Article Text

Abstract
Background Dendritic cells (DCs) are a promising therapeutic target in cancer immunotherapy given their ability to prime antigen-specific T cells, and initiate antitumor immune response. A major obstacle for DC-based immunotherapy is the difficulty to obtain a sufficient number of functional DCs. Theoretically, this limitation can be overcome by using induced pluripotent stem cells (iPSCs); however, therapeutic strategies to engage iPSC-derived DCs (iPSC-DCs) into cancer immunotherapy remain to be elucidated. Accumulating evidence showing that induction of tumor-residing DCs enhances immunomodulatory effect of radiotherapy (RT) prompted us to investigate antitumor efficacy of combining intratumoral administration of iPSC-DCs with local RT.
Methods Mouse iPSCs were differentiated to iPSC-DCs on OP9 stromal cells expressing the notch ligand delta-like 1 in the presence of granulocyte macrophage colony-stimulating factor. Phenotype and the capacities of iPSC-DCs to traffic tumor-draining lymph nodes (TdLNs) and prime antigen-specific T cells were evaluated by flow cytometry and imaging flow cytometry. Antitumor efficacy of intratumoral injection of iPSC-DCs and RT was tested in syngeneic orthotopic mouse tumor models resistant to anti-PD-1 ligand 1 (PD-L1) therapy.
Results Mouse iPSC-DCs phenotypically resembled conventional type 2 DCs, and had a capacity to promote activation, proliferation and effector differentiation of antigen-specific CD8+ T cells in the presence of the cognate antigen in vitro. Combination of in situ administration of iPSC-DCs and RT facilitated the priming of tumor-specific CD8+ T cells, and synergistically delayed the growth of not only the treated tumor but also the distant non-irradiated tumors. Mechanistically, RT enhanced trafficking of intratumorally injected iPSC-DCs to the TdLN, upregulated CD40 expression, and increased the frequency of DC/CD8+ T cell aggregates. Phenotypic analysis of tumor-infiltrating CD8+ T cells and myeloid cells revealed an increase of stem-like Slamf6+ TIM3− CD8+ T cells and PD-L1 expression in tumor-associated macrophages and DCs. Consequently, combined therapy rendered poorly immunogenic tumors responsive to anti-PD-L1 therapy along with the development of tumor-specific immunological memory.
Conclusions Our findings illustrate the translational potential of iPSC-DCs, and identify the therapeutic efficacy of a combinatorial platform to engage them for overcoming resistance to anti-PD-L1 therapy in poorly immunogenic tumors.
- dendritic cells
- radioimmunotherapy
- vaccination
- adaptive immunity
- programmed cell death 1 receptor
Data availability statement
Data are available upon reasonable request.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Dendritic cells (DCs) play a critical role in the initiation of antitumor immune responses and thus have been considered to be a potential target in cancer immunotherapy.1–3 Clinically, monocytes obtained from patients have been commonly used as a source of DCs.2–4 However, despite favorable toxicity, tolerability and immunogenicity profiles with monocyte-derived DC vaccination, objective clinical responses remain low in patients.4 Naturally occurring (primary) DCs harbor superior antigen presentation, priming and migratory capabilities compared with in vitro-generated monocyte-derived DCs.3 5 6 However, circulating DCs are rare (<1.0%) in peripheral blood mononuclear cells (PBMCs), and isolating a sufficient number of natural DCs requires multiple large-scale PBMC isolation via leukapheresis for repeated vaccine doses in a therapeutic setting.3 5 6
This limitation of DC-based therapy can be theoretically overcome by using pluripotent stem cells (PSCs) such as embryonic stem cells and induced pluripotent stem cells (iPSCs) as an unlimited source of DCs. Accumulating evidence suggests that mouse and human PSC-derived DCs are morphologically, phenotypically, and functionally comparable with myeloid DCs differentiated from bone marrow (BM) hematopoietic progenitors.7–12 Antigen-loaded mouse and human PSC-derived DCs can activate antigen-specific T cells in vitro, and establish protective immunity against tumor challenge in vivo.9–13 Furthermore, genetically modified iPSC-derived DCs (iPSC-DCs) have shown therapeutic efficacy against established tumors, suggesting the utility of iPSC-DCs in cancer immunotherapy.10 11 14 However, in vivo therapeutic potential of iPSC-DCs against cancer has been demonstrated in the immunogenic tumor models9 14 and/or by using iPSC-DCs expressing the virally transduced cognate antigen such as ovalbumin (OVA), hgp100 and carcinoembryonic antigen.10 11 Thus, the therapeutic strategies to use iPSC-DCs in cancer immunotherapy for poorly immunogenic ‘cold’ tumors remain elusive.
Local radiotherapy (RT) is a longstanding pillar of cancer treatment, which has been used for patients with various stages of cancer. Although the main mechanisms of the tumor reduction is the induction of irreversible DNA damage to the tumor cells,15 RT has also been shown to elicit immunomodulatory effects on the tumor microenvironment (TME).16 17 Moreover, there is a growing body of evidence that local irradiation triggers immunogenic cell death,18 and generates inflammatory cytokines that promote the ability of tumor-residing DCs to cross-present released antigens to T cells.19 20 Therefore, RT has been considered as an attractive partner for DC-based cancer immunotherapy.21 22
In view of these observations, we explore the capacity of in situ iPSC-DC administration to enhance immunogenicity of RT against poorly immunogenic tumors. To this end, we evaluate the therapeutic efficacy of in situ iPSC-DC delivery and RT in two histologically distinct syngeneic mouse models of poorly T cell-inflamed tumors that are refractory to PD-1 ligand 1 (PD-L1) blockade therapy. Our data demonstrate that RT increases trafficking of intratumorally injected iPSC-DCs to the tumor-draining lymph nodes (TdLNs), augments the priming of tumor-specific CD8+ T cells, and elicits synergistic antitumor immune responses. Furthermore, this multimodal intralesional therapy controls growth of untreated distant tumors, renders poorly immunogenic tumors responsive to PD-L1 blockade, and establishes systemic tumor-specific immunological memory.
Methods
Mice
Female C57BL/6 mice and Pmel-1 T-cell receptor (TCR)-transgenic mice (B6.Cg Thy1a-Tg(TcraTcrb)8Rest/J) were purchased from the Jackson Laboratories. OT-I Rag2−/− mice have been previously described.23 OT-I and Pmel-1 mice were bred in-house. All mice were age matched (7–10 weeks old) at the beginning of each experiment and maintained under specific pathogen-free conditions and housed in the Laboratory Animal Resources facility. All animal studies were conducted in accordance with and approved by the Institutional Animal Care and Use Committee at the Roswell Park Comprehensive Cancer Center.
