared between prewash supernatants (a) and postwash supernatants (b). (C) Cytokine release was compared between GD2-BsAb plus T cells (c), GD2-EATs (d) and unarmed T cells after exposure to target antigen. (D) After intravenous injection of GD2-EATs (armed with 10 µg of GD2-BsAb/2×107 cells) or GD2-BsAb (10 µg) plus unarmed T cells (2×107 cells) into osteosarcoma PDX-bearing mice, serum Th1 cytokine levels were measured at different time points. *P<0.05, **P<0.01, ***P<0.001, and ****P<0.0001. BsAb, bispecific antibody; EATs, ex vivo armed T cells; GD2, disialoganglioside; IFN, interferon; IL, interleukin; PBS, phosphate-buffered saline; PDX, patient-derived tumor xenograft; Th1, T helper 1; TNF, tumor necrosis factor.
EATs facilitate T cell trafficking into tumors
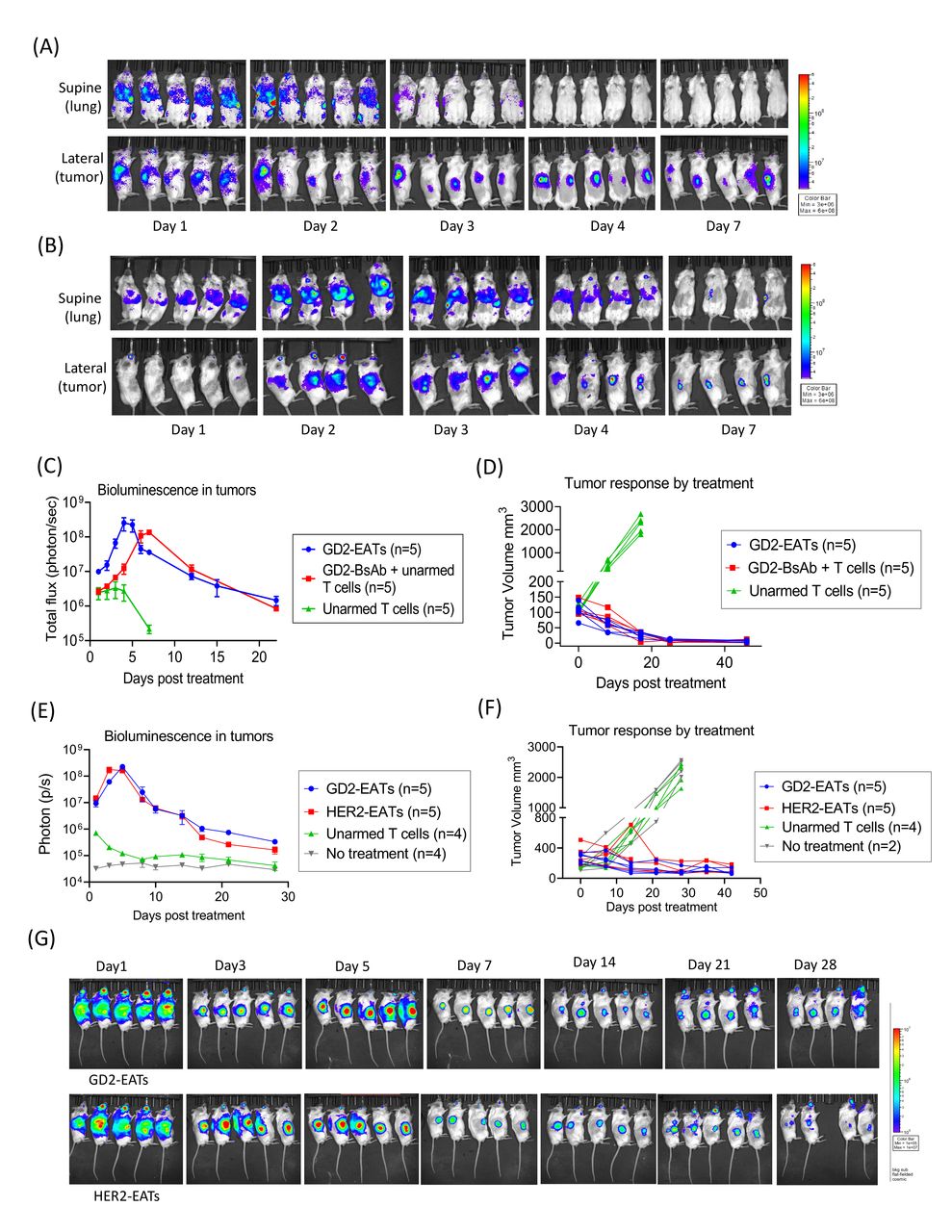
T cell trafficking and persistence in tumors play an essential role in tumor immunity. To quantitate how efficiently EATs traffic into tumors and persist in vivo, we generated luciferase transduced T cells and armed with GD2-BsAb. Luc(+) GD2-EATs or Luc(+) unarmed T cells were intravenously administered into neuroblastoma PDX-bearing mice. Without GD2-BsAb arming, Luc(+) T cells did not localize to tumors and dissipated. In contrast, Luc(+) GD2-EATs rapidly trafficked into GD2(+) tumors following a transient sequestration in the lungs on day 1. The luciferase signals (total flux) of GD2-EATs in tumors increased over time to peak on day 4 (figure 4A,C). On the other hand, Luc (+) unarmed T cells with intravenous GD2-BsAb showed delayed egress from the lungs and the liver, and the bioluminescence signals in tumors peaked around day 7 (figure 4B,C). As tumors regressed, the total bioluminescence of Luc(+) GD2-EATs also diminished (figure 4C,D). In a second set of EAT trafficking study using osteosarcoma PDX model, EATs persisted longer with residual tumors (figure 4E,F). The bioluminescence of Luc(+) GD2-EATs or Luc(+) HER2-EATs in tumors peaked around day 5 and diminished slowly with still detectable signals over 1 month (figure 4G).