Cell lines
Generation of mouse embryonic fibroblast-derived iPSC lines, 2A-4F-118 and 2A-4F-136, was previously described.24 OP9 and OP9 cells expressing a notch ligand, delta-like-1 (OP9-DL1) cells were purchased from RIKEN (Japan). The iPSC lines were maintained in Gibco Dulbecco's Modified Eagle Medium (DMEM) containing 15% fetal bovine serum (FBS) (Sigma-Aldrich), 1% non-essential amino acid (NEAA) (Gibco), 2 mM L-glutamine (Gibco), 0.5% penicillin/streptomycin (Gibco), 55 µM 2-mercaptoethanol (2-ME) (Gibco), 0.5 µM PD0325901 (mitogen-activated protein kinase inhibitor) (Stemgent), 3 µM CHIR99021 (glycogen synthase kinase-3 inhibitor) (Stemgent), and 1000 U/mL of leukemia inhibitory factor (Millipore) on mitomycin-C-treated SNL cells (Cell Biolabs) as described.25 SNL cells were cultured in DMEM containing 7% FBS (Sigma-Aldrich), 2 mM L-glutamine, and 0.5% penicillin/streptomycin. OP9 and OP9-DL1 cells were cultured on gelatin-coated dishes in OP9 medium: αMEM supplemented with 20% non-heat inactivated FBS (Biowest), 0.5% penicillin/streptomycin and 2.2 g/L of sodium bicarbonate (Sigma-Aldrich).
B16-F10 (B16) tumor cell lines were purchased from the American Type Culture Collection (ATCC), authenticated at ATCC and maintained. The AT-3, B16-OVA, and MC38 cell lines were gift from Drs Scott Abrams, Sharon Evans (Roswell Park Comprehensive Cancer Center), and Weiping Zou (University of Michigan), respectively. AT-3 tumor cells expressing green fluorescent protein (GFP) (AT-3-GFP) were described.26 B16, B16-OVA and MC38 cells were cultured in RPMI 1640 (Gibco) supplemented with 10% FBS, 1% NEAA, 2 mM L-glutamine, 0.5% penicillin/streptomycin, and 55 µM 2-ME. AT-3 and AT-3-GFP cells were cultured in DMEM (Gibco) supplemented with 10% FBS (Sigma-Aldrich), 1% NEAA, 2 mM L-glutamine, 1 mM sodium pyruvate, 15 mM 4-(2-hydroxyethyl)-1-piperazine ethanesulfonic acid, 0.5% penicillin/streptomycin, and 55 µM 2-ME. These cell lines were authenticated by morphology, phenotype and growth, and routinely screened for Mycoplasma, and were maintained at 37°C in a humidified 5% (B16, B16-OVA and MC38) or 7% (AT-3, AT-3-GFP) CO2 atmosphere.
Generation of DCs from iPSCs
The differentiation of mouse iPSCs to DCs is composed of three steps as previously described,12 27 but step 2 was slightly modified in our laboratory. In brief, iPSCs (1×105 cells) were seeded onto OP9 cell layers in 10 cm dishes and cultured in OP9 media (step 1). After 7 days of incubation, cells were collected, suspended with OP9 media containing 100 ng/mL of mouse granulocyte macrophage colony-stimulating factor (mGM-CSF) (Peprotech), and 1×106 cells were seeded onto a newly prepared OP9-DL1 cell layer (step 2). On day 14, loosely adherent cells were collected by pipetting, and 5×105 cells were transferred to new dish coated by 10% poly 2-hydroxyethyl methacrylate (pHEMA) (Sigma-Aldrich) without feeder cells and cultured in RPMI 1640 containing 10% FBS, 100 ng/mL of mGM-CSF and 55 µM of 2-ME for another 14 days (step 3). On day 28, the non-adherent cells were collected and used for further analysis or in vivo study.
Generation of BM-derived DCs
Mouse BM-derived DCs (BM-DCs) were generated as described.27 BM cells from flushed marrow cavities of femurs and tibiae of female C57BL/6 mice aged 6–8 weeks old were cultured in RPMI 1640 supplemented with 10% heat-inactivated FBS (Sigma-Aldrich), 2 mM L-glutamine, 55 µM 2-ME, 0.5% penicillin/streptomycin, and 20 ng/mL mGM-CSF in petri dishes with 100 mm diameter (Falcon #351029, BD Biosciences) at 2×106 cells/10 mL. At day 3, 10 mL of media containing 20 ng/mL mGM-CSF were added to the plates. At days 6 and 8, half of the culture supernatant was changed with media containing 20 ng/mL mGM-CSF. Non-adherent cells were collected on day 10.
In vitro activation of Pmel-1 T cells by iPSC-DCs
CD8+ cells were isolated from Pmel-1 splenocytes using EasySep Mouse CD8α Positive Selection Kit (STEMCELL Technologies). The isolated cells were labeled with CellTrace carboxyfluorescein succinimidyl ester (CFSE) (Thermo Fisher Scientific), and seeded onto 24-well plate (2×106 cells/well) in media containing interleukin (IL)-2 (60 IU/mL) (Prometheus Laboratories) with H-2Db-restricted epitope of the influenza nucleoprotein (NP) peptide, NP366–374 (ASNENMETM; GenScript) (1 µM) or human (h)gp10025–33 peptide (KVPRNQDWL: GenScript) (1 µM). Then, iPSC-DCs (1×106) stimulated with mouse IL-4 (mIL-4) (Peprotech) (10 ng/mL), mouse tumor necrosis factor alpha (mTNFα) (Peprotech) (5 ng/mL), and lipopolysaccharides (LPS) (Sigma-Aldrich) (0.5 µg/mL) for 2 days were added to each well and co-cultured for 2 days. In vitro-activated Pmel-1 CD8+ T cells were evaluated for proliferation, surface markers, and cytokine production against NP366–374 and hgp10025–33 peptides.
Therapy of transplanted tumors
AT-3 (5×105) tumor cells were implanted into the left fourth mammary gland under anesthesia with isoflurane. B16 (5×105) tumor cells were injected subcutaneously on the left flank. To test the establishment of immunological memory, mice that had durable complete responses to the treatment and fresh naïve C57BL/6 mice were injected subcutaneously with AT-3 (5×105) and MC38 (5×105) tumor cells in the right flank and on the back, respectively. To generate mice bearing bilateral tumors, AT-3 (5×105) tumor cells were implanted into the left fourth mammary gland (primary tumors) under anesthesia with isoflurane, and AT-3 (5×105) tumor cells were injected in the right flank (distant tumors) subcutaneously 2 days after the inoculation for primary tumors. iPSC-DCs (1×106 cells) in 50 µL phosphate buffered saline (PBS) were injected intratumorally every 4 days. Irradiation of mammary and subcutaneous tumors was conducted as recently described.26 Briefly, the mice were anesthetized with isoflurane and positioned for exposure to radiation (9 Gy) under a 2 mm thick lead shield containing 1 cm2 hole, limiting exposure to the tumors. Irradiation was performed with an orthovoltage X-ray machine (Philips RT250, Philips Medical Systems) at 75 kV using a 1×2 cm cone. Radiation was given 1 day after first DC injection and repeated every 6 days. Anti-PD-L1 antibody (Ab) (clone 10F.9G2, BioXCell) or rat IgG2b Ab (clone LTF-2, BioXCell) was injected intraperitoneally at a dose of 200 µg/mouse every 3 days starting on the day first iPSC-DCs were injected. Tumor growth was measured three times a week, and the volumes were calculated by determining the length of short (l) and long (L) diameters (volume=l2×L/2). Experimental endpoints were reached when tumors exceeded 20 mm in diameter or when mice became moribund and showed signs of lateral recumbency, cachexia, lack of response to noxious stimuli, or observable weight loss.
Flow cytometry
Single-cell suspensions of blood and tumors were prepared for flow cytometric analysis. Red blood cells were lysed using ACK Lysis Buffer (Life Technologies). Cells were incubated with Abs in PBS containing 2% FBS for 20 min at room temperature after blockade by anti-CD16/CD32 (BD Biosciences). The following Abs were used: anti-CD8 (clone 53-6.7, BD Biosciences), anti-CD45 (clone 30-F11, Invitrogen), anti-CD90.1 (clone Ox-7, BioLegend), anti-programmed cell death protein 1 (PD-1) (clone 29F.1A12, BioLegend), anti-CD127 (clone A019D5, BioLegend), anti-interferon (IFN) regulatory factor-4 (IRF4) (clone IRF4.3E4, BioLegend), anti-IRF8 (clone V3GYWCH, Thermo Fisher Scientific), anti-Slamf6 (clone 330-AJ, BioLegend), anti-T cell immunoglobulin and mucin domain-containing protein 3 (TIM3) (clone B8.2C12, BioLegend), anti-IFNγ (clone XMG1.2, Thermo Fisher Scientific), anti-TNFα (clone MP6-XT22, Thermo Fisher Scientific), anti-IL-2 (clone JES6-5H4, BD Biosciences), anti-CD62L (clone MEL-14, BioLegend), anti-CD25 (clone PC61, BioLegend), anti-Ly6C (clone HK1.4, BioLegend), anti-CD11b (clone M1/70, BD Biosciences), anti-CD11c (clone HL3, BD Biosciences), anti-I-Ab (clone AF6-120.1, BioLegend), anti-XCR1 (clone ZET, BioLegend), anti-CD24 (clone M1/69, BD Biosciences), and anti-F4/80 (clone BM8, BioLegend). LIVE/DEAD Fixable Near-IR Dead Cell Stain Kit (Thermo Fisher Scientific)-stained cells were excluded from the analysis. To identify apoptotic cells, cells were stained with annexin V, Alexa Fluor 488 conjugate (#A13201, Invitrogen). For analysis of IRF4 and IRF8 expression in iPSC-DCs, iPSC-DCs (1×106) were stimulated with mIL-4 (10 ng/mL), mTNFα (5 ng/mL), and LPS (0.5 µg/mL) for 2 days. Cells were harvested, permeabilized with True-Nuclear Transcription Factor Buffer Set (BioLegend) and stained with anti-IRF4 and IRF8 Abs. For intracellular staining of cytokines, in vitro-activated Pmel-1 CD8+ T cells were co-cultured with NP366–374 (1 µM) or hgp10025–33 (1 µM) and splenocytes from C57BL/6 mice in the presence of Brefeldin A (BD Biosciences) for 5 hours. Cells were harvested, permeabilized with Fixation and Permeabilization Kit (BD Biosciences) and stained with anti-IFNγ, TNFα, and IL-2 Abs. Samples were analyzed using LSRII or LSRFortessa (BD Biosciences) with FlowJo software (TreeStar).
Analysis of in vivo DC trafficking and imaging flow cytometry
iPSC-DCs (1×106 per 50 µL) labeled with 5-(and-6)-(((4-chloromethyl) benzoyl) amino) tetramethylrhodamine (CellTracker Orange CMTMR, #C2927, Invitrogen) were injected into the established orthotopic AT-3 or AT-3-GFP tumors in the left fourth mammary gland. After 24 hours, mice were untreated or treated with RT (9 Gy). TdLNs (left inguinal LN) were harvested 24 hours after RT for flow cytometry or imaging flow cytometry. ImageStream-MKII was used for imaging flow cytometry (Luminex Corporation) which enables spatially correlated image analysis of spectrally separated cell imagery. Data were analyzed using IDEAS software (Luminex Corporation). Immunophenotypically defined cell aggregates were identified as described.26
Statistical analysis
Statistical analysis was performed using two-tailed Student’s t-test or Mann-Whitney U test (for comparisons between two groups), or the Mantel-Cox method (log-rank test) for survival analysis using GraphPad Prism V.9.02 (GraphPad Software). Data are presented as mean±SEM.