EATs showed faster tumor homing kinetics than BsAb-directed unarmed T cells, rapidly bypassing lung sequestration. Luciferase transduced T cells were expanded and armed with BsAb (Luc(+) GD2-EATs). Luc(+) GD2-EATs (10 µg of GD2-BsAb/2×107 T cells) or Luc(+) unarmed T cells (2×107 cells) with or without GD2-BsAb (10 µg) were administered intravenously into GD2(+) neuroblastoma PDX mice when the average tumor volume reached 100 mm3. (A) Bioluminescence images of GD2-EATs trafficking into tumors over days. (B) Bioluminescence images of GD2-BsAb directed unarmed T cell trafficking into tumors over days. (C) Quantitation of T cell infiltration into tumors over time measured by bioluminescence (n=5 mice/group) expressed as total flux or radiance (photons/s) per pixel integrated over the tumor contour (ROI). (D) Tumor growth curves of individual mice treated with GD2-EATs, GD2-BsAb plus unarmed T cells, or unarmed T cells. To test the in vivo persistence of target antigen-specific EATs, Luc(+) GD2-EATs (armed with 10 µg of GD2-BsAb/2×107 T cells) or Luc(+) HER2-EATs (10 µg of HER2-BsAb/2×107 T cells) were intravenously administered into osteosarcoma TEOS1C PDX-bearing mice. Additional two doses of non-luciferase transduced GD2-EATs or HER2-EATs were administered on day 7 and day 14. (E) Quantitation of bioluminescence of Luc(+) EATs in tumors post-treatment. (F) Tumor growth curves of individual mice treated with GD2-EATs, HER2-EATs, or unarmed T cells, or no treatment. (G) Bioluminescence images of Luc(+) GD2-EATs (upper) or Luc(+) HER2-EATs (lower) in tumors over time. Bioluminescence of Luc(+) EATs was detected over 28 days postinjection. BsAb, bispecific antibodies; EATs, ex vivo armed T cells; GD2, disialoganglioside; HER2, human epidermal growth factor receptor 2; PDX, patient-derived tumor xenograft; ROI, region of interest.
EATs showed potent antitumor activity with minimal toxicities in vivo
Autologous GD2-EATs were equally effective in vivo
To remove allogeneic T cell-induced graft-versus-xenograft effect in humanized mouse models,30 31 we tested autologous T cells derived from the same donor of tumor xenografts and evaluated the antitumor response of EATs (online supplemental figure 4). Autologous GD2-EATs significantly suppressed tumor growth, demonstrating the antitumor response of EATs is independent from the allogeneic ‘graft-versus-cancer’ effect, which has been a confounding factor for efficacy and toxicity assessment in humanized mouse models. Since autologous T cell-PDX pairs were in short supply, we proceeded with the rest of EAT studies using random donor T cells.
EATs exerted significant antitumor effects in a variety of tumor targets
In vivo antitumor effects of EATs were tested in a panel of xenograft mouse models. Four to six doses of EATs (10 µg of each BsAb/2×107 cells) were administered intravenously with a frequency of two doses per week. GD2-EATs were tested against neuroblastoma PDXs (Piro20Lung), neuroblastoma cell line (IMR32Luc) xenografts, and melanoma cell line (M14Luc) xenografts, respectively (online supplemental figure 5). HER2-EATs were tested against osteosarcoma PDXs (TEOS1C), breast cancer PDXs (M37), and osteosarcoma cell line (143BLuc) xenografts (online supplemental figure 6). Beyond GD2 and HER2, EATs targeting other cancer antigens including STEAP1 (six transmembrane epithelial antigen prostate-1) on Ewing sarcoma cell line (TC71), PSMA (prostate membrane antigen) on prostate cancer cell line (LNCaP-AR), and EGFR (epidermal growth factor receptor) on osteosarcoma cell line (143B) were tested against each target cell line xenograft (online supplemental figure 7); in each instance, EATs exerted significant antitumor effects, without weight loss or adverse events during follow-up period (online supplemental figure 8).
Factors determining in vivo efficacy of EATs