Results
Generation of functional DCs from mouse iPSCs
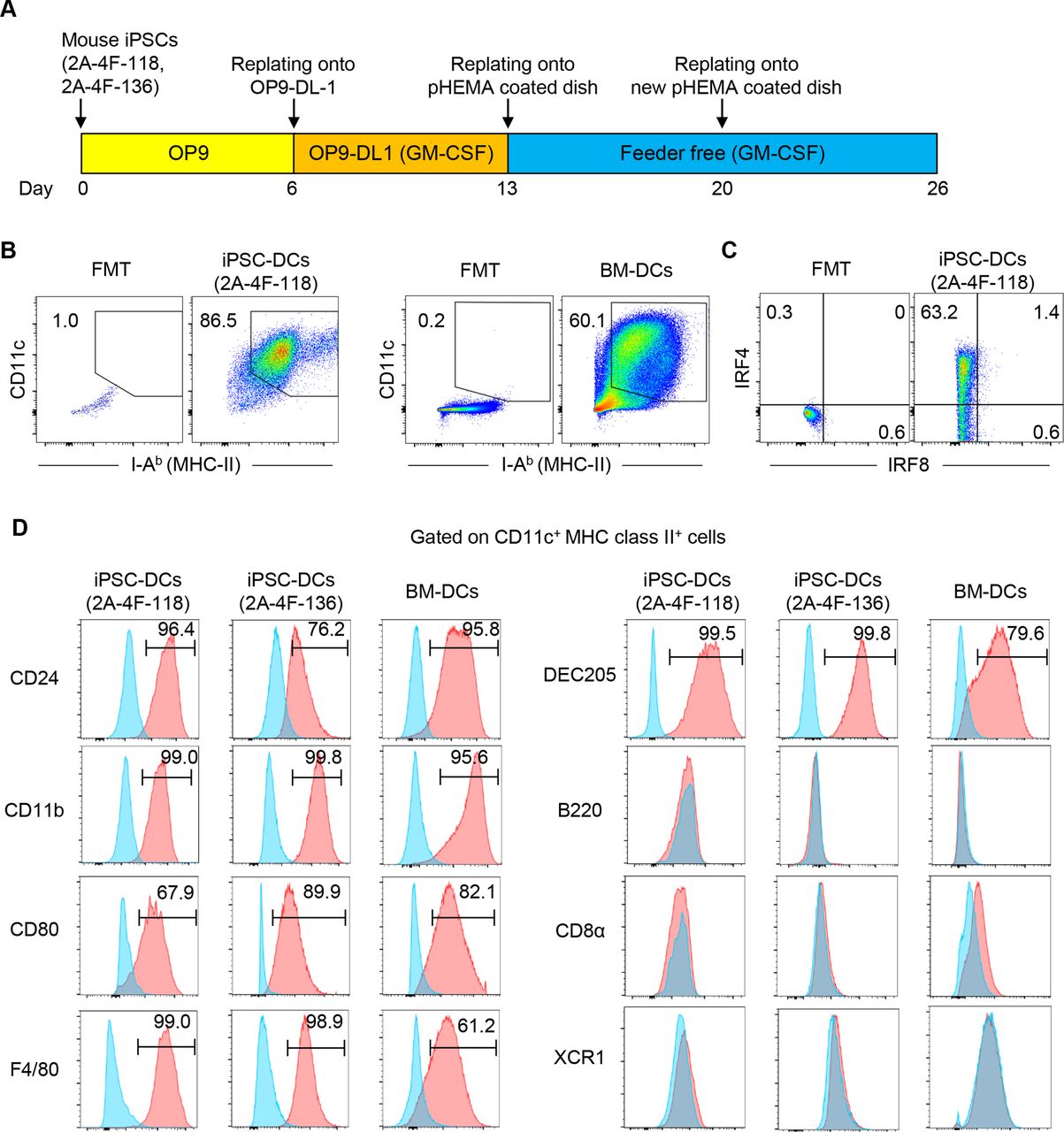
In this study, we aimed to generate DCs from previously established mouse embryonic fibroblasts-derived iPSC clones, 2A-4F-118 and 2A-4F-136.24 First, we tested whether mouse iPSCs could differentiate into functional DCs using the previously established protocol12 with some modifications based on the emerging evidence that notch signaling facilitates in vitro generation of mouse and human DCs28 29 (figure 1A). In this study, we differentiated mouse iPSCs on OP9 feeder layers for 6 days (step 1). On day 6, cells were harvested and transferred onto OP9-DL1,30 and cultured in the presence of GM-CSF for 7 days (step 2). On day 13, loosely adherent cells were recovered and transferred to a dish coated with pHEMA to create a hydrophobic surface and prevent cell adhesion. On day 26, the majority of the floating iPSC-derived cells were positive for CD11c and Major Histocompatibility Complex (MHC) class II similar to BM-derived cells differentiated in the presence of GM-CSF (figure 1B). Mouse iPSC-derived cells expressed CD24, CD11b, F4/80, CD80 and IRF4, but not B220, CD8α, XCR1 and IRF8, resembling conventional type 2 DCs (cDC2) (figure 1C,D).31 32 Mouse iPSC-DCs also expressed DEC205, which is an endocytic receptor of DCs and mediates cross-presentation of antigens onto MHC class I.33 34 Hereafter we focus on clone 2A-4F-118 for further experiments.

Generation of dendritic cells (DCs) from mouse induced pluripotent stem cells (iPSCs). (A) Schematic illustration showing the generation of DCs from iPSCs. (B) Phenotypic analysis of iPSC-derived cells on day 26, and bone marrow-derived cells on day 10 differentiated in the presence of GM-CSF in vitro. (C) Representative flow cytometric plots showing IRF4 and IRF8 expression of CD11c+ MHC class II+ iPSC-derived DCs (iPSC-DCs). (D) Phenotypic analysis of CD11c+ MHC class II+ cells. Representative histogram showing CD24, DEC205, CD80, CD11b, B220, CD8α, XCR1 and F4/80 expressions on CD11c+ MHC class II+ 2A-4F-118 iPSC-DCs, 2A-4F-136 iPSC-DCs and bone marrow-derived DCs (BM-DCs) (red). Isotype-matched controls are shown in blue. Number denotes per cent positive cells for each marker. Data shown are representative of three independent experiments. FMT, fluorescence minus two; GM-CSF, granulocyte macrophage colony-stimulating factor; IRF4/8, interferon regulatory factor-4 and 8; MHC, Major Histocompatibility Complex; OP9-DL-1, OP9 cells expressing a notch ligand, delta-like-1; pHEMA, poly 2-hydroxyethyl methacrylate.