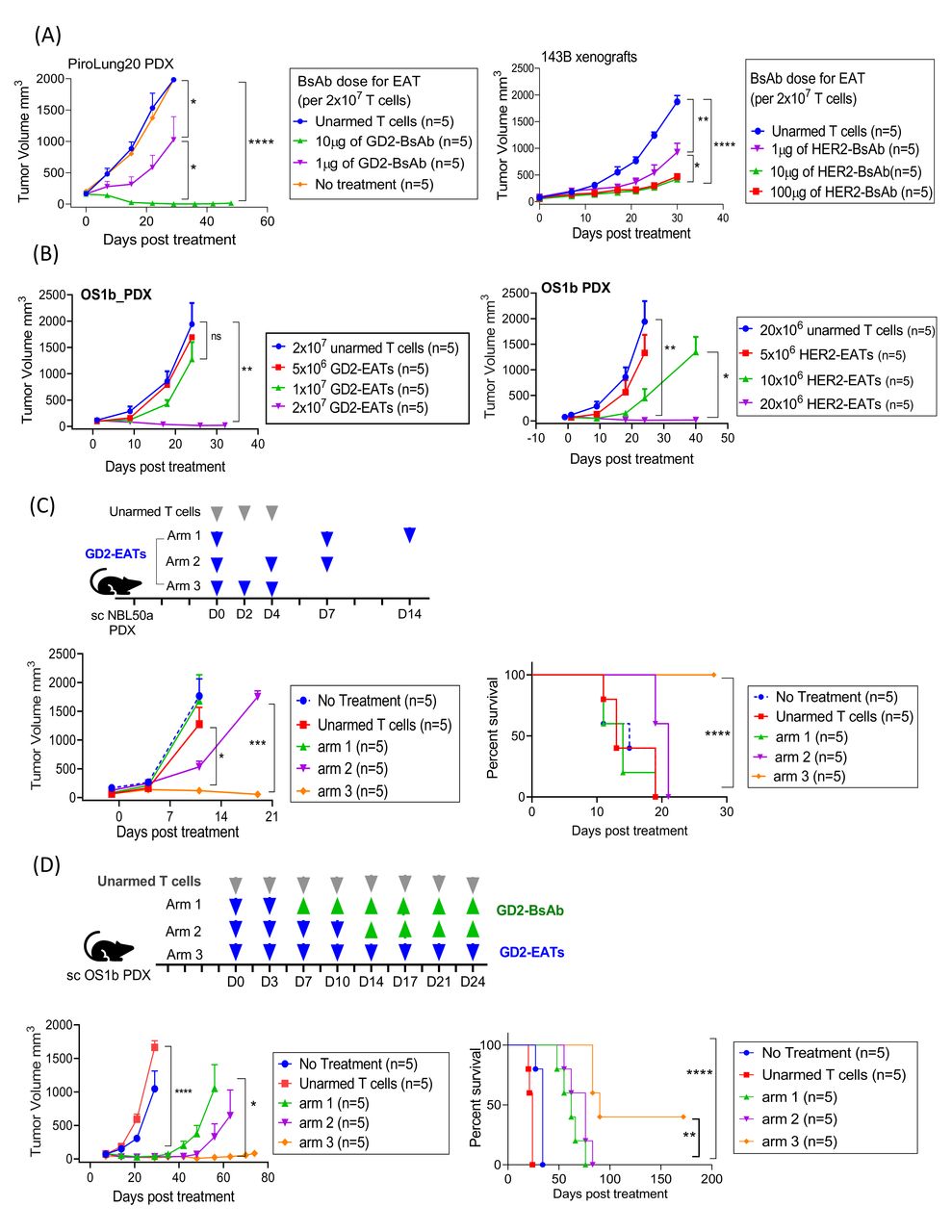
BsAb dose for arming
In vitro tumor cell killing by EATs showed the best efficacy at arming doses between 0.05 µg and 5 µg of BsAb/1×106 T cells, corresponding to BsAb surface densities between 500 and 20,000 molecules per T cell. In vivo studies of EATs suggest minimum requirement of BsAb for arming (>1 µg of BsAb/2×107 T cells (0.05 µg/1×106 cells)) for consistent treatment outcome (figure 5A). When delivered by EATs, 1 µg of BsAb was substantially inferior to 10 µg or 100 µg of BsAb. In contrast to separate BsAb injection, where high-dose BsAb (100 µg of HER2-BsAb) showed inferior antitumor effect compared with low-dose BsAb (p=0.0056) (online supplemental figure 9A), 100 µg of HER2-BsAb was equally effective as 10 µg of HER2-BsAb when delivered by EATs (p=0.2448).

Factors determining the in vivo efficacy of EATs. (A) The effect of BsAb dose on in vivo efficacy of EATs. 2×107 of T cells were armed with increasing dose of GD2-BsAb or HER2-BsAb and their antitumor effects were compared. (B) The effect of EAT cell number on the in vivo efficacy of EATs. At a fixed arming dose of 0.5 µg of BsAb/1×106 cells, increasing numbers of GD2-EAT or HER2-EAT (5×106 cells, 1×107 cells, and 2×107 cells) were administered. (C) The effect of EAT treatment schedule on the in vivo efficacy of EATs. GD2-EATs (2 µg of GD2-BsAb/2×107 cells) were administered as low intensity (1 dose/week in arm 1), standard (2 doses/week in arm 2), or dose-dense (3 doses/week in arm 3) and their in vivo antitumor response and survival were compared. (D) The effect of total number of doses of EATs on long-term tumor control and survival. Arm 1, GD2-EATs (10 µg of GD2-BsAb/2×107 cells) were administered twice weekly for 1 week, and GD2-BsAb (10 µg/dose) were followed twice weekly for 3 weeks; arm 2, four doses of GD2-EATs followed by four doses of GD2-BsAb; arm 3, eight doses of GD2-EATs. *P<0.05, **P<0.01, ***P<0.001, and ****P<0.0001. BsAb, bispecific antibody; EATs, ex vivo armed T cells; GD2, disialoganglioside; HER2, human epidermal growth factor receptor 2; PDX, patient-derived tumor xenograft; sc, subcutaneous.
To investigate the BsAb dose and T cell response relationship, CD3(+) T cells were incubated with or without target cells at 37°C for 4 hours in the presence of increasing BsAb dose (5×10–5 µg to 50 µg of HER2-BsAb/1×106 T cells). T cells were analyzed for activation (CD69), exhaustion (TIM-3), activation induced T cell apoptosis and death (annexin V and 7-aminoactinomycin D, 7-AAD) and compared with HER2-EATs under identical conditions (online supplemental figure 9B). While CD69 and TIM-3 expression rose when HER2-BsAb was above 0.05 µg/1×106 T cells, 7-AAD or annexin V expression started to increase above 0.5 µg/1×106. All four markers were consistently and significantly lower for HER2-EATs when compared with HER2-BsAb plus T cells, in agreement with the in vivo antitumor effect of high-dose BsAb when delivered as soluble BsAb versus EATs.
Infused EAT cell number
Next, we evaluated the effect of infused EAT cell number on antitumor response in vivo (figure 5B). At a fixed arming dose of 0.5 µg of BsAb/1×106 cells, different cell numbers of GD2-EAT or HER2-EAT (5×106 cells, 1×107 cells, and 2×107 cells) were administered twice weekly for 2 weeks. The antitumor effect consistently increased with the cell number of EATs infused. The in vivo antitumor effect of 2×107 of GD2-EATs was significantly superior to 5×106 or 1×107 of GD2-EATs (p=0.0018 and p=0.0366, respectively). The effect of 2×107 of HER2-EATs was also superior to 5×106 or 1×107 of HER2-EATs (p=0.0077 and p=0.0340, respectively). This antitumor response was correlated with the percentage of human CD45(+) TILs, which was obvious with 2×107 of GD2-EATs, but negligible with 5×106 of GD2-EATs (online supplemental figure 10).
Treatment schedule
To evaluate the impact of treatment interval on the in vivo efficacy of EATs, we tested three different schedules: arm 1, low intensity (1 dose/week); arm 2, standard (2 doses/week); or arm 3, dose-dense (3 doses/week) (figure 5C). GD2-EATs were fixed at 2 µg GD2-BsAb/2×107 cells. Dose-dense schedule contributed to improved tumor control over standard or low-intensity treatment schedules. Arm 3 showed the most potent antitumor effect against rapidly growing neuroblastoma PDXs compared with arm 2 (standard) or arm 1 (low-intensity schedules) (p=0.0276 and p=0.0001, respectively), which translated into survival benefit (both p<0.0001).
Supplemental EATs enabled long-term remission, prolonging survival
To test the effect of repeat EAT dosing on long-term remission and whether supplemental BsAb could eliminate the necessity of repeat EAT, we tested three different treatment schedules on osteosarcoma PDXs (figure 5D and online supplemental figure 11A): arm 1, two doses of EATs followed by six doses of intravenous BsAb; arm 2, four doses of EATs followed by four doses of BsAbs; and arm 3, eight doses of EATs. In contrast to the rapid tumor growth with no treatment or eight doses of unarmed T cells, two doses of GD2-EATs or HER2-EATs successfully ablated tumors. However, additional doses of EATs were beneficial in maintaining long-term remission. Contrary to the mice treated with two doses of GD2-EATs showing short-term response, those treated with eight doses of GD2-EATs, two of five mice showed sustained remission past 6 months, demonstrating statistically significant survival benefit over two doses or four doses of GD2-EATs supplemented with identical doses of GD2-BsAb (p=0.0018 and p=0.0089, respectively). Eight doses of HER2-EATs also showed a benefit in sustaining long-term remission over two doses or four doses of HER2-EATs supplemented with HER2-BsAb (p=0.0018 and p=0.0494, respectively) (online supplemental figure 11B). When we analyzed the relapsed tumors, those treated with GD2-EATs lost their GD2 expression (online supplemental figure 12), suggesting that tumor antigen downregulation or loss following treatment led to immune evasion.
Expanding the clinical utility of EAT therapy
EATs retain antitumor properties after cryopreservation
We tested EATs for their viability, BsAb surface density, and tumoricidal activities after a freeze-thaw process. After thawing at 37°C, EATs remained over 85% viable, irrespective of whether they were frozen for 2 hours at −80°C or up to 12 months in liquid nitrogen. When the thawed EATs were stained with anti-idiotype antibody or anti-human IgG Fc antibody, BsAb surface density remained comparable with freshly armed EATs (fresh EATs) by MFIs (figure 6A). The CD4:CD8 ratio was preserved after freeze-thaw (online supplemental figure 13A). However, the thawed CD4(+) GD2-EATs expressed more apoptosis marker than CD8(+) GD2-EATs compared with freshly armed GD2-EATs. Although the maximal killing of target cells did diminish after freeze-thaw (50% of maximal killing efficacy of fresh EATs) due to not enough recovery time after thawing, antigen specificity and the potency of EAT were maintained (figure 6B).
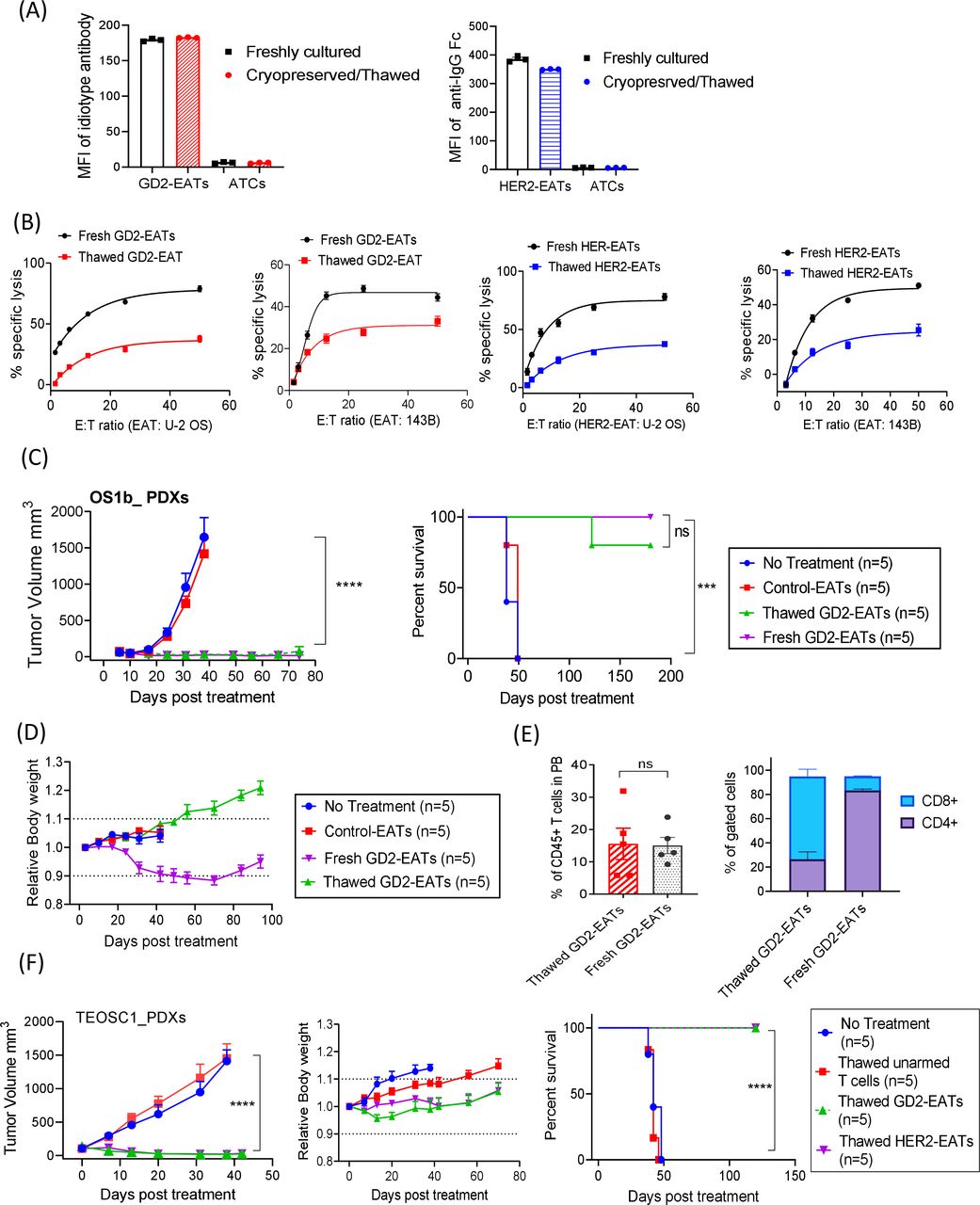