Antigen-specific T cell activation, proliferation and cytokine production by iPSC-DCs
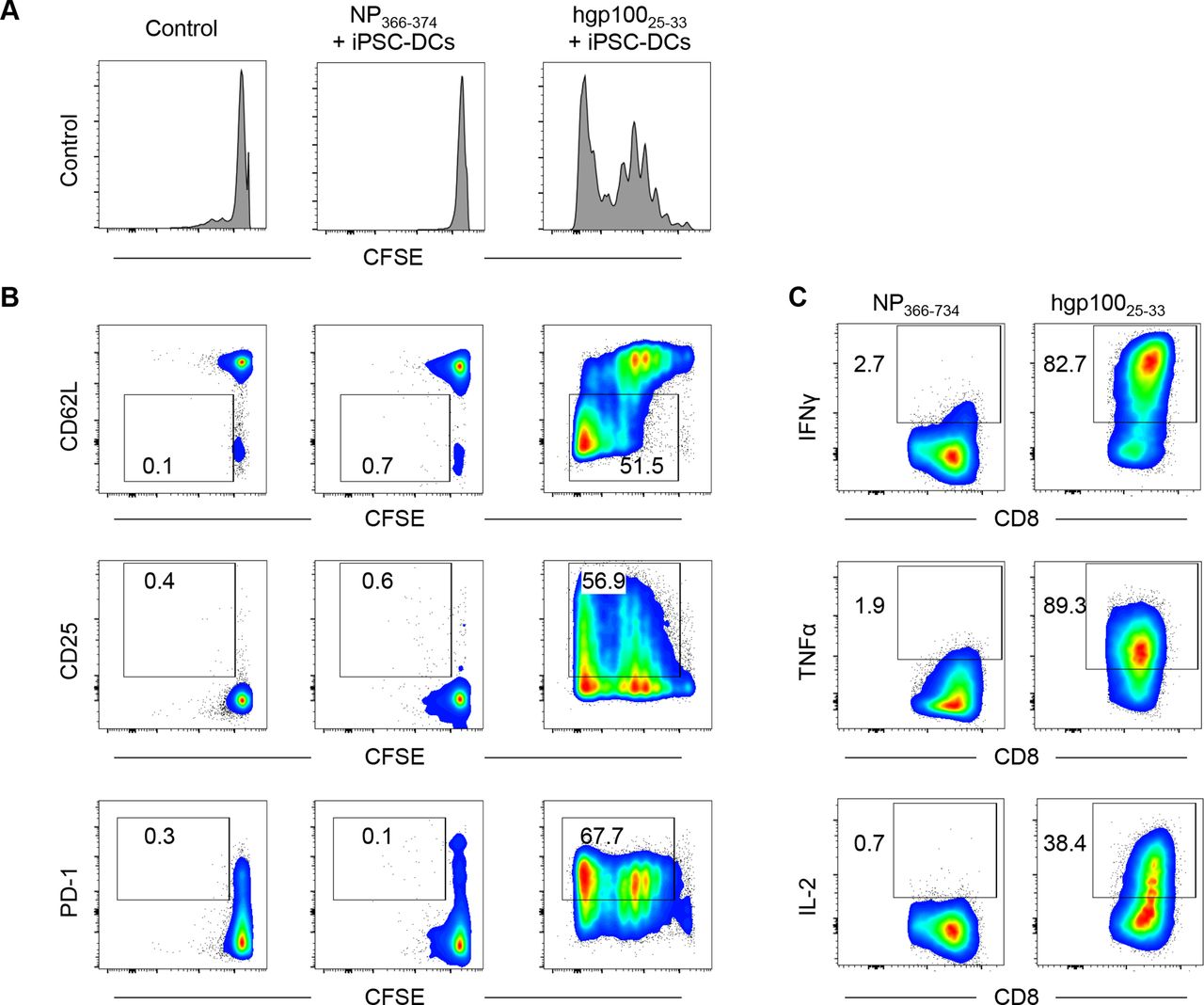
Next, we tested whether iPSC-DCs could activate antigen-specific CD8+ T cells in vitro. To this end, we labeled Pmel-1 CD8+ T cells that can recognize hgp10025–33 with CFSE, and co-cultured them with iPSC-DCs in the presence of hgp10025–33. NP epitope NP366–374 was used as an irrelevant control peptide. Pmel-1 CD8+ T cells vigorously proliferated in response to hgp10025–33 but not to NP366–374 (figure 2A). Phenotypic analysis revealed that hgp100 but not NP peptide pulsed-iPSC-DCs downregulated L-selectin (CD62L) and upregulated CD25 and PD-1 in proliferating Pmel-1 CD8+ T cells (figure 2B). Furthermore, Pmel-1 CD8+ T cells activated by hgp100-pulsed iPSC-DCs had a capacity to produce effector cytokines, IFNγ, TNFα and IL-2 against hgp10025–33 but not NP366–374 (figure 2C). Collectively, these findings indicate effective activation and effector differentiation of antigen-specific CD8+ T cells by iPSC-DCs.

iPSC-derived DCs (iPSC-DCs) activate antigen-specific CD8+ T cells in vitro. (A,B) Pmel-1 T cells (CD90.1+ CD8+) were isolated from splenocytes by CD8α positive selection, labeled with CFSE, and co-cultured with iPSC-DCs in the presence of influenza nucleoprotein (NP) epitope NP366–374 or hgp10025–33 and IL-2 (60 IU/mL). Two days later, cells were harvested for flow cytometric analysis. (A) Representative histogram showing CFSE dilution in Pmel-1 T cells. (B) Representative flow cytometric plots showing CD62L, CD25, and PD-1 expression in Pmel-1 T cells. Numbers denote per cent dividing CD62L−, CD25+, or PD-1+ cells. (C) Activated Pmel-1 T cells from the experiment (A,B) were co-cultured with hgp10025–33 or NP366–374 in the presence of antigen-presenting cells (splenocytes from C57BL/6 mice) for another 2 days. Representative flow cytometric plots show intracellular production of IFNγ, TNFα, and IL-2 in Pmel-1 T cells activated by iPSC-DCs. Numbers denote per cent positive cells. Data shown are representative of two independent experiments. CFSE, carboxyfluorescein succinimidyl ester; IFNγ, interferon gamma; IL-2, interleukin 2; PD-1, programmed cell death protein 1; TNFα, tumor necrosis factor alpha.