Cryopreserved EATs retained target antigen-specific cytotoxicity and exerted a comparable antitumor activity. (A) Geometric mean fluorescence intensities of BsAb on GD2-EATs (0.5 µg of GD2-BsAb/106 cells) and HER2-EATs (0.5 µg of HER2-BsAb/106 cells) before and after cryopreservation. (B) Antibody-dependent T cell-mediated cytotoxicity assay of GD2-EATs and HER2-EATs against GD2(+) and HER2(+) osteosarcoma cell lines before and after cryopreservation (n=6). (C) In vivo antitumor response of thawed GD2-EATs. T cells were first armed with GD2-BsAb (10 µg of BsAb/2×107 cells) before cryopreservation. These cells were then thawed and administered into mice bearing GD2(+) osteosarcoma PDX to compare their in vivo antitumor potency with freshly armed GD2-EATs derived from the same donor. Four to six doses of cryopreserved (thawed) GD2-EATs or freshly armed (fresh) GD2-EATs were administered intravenously twice per week. (D) Relative body weights over time after treatment with fresh or thawed GD2-EATs. (E) Flow cytometry analyses of peripheral blood (PB) T cells in the mice treated with fresh or thawed GD2-EATs on day 35 after treatment. (F) In vivo antitumor response of thawed GD2-EATs or HER2-EATs against telangiectatic osteosarcoma PDXs. T cells were first armed with GD2-BsAb or HER2-BsAb (10 µg of BsAb/2×107 cells) before cryopreservation. Six doses of GD2-EATs or HER2-EATs were given twice a week for 3 weeks with SC interleukin-2. Both thawed EATs significantly suppressed tumor growth without weight loss, improving survival (p<0.0001). ***P<0.001, ****P<0.0001. ATC, activated T cells; EATs, ex vivo armed T cells; E:T, effector to target cell ratio; GD2, disialoganglioside; HER2, human epidermal growth factor receptor 2; PDX, patient-derived tumor xenograft.
In vivo antitumor activities of thawed EATs were evaluated in two different osteosarcoma PDX models. Thawed GD2-EATs exerted potent and comparable antitumor effect to fresh GD2-EATs (p=0.1633) and significantly prolonged survival compared with controls (p=0.0002) (figure 6C). Four of five mice treated with thawed GD2-EATs showed long-term remission past 6 months post-treatment. IHC staining of the tumors treated with thawed GD2-EATs showed diffuse infiltration of CD4(+) T cells as well as CD8(+) T cells (online supplemental figure 13B), supporting their intact homing properties. Interestingly, while the mice treated with fresh GD2-EATs developed mild to moderate GVHD 1–2 months post-treatment, the mice treated with thawed GD2-EATs did not present clinical signs of GVHD throughout the entire follow-up period, maintaining body weight, good coat condition and general activity (figure 6D). When blood samples were analyzed on day 45 post-treatment (figure 6E), the fresh GD2-EATs-treated mice showed CD4(+) T cell predominance with signs of xenogeneic GVHD, but the thawed GD2-EATs-treated mice displayed a predominance of CD8(+) T cells without signs of GVHD, correlating with their clinical manifestations. The comparable in vivo potency of thawed EATs was confirmed in the second tumor model (figure 6F); both thawed GD2-EATs and thawed HER2-EATs exerted significant antitumor effect against osteosarcoma PDXs (p<0.0001) and improved survival (p<0.0001) compared with controls. All tumors regressed without toxicities, and there were no signs of GVHD or tumor relapse past 4 months post-treatment.
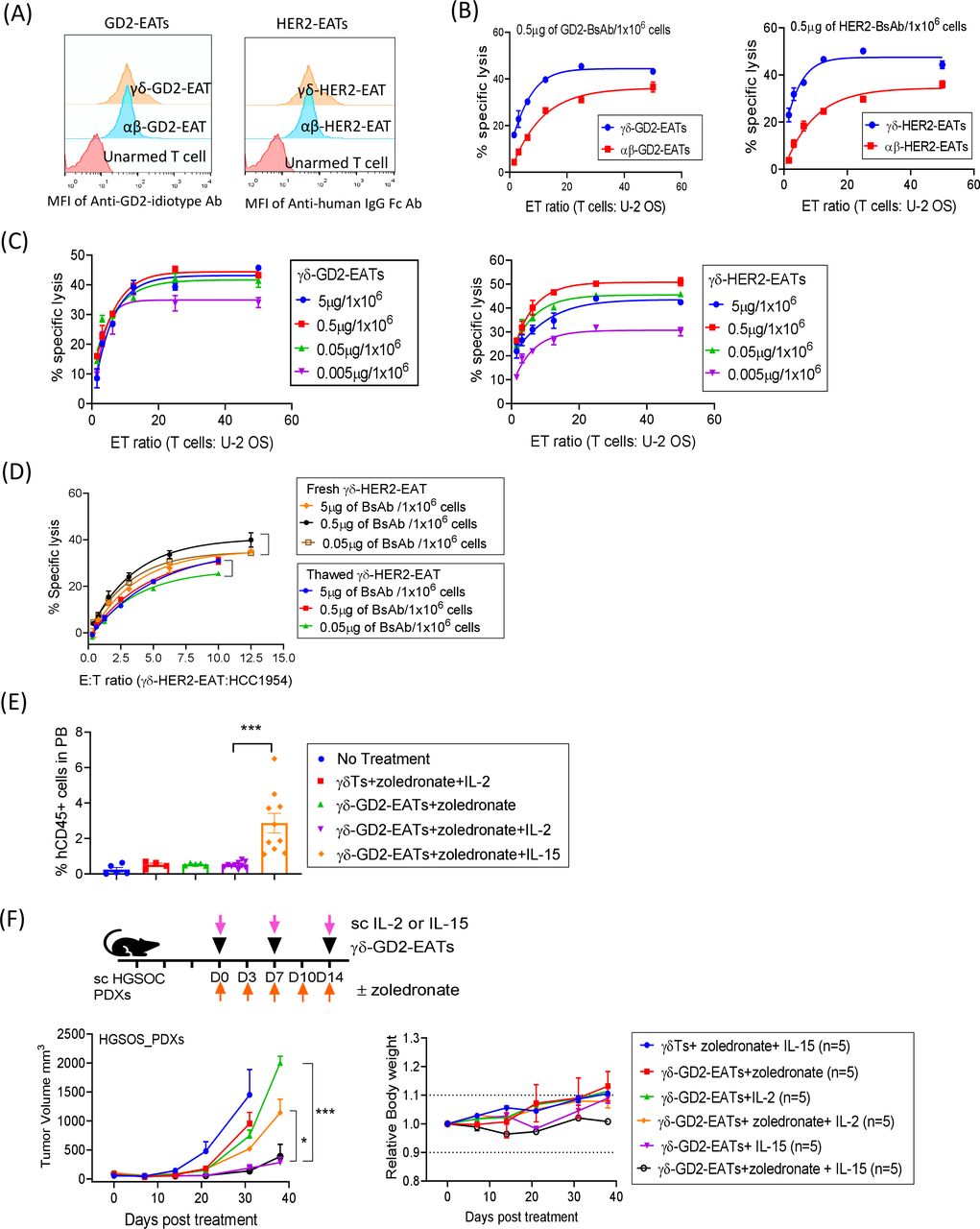
γδ T cells are equally effective as αβ T cells
Since CD3 is present on diverse subpopulations of T cells, all kinds of effector T cells should be able to arm T-BsAb. We tested the potential applicability of EATs using γδ T cells known to have a reduced alloreactivity with potential as an allogeneic ‘off-the-shelf’ T cell source.32 After expansion from fresh PBMCs with IL-2 and zoledronate for 12 days, most of CD3(+) T cells were γδ TCR(+) (online supplemental figure 14A). Majority of the γδ T cells were CD3 positive and CD4 and CD8 double negative, contrasting with T cells expanded using CD3/CD28 beads, where majority were αβ TCR positive and CD4 or CD8 positive (online supplemental figure 14B). After arming γδ T cells with GD2-BsAb (γδ-GD2-EATs) or HER2-BsAb (γδ-HER2-EATs), surface BsAb density was measured by flow cytometry, and their MFIs were comparable with those of αβ-EATs (figure 7A). In the presence of GD2-BsAb or HER2-BsAb, γδ T cells mediated potent tumoricidal activity in vitro, exceeding the efficacy of αβ-EATs (figure 7B). The maximal cytotoxicity of γδ-GD2-EATs and γδ-HER2-EATs was achieved at arming doses between 0.05 µg and 5 µg of BsAb/1×106 T cells (figure 7C), where antigen-specific cytotoxicity was maintained after freeze and thaw (figure 7D).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
EAT as a versatile platform to harness γδ T cells. (A) Flow cytometry of GD2-BsAb or HER2-BsAb on γδ T cells (γδTs) or unselected polyclonal T cells (mostly αβ T cells (αβTs)) after arming with GD2-BsAb or HER2-BsAb. (B) ADTC assays of γδ-GD2-EATs and γδ-HER2-EATs compared with αβ-GD2-EATs and αβ-HER2-EATs. Non-specific tumor cell killing by unarmed γδ T cells and unarmed αβ T cells (background) were subtracted. (C) ADTC assay of γδ-GD2-EATs and γδ-HER2-EATs at increasing effector to target ratios (E:T ratios) and at increasing BsAb arming doses. (D) In vitro cytotoxicity against breast cancer cell line HCC1954 was compared between fresh γδ-HER2-EATs and thawed γδ-HER2-EATs at increasing E:T ratios and at increasing BsAb arming doses. (E) Flow cytometry analyses of peripheral blood T cells after second dose of γδ-GD2-EATs plus zoledronate and supplementary IL-2 or IL-15. (F) Three doses of γδ-GD2-EATs (10 µg of GD2-BsAb/2×107 cells) were administered with supplementary IL-2 or IL-15 to treat osteosarcoma PDXs and the antitumor response was compared among groups. *P<0.05, ***P<0.001. ADTC, antibody-dependent T cell-mediated cytotoxicity; BsAb, bispecific antibodies; EATs, ex vivo armed T cells; γδ T cells, gamma delta T cells; IL, interleukin; MFI, mean fluorescence intensity; PDX, patient-derived tumor xenograft.