Synergistic antitumor efficacy of in situ iPSC-DC administration and local RT against established poorly immunogenic tumors
Given the capacity of iPSC-DCs to activate and expand antigen-specific CD8+ T cells in vitro, we next investigated in vivo antitumor efficacy of iPSC-DCs. Local irradiation to the tumor can induce immunogenic tumor cell deaths, resulting in the release of tumor-associated antigens (TAAs), which are uptaken, processed and presented to T cells by DCs.15–21 Our previous work suggests that a combination of intratumoral BM-DC injections and RT synergistically increases antitumor efficacy compared with administration of BM-DCs or RT alone in preclinical models.22 These observations prompted us to test in vivo antitumor efficacy of iPSC-DCs in combination with RT, using two syngeneic orthotopic mouse models of poorly T cell-inflamed tumors, AT-3 triple negative breast cancer and B16 melanoma, which display primary resistance to PD-1/PD-L1 blockade.26 Although each single therapy of in situ iPSC-DC injection or RT exhibited modest tumor growth delay and improved survival in AT-3 bearing mice, combination of these markedly delayed tumor growth and prolonged survival, indicating synergistic antitumor efficacy of this combination therapy (figure 3A, online supplemental figure 1A). Furthermore, we also observed the synergistic antitumor reactivity of the combined therapy against rapidly growing B16 tumors (figure 3, online supplemental figure 1B)
Supplemental material
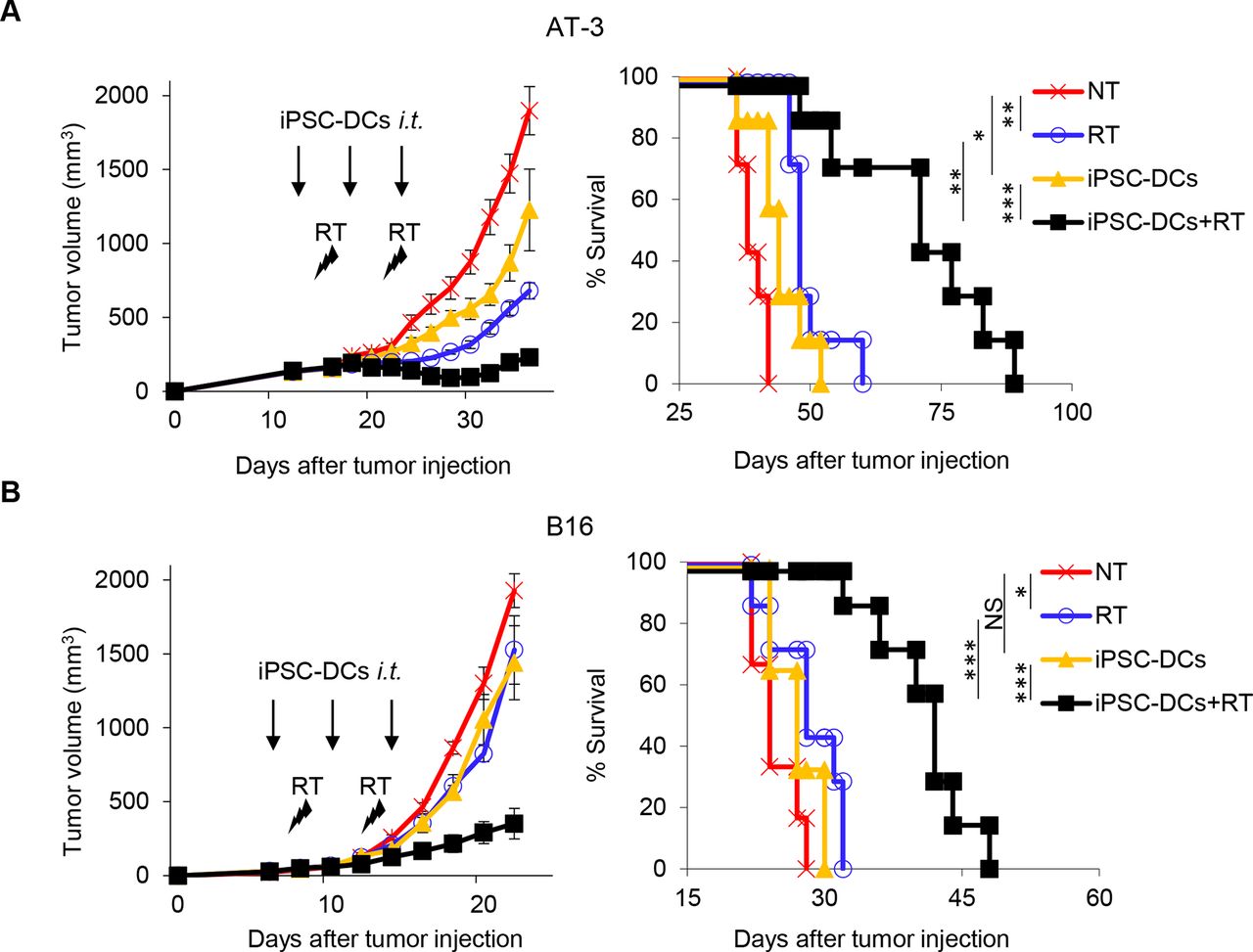
Synergistic antitumor efficacy of in situ iPSC-DC administration and local radiotherapy (RT) against poorly immunogenic tumors. (A,B) Tumor volume curves (mean) and survival curves in AT-3 (n=7) (A) and B16 (n=6–7) (B) tumor-bearing mice in different treatment as indicated. NT, non treatment. Individual tumor volume curves are shown in online supplemental figure 1. NS not significant, *p<0.05, **p<0.01, ***p<0.001 by a log-rank test. Data shown are representative of two independent experiments. Mean±SEM. iPSC-DCs, induced pluripotent stem cell-derived dendritic cells; i.t., intratumorally.
RT augments trafficking of intratumorally injected iPSC-DCs to the TdLN, upregulates CD40 expression, and increases the frequency of DC/CD8+ T cell aggregates
Higher density of CD8+ T cells in the TME correlates with better prognosis in various types of cancers including melanoma and breast cancer,35 and response to anti-PD-1/PD-L1 therapy.36 In contrast, high frequency of tumor-infiltrating myeloid cells and tumor-associated macrophages (TAMs) associates with poor prognosis and resistance to immune checkpoint blockade.26 37 38 Therefore, we examined by flow cytometry analysis whether combined therapy could alter the frequency of tumor-infiltrating CD8+ T cells and CD11b+ myeloid cells (online supplemental figure 2A). Despite increased antitumor efficacy, we found that the frequency of CD8+ tumor-infiltrating lymphocytes (TILs) and a CD8+ T cell to CD11b+ myeloid cell ratio were unchanged by the combination therapy (online supplemental figure 2B). We then evaluated the myeloid compartment of the TME (online supplemental figure 3A). The frequency of DCs was increased in mice treated with intratumoral injection of iPSC-DCs alone (online supplemental figure 3B). However, adding RT to in situ iPSC-DC administration decreased the frequency of DCs in the TME. The frequency of TAMs was decreased by the combination therapy. These findings suggest that the majority of injected iPSC-DCs remained in situ, and that RT may have facilitated their trafficking to the TdLN.
To investigate this notion, we took two complementary approaches. First, mice bearing AT-3 tumors were intratumorally injected with 1×106 fluorescently labeled iPSC-DCs, and were treated with or without RT (figure 4A). Twenty-four hours later, tumors and TdLN were analyzed for flow cytometry (online supplemental figure 4). The frequency of iPSC-DCs was lower in the tumor and higher in the TdLN in RT-treated mice compared with mice which did not receive RT (figure 4B). We also examined the viability of iPSC-DCs in the tumor and TdLN by flow cytometry (online supplemental figure 4). We did not observe many apoptotic iPSC-DCs in the tumor while 40%–60% of iPSC-DCs in the TdLN express annexin V regardless of local RT (figure 4C).