We next tested the in vivo efficacy of γδ-EATs and compared with corresponding αβ-EATs (online supplemental figure 15A). With supplementary IL-2, γδ-GD2-EATs and γδ-HER2-EATs did not produce significant antitumor response irrespective of additional zoledronate. Flow cytometry analyses of T cells in the blood and tumors after γδ-EAT treatment presented substantially fewer human CD45(+) T cells in the circulation and in the tumors compared with αβ-EATs (p=0.0006 and p=0.0007, respectively), suggesting poor in vivo survival of γδ-EATs (online supplemental figure 15B).
However, when exogenous IL-15 (as IL15Rα-IL15 complex) instead of IL-2 was used as T cell survival cytokine, the in vivo frequencies of γδ-EATs significantly increased (figure 7E). γδ T cells expanded from fresh PBMCs using 2 µM of zoledronate plus 30 ng/mL of IL-15 for 12–14 days were armed with GD2-BsAb or HER2-BsAb and administered intravenously into osteosarcoma PDX-bearing mice, with 5 µg of subcutaneous IL-15 or 1000 IU of IL-2 (figure 7F and online supplemental figure 16A). While γδ-GD2-EATs and γδ-HER2-EATs sustained with IL-2 failed to suppress tumor, same γδ-EATs sustained with IL-15 exerted significant antitumor effects without toxicities or weight loss during follow-up period. The PDXs treated with γδ-GD2-EATs plus IL-15 showed a diffuse T cell infiltration into tumors, in contrast to tumors treated by γδ-GD2-EATs plus IL-2 or unarmed γδ-EATs plus IL-15 (online supplemental figure 16B). Furthermore, cryopreserved γδ-EATs showed strong in vivo antitumor effect when supplemented with IL-15 and zoledronate (online supplemental figure 17A). Thawed γδ-STEAP1-EATs exerted a significant antitumor response against STEAP1(+) Ewing sarcoma PDXs in vivo. These findings implicate the potential applicability of IL-15 and allogeneic γδ T cells for IgG-[L]-scFv-platformed BsAb arming for the treatment of human cancers.
Discussion
For successful BsAb-directed T cell immunotherapy, adequate number and quality of tumor-directed T cells are required. Unfortunately, autologous T cells of patients with cancer are often dysfunctional, exhausted, or compromised by chemotherapy or the cancer itself.33 Here we show that ex vivo expanded T cells armed with BsAb of IgG-[L]-scFv platform can be transformed into living drugs producing robust antitumor response. EATs produced significantly less TNF-α compared with direct BsAb injection, while infiltrating tumors faster to produce robust tumoricidal activity. EATs mediate significant antitumor effect in an autologous system, excluding the allogeneic T cell effect in the humanized mouse models. The feasibility of cryopreservation should allow ample time for detailed quality control of EATs before release, facilitating product storage and distribution. By harvesting autologous T cells before intensive chemotherapy or using healthy third-party or haploidentical donor’s alternative effector T cells with reduced alloreactivity, the clinical potential of this approach can be further expanded.
For BsAb armed T cell immunotherapy, the BsAb structural format was critical in order to drive T cells to infiltrate ‘cold’ tumors to exert their antitumor effect. Despite similar antitumor properties among BsAb armed T cells in vitro, only ‘EATs’, T cells armed with the IgG-[L]-scFv-platformed BsAb, showed strong infiltration into tumors to produce durable antitumor responses which translated into prolonged survival. Although genetically engineered T cells expressing CD19-BiTE or CAR T cells secreting EGFR-BiTE had shown success in preclinical models,34 35 T cells ex vivo armed with GD2-BiTE have never been as successful. This may attribute to the short half-life of BiTE or the nature of the tumor target, partly explaining why there have been no successful reports of armed T cells using BiTE despite the clinical success of Blincyto.36 Besides, protein size, distance of the epitope to the membrane, purity, and binding affinities to tumor and to CD3 T cells strongly affect the biodistribution and in vivo antitumor activities of T-BsAb.37–41 Since all GD2-BsAb platforms were built using the same anti-GD2 and anti-CD3 VH and variable light chain (VL) sequences, monovalent binding to CD3 or to target in monomeric BiTE and IgG heterodimer (online supplemental table 3) might affect the stability of BsAb armed T cells in vivo. However, despite the multivalency in dimeric BiTE, BiTE-Fc, IgG-[H]-scFv, and IgG conjugate, none of these platforms was able to achieve the potency of IgG-[L]-scFv, implicating additional factors besides binding avidity. We previously described the critical importance of interdomain distance and cis-configuration of BsAb,14 now confirmed with these extensive series of EAT-based experiments. The mechanism behind the in vivo superiority of the IgG-[L]-scFv format is likely to be more complex.
One of the major toxicities of T cell-based immunotherapy is CRS,42 typically associated with surge of IFN-γ, IL-6, and TNF-α, although elevations of IL-2, granulocyte-macrophage colony-stimulating factor (GM-CSF), IL-10, IL-8, and IL-5 have also been reported.43 44 IL-6 and TNF-α have been implicated as the central mediators of CRS.42 When the anti-CD28 antibody TGN1412 was administered to healthy subjects, CRS developed in an hour, coinciding with peak cytokine levels: TNF-α, as an initial trigger, was followed by INF-γ and IL-10, IL-8, IL-6, IL-4, IL-2, and IL-1β.45 When BsAb engages polyclonal T cells to undergo synchronous activation, TNF-α acts as the initial signal for monocyte activation, resulting in release of IL-6 and IL-1, preventable by anti-TNF-α antibodies without compromising antitumor activity.46 However, the absence of cytokine storm in clinical studies of BsAb armed T cell is notable.12 13 47 T cells exposed to BsAb produced TNF-α during the initial 20 min of BsAb arming; after washing, EATs released significantly less cytokines, particularly TNF-α, in vitro and in vivo without affecting their trafficking ability or tumoricidal activity.
We also showed that thawed EATs maintained the viability and their affinity to target antigens and to CD3(+) T cells. The cryopreservation did not affect the frequency of CD4 or CD8 T cells in EATs. The thawed GD2-EATs preserved their target antigen specificity and were effective in vitro and in vivo. When thawed, the cryopreserved CD4(+) GD2-EATs did have higher levels of apoptosis marker than CD8(+) GD2-EATs, which was consistent with the in vivo dominance of CD8(+) T cell population following treatment. CD4(+) T cell dysfunction including reduced IFN-γ production has been reported in the cryopreserved product of T cells, PBMCs or cord blood.48–50 To improve antitumor activity and to reduce treatment-related toxicity, optimizing cryopreservation protocols for EATs would be the next front.
Another limitation of CAR T cell therapy is the need for autologous T cells. Alternative effector T cells with low alloreactivity including human leukocyte antigen (HLA)-identical donor’s activated T cells,51 52 clonally committed Epstein-Barr virus (EBV)-specific cytotoxic T cells,53 and innate-like lymphocytes such as γδ T cells or natural killer T (NKT) cells could be potential ‘off-the-shelf’ cytotherapy products. When obtained from healthy donors, the probability of successful large-scale ex vivo expansion of effector cells with consistent product quality should be high. γδ T cells are known to have innate antitumor properties and significantly reduced alloreactivity or ‘on-target off-tumor’ toxicities.32 In our studies, γδ-EATs could suppress osteosarcoma xenografts in the presence of the IL15Rα-IL15 cytokine complex without toxicities. IL-15 enhanced proliferation of T cells in vivo and improved potency of T cell-based immunotherapy.54 55 For γδ T cell treatment, poor in vivo survival has been a major challenge. Supplementary zoledronate and IL-15/IL-15Ra enabled γδ-EATs to survive and to proliferate in vivo to exert significant antitumor effects. By employing optimal cytokines to prolong in vivo survival,56 γδ-EATs deserve to be explored in more depth. EATs using γδ T cells should provide a powerful platform to harness third-party or haploidentical donor’s effector T cells with minimal graft-versus-host side effects, with the additional benefit of cytotoxic mechanisms through TNF-related apoptosis-inducing ligand (TRAIL), NKG2D receptor, and FcγRIII (CD16).57 58
Neurotoxicity is the other major hurdle that has plagued CAR T cell and BsAb.4 Since GD2 is expressed in mouse brain, several studies have reported neurotoxicity in mice when CAR T cells were used.59 Although we did not directly compare EATs and CAR T cells in this manuscript, we previously compared the efficacy of GD2-CAR T cells and GD2 IgG-[L]-scFv BsAb built with identical anti-GD2 IgG sequence.60 GD2-BsAb-driven T cells were more efficient than GD2-CAR T cells in vivo. More importantly, while GD2-CAR T cells caused neurological damage, neither GD2-BsAb nor GD2-EATs caused any neurotoxicity in mouse models.61
In conclusion, ex vivo arming of T cells with IgG-[L]-scFv BsAb is a potential strategy beyond CAR T cell technology to use proteins instead of genes to modify T cells. While avoiding CRS and neurotoxicity, the potential to use cryopreserved alternative effector T cells other than αβ T cells should expand the clinical utility of EATs.
Data availability statement
All data relevant to the study are included in the article or uploaded as supplementary information.
Ethics statements
Ethics approval
All animal experiments were approved by the Memorial Sloan Kettering’s Institutional Animal Care and Use Committee (IACUC) and were executed according to the ACUC guidelines. Patient-derived tumor xenografts were established with MSKCC IRB approval.
Acknowledgments
We would like to especially thank Alan W Long, Tsung-Yi Lin, See Liang Ng, and Steven Tsung-Yi Lin for their valuable comments on earlier drafts. Steven Tsung-Yi Lin also designed and validated BsAbs to STEAP1 and PSMA. Hong-fen Guo tested the purity and stability of the BsAbs using HPLC and SDS-PAGE and analyzed endotoxin released by BsAb. Yi Feng stained fresh frozen tumor tissues with hu3F8 antibody. Dr Andrew Kung, Dr Filemon DeLa Cruz, and Dr Sarat Chandarlapaty and Dr Elisa De Stanchina provided PDXs for these studies. Archana Thakur provided the Herceptin and hu3F8 chemical conjugates. Technical service was provided by the MSK Animal Imaging Core Facility, Antitumor Assessment Core Facility, and Molecular Cytology Core Facility.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors JAP and N-KVC designed the experiments, interpreted and analyzed the results, and wrote the manuscript. LL provided hu3F8 IgG chemical conjugates and Herceptin IgG chemical conjugates. BS designed and validated the diverse formats of anti-GD2-BsAb. HX designed and validated the anti-HER2 BsAb and tested each BsAb’s affinity to CD3 and to each target antigen.
Funding This work was supported by funds to N-KVC from R01 CA 182526, Enid A Haupt Endowed Chair, the Robert Steel Foundation, Kids Walk for Kids with Cancer, as well as sponsored research fund from Y-mAbs Therapeutics. Technical service provided by the MSK Animal Imaging Core Facility, Antitumor Assessment Core Facility, and Molecular Cytology Core Facility was supported in part by the NCI Cancer Center Support Grant P30 CA008748. LL was supported in part by the Marion McNulty Weaver and Malvin C Weaver Professor of Oncology Endowed Chair, R01 CA 182526, and NCI Cancer Center Support Grant P30 CA044579 at UVA.
Competing interests Both MSK and N-KVC have financial interest in Y-mAbs, Abpro-Labs and Eureka Therapeutics. N-KVC reports receiving commercial research grants from Y-mAbs Therapeutics and Abpro-Labs. N-KVC was named as inventor on multiple patents filed by MSK, including those licensed to Y-mAbs Therapeutics, Biotec Pharmacon, and Abpro-Labs. N-KVC is a SAB member for Abpro-Labs and Eureka Therapeutics. LL is cofounder of Transtarget and is a SAB member for Rapa Therapeutics. JAP has no disclosures to report.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.