RT augments trafficking of intratumorally injected iPSC-DCs to the TdLN, upregulates CD40 expression, and increases the frequency of DC/CD8+ T cell aggregates. (A) Experimental set-up. Mice bearing AT-3 or AT-3-GFP tumors had an intratumoral iPSC-DC injection with or without local RT (9 Gy). Gating strategy for identifying fluorescently (CTO: CellTracker Orange)-labeled iPSC-DCs by flow cytometry and imaging flow cytometry is shown in online supplemental figure 4 and online supplemental figure 5, respectively. FACS, fluorescence-activated cell sorting. (B) Frequency of iPSC-DCs in the tumor and TdLN in different treatment groups as indicated (n=6). (C) Representative flow cytometric plots showing annexin V and near-IR expression of iPSC-DCs in the tumor and TdLN. The data panel shows the frequency of annexin V+ iPSC-DCs in the tumor and TdLN (n=3–6). Data with total iPSC-DCs >10 from the experiment (B) were evaluated. (D–G) Representative dot plots and frequencies of GFP+ iPSC-DCs as single cells or engaged in cell aggregates (D), CD40+ CD11c+ cells in iPSC-DCs (E), GFP+ iPSC-DC in direct contact with CD8+ T-cell (F), and GFP+ iPSC-DC-XCR1+ DC in direct contact with CD8+ T-cell (G) analyzed by imaging flow cytometry. Representative images are shown in online supplemental figure 6. Each dot represents biologically independent mice (B–G). One-way ANOVA with Tukey’s multiple comparisons (C) and two-tailed unpaired t-test (B, D–G). Mean±SEM. ANOVA, analysis of variance; GFP, green fluorescent protein; iPSC-DCs, induced pluripotent stem cell-derived dendritic cells; IR, infrared; i.t., intratumorally; RT, radiotherapy; TdLN, tumor-draining lymph node.
To further confirm the trafficking of iPSC-DCs to the TdLN, we examined the frequency of injected iPSC-DCs in mice bearing AT-3-GFP tumors by imaging flow cytometry (online supplemental figure 5). Consistent with the finding from flow cytometric analysis, RT enhanced migratory capacity of iPSC-DCs (online supplemental figure 6A) with increased frequencies of GFP+ iPSC-DCs (figure 4D, online supplemental figure 6B) and CD40+ iPSC-DCs (figure 4E, online supplemental figure 6C) in TdLN. Furthermore, RT-treated mice exhibited increased frequencies of GFP+ iPSC-DCs and GFP+ iPSC-DC-XCR1+ DCs in contact with CD8+ T cells in TdLN (figure 4F,G and online supplemental figure 6D,E). Taken together, these results suggest that RT augments trafficking of intratumorally injected iPSC-DCs, upregulates CD40 and facilitates a cross-talk with other DC subsets and CD8+ T cells in TdLN.
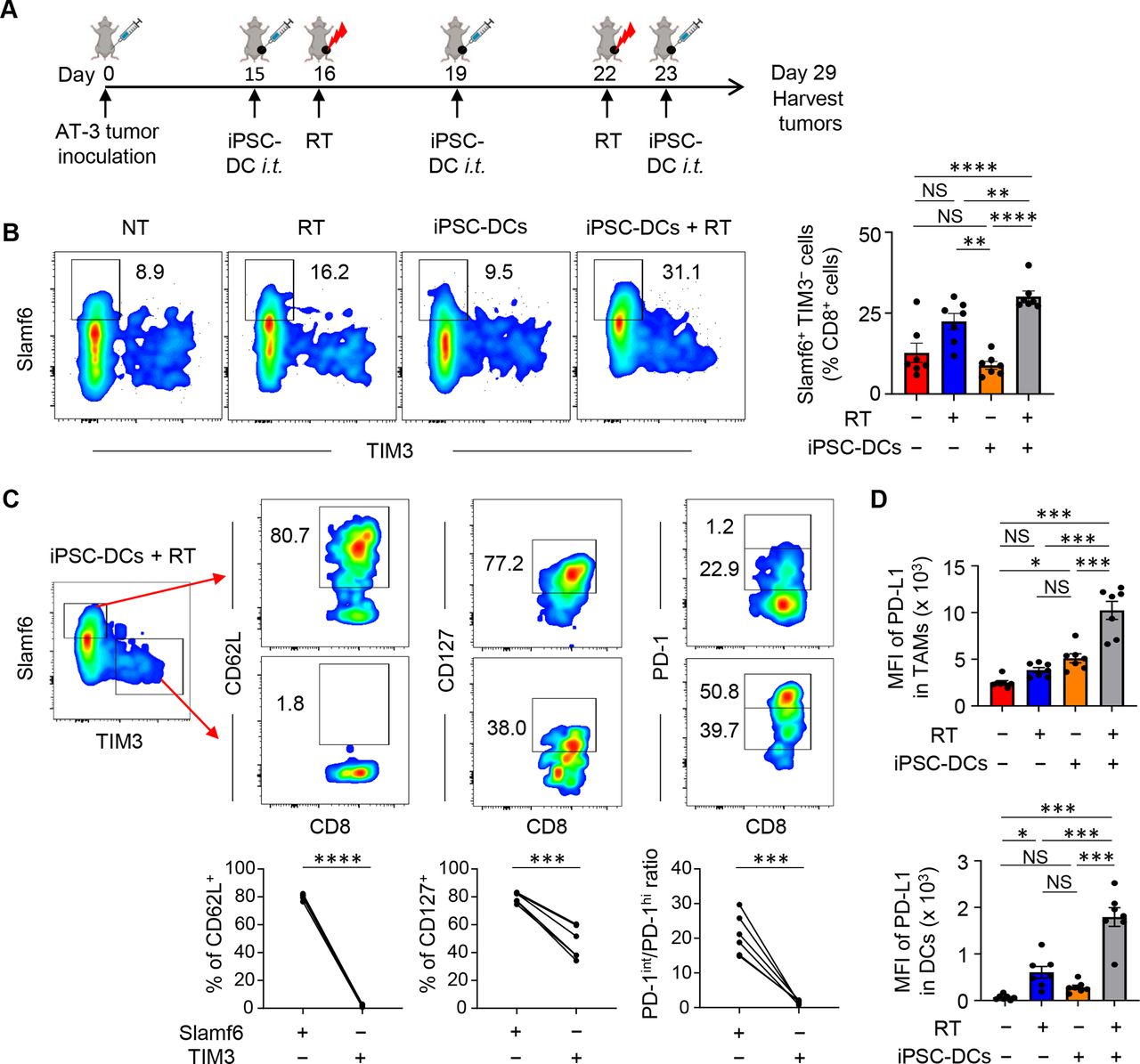
Combined in situ administration of iPSC-DCs and local RT increases Slamf6+ TIM3− CD8+ TILs
Emerging evidence has indicated that intratumoral Slamf6+ PD-1 intermediate (PD-1int) TIM3− progenitor exhausted CD8+ T cells with stem-like properties but not Slamf6− PD-1 high (PD-1hi) TIM3+ terminally exhausted CD8+ T cells mediate cellular expansion and tumor control in response to anti-PD-1/PD-L1 therapy.39 40 Therefore, we evaluated Slamf6 and TIM3 expression in CD8+ TILs in mice bearing AT-3 tumors (figure 5A). We found that the frequency of Slamf6+ TIM3− CD8+ TILs was higher in CD8+ TILs from mice treated with combined therapy of in situ injection of iPSC-DCs and RT (figure 5B). This subset expressed higher levels of CD62L and CD127 with an increased ratio of PD-1int to PD-1hi cells compared with Slamf6− TIM3+ CD8+ TILs (figure 5C), suggesting that combined therapy facilitated the infiltration of the stem-like progenitor exhausted subpopulation.

A combination of in situ iPSC-DC injection and RT increases stem-like progenitor exhausted CD8+ T cells and PD-L1 expression in myeloid cells in the tumor. (A) Experimental set-up. (B) Phenotypic analysis of CD8+ T cells among CD45+ cells in AT-3 tumors in different treatment groups as indicated. Numbers denote per cent Slamf6+ TIM3− cells. Representative flow cytometric plots showing expression of Slamf6 and TIM3 in CD8+ TILs. The data panel shows the frequency of Slamf6+ TIM3− cells in CD8+ TILs (n=7). (C) Phenotypic characterization of Slamf6+ TIM3− and Slamf6− TIM3+ CD8+ TILs (n=6). (D) PD-L1 expression (MFI: median fluorescence intensity) of Ly6c− CD11c+ class II+ F4/80hi CD24− tumor-associated macrophages (TAMs) (upper) and Ly6c− CD11c+ class II+ F4/80lo CD24+ DCs (lower) in AT-3 tumors (n=7). Gating strategy for identifying TAMs and DCs is shown in online supplemental figure 3. NS not significant, *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001 by one-way ANOVA with Tukey’s multiple comparisons (B,D) and two-tailed paired t-test (C). Each dot represents biologically independent mice (B,D). Data shown are representative of two independent experiments. Mean±SEM. ANOVA, analysis of variance; iPSC-DC, induced pluripotent stem cell-derived dendritic cells; i.t., intratumorally; PD-1, programmed cell death protein 1; PD-L1, PD-1 ligand 1; RT, radiotherapy; TILs, tumor-infiltrating lymphocytes; TIM3, T cell immunoglobulin and mucin domain-containing protein 3.
PD-L1 can be adaptively induced in the TME by inflammatory factors secreted by tumor-specific T cells.36 Therefore, we examined whether combined therapy would increase PD-L1 expression in tumor-residing myeloid cells. PD-L1 expression was synergistically upregulated in TAMs and DCs by in situ injection of iPSC-DCs and RT (figure 5D).
Combined in situ administration of iPSC-DCs and local RT increases antigen-specific CD8+ T cell infiltrates
Findings of increased PD-L1 expression in the TME prompted us to hypothesize that combined therapy would facilitate the priming of antigen-specific CD8+ T cells that infiltrate into the TME. To this end, we used an adoptive transfer system of OVA-specific TCR transgenic OT-I CD8+ T cells into mice bearing OVA-expressing B16 (B16-OVA) tumors. The frequency of infused OT-I CD8+ T cells (CD90.1) in C57BL/6 mice (CD90.2) was evaluated after treatment with intratumoral iPSC-DC administration and/or RT (figure 6A). We found that in situ injection of iPSC-DCs and RT increased the frequency of tumor-infiltrating OT-I CD8+ T cells (figure 6B). Increased frequency of adoptively transferred Pmel-1 CD90.1+ CD8+ T cells was also observed in C57BL/6 mice bearing B16 tumors treated with the combined therapy (online supplemental figure 7). Notably, higher frequency of Slamf6+ TIM3− CD8+ T cells was identified in infiltrating OT-I T cells compared with endogenous (CD90.2) CD8+ T cells (figure 6C), suggesting that the combined therapy triggers an increase of progenitor exhausted tumor-specific T cells in the tumor. Of note, we evaluated function of iPSC-DCs in comparison with BM-DCs in vivo (figure 6A), and found that iPSC-DCs and BM-DCs demonstrated equivalent antitumor efficacy (figure 6D) and capacity of increasing tumor-specific Slamf6+ TIM3− CD8+ TILs (figure 6E,F) with local RT.

In situ injection of iPSC-DCs with local RT increases antigen-specific CD8+ T cell infiltrates in the tumor. (A) Experimental set-up. (B) Representative flow cytometric plots showing OT-I T cells (CD90.1+ CD8+) in CD45+ cells in tumors. Data panels show numbers (/g) (tumor) of OT-I T cells (n=3). (C) Representative flow cytometric plots showing expression of Slamf6 and TIM3 in OT-I (CD90.1+) and endogenous (CD90.2+) CD8+ TILs. Numbers denote per cent Slamf6+ TIM3− T cells. (D) Tumor growth curves (mean) (left) and tumor weight (right) of B16-OVA tumor-bearing mice in different treatment as indicated. (n=5). (E) Numbers (/g) (tumor) of OT-I T cells (n=5). Each dot represents biologically independent mice (B, D–F). NS not significant, *p<0.05, **p<0.01, ***p<0.001, one-way ANOVA with Tukey’s multiple comparisons (B,D,E) and two-tailed paired (C) and unpaired (F) t-test. Mean±SEM. ANOVA, analysis of variance; BM-DCs, bone marrow-derived dendritic cells; iPSC-DC, induced pluripotent stem cell-derived dendritic cells; i.t., intratumorally; OVA, ovalbumin; RT, radiotherapy; TILs, tumor-infiltrating lymphocytes; TIM3, T cell immunoglobulin and mucin domain-containing protein 3.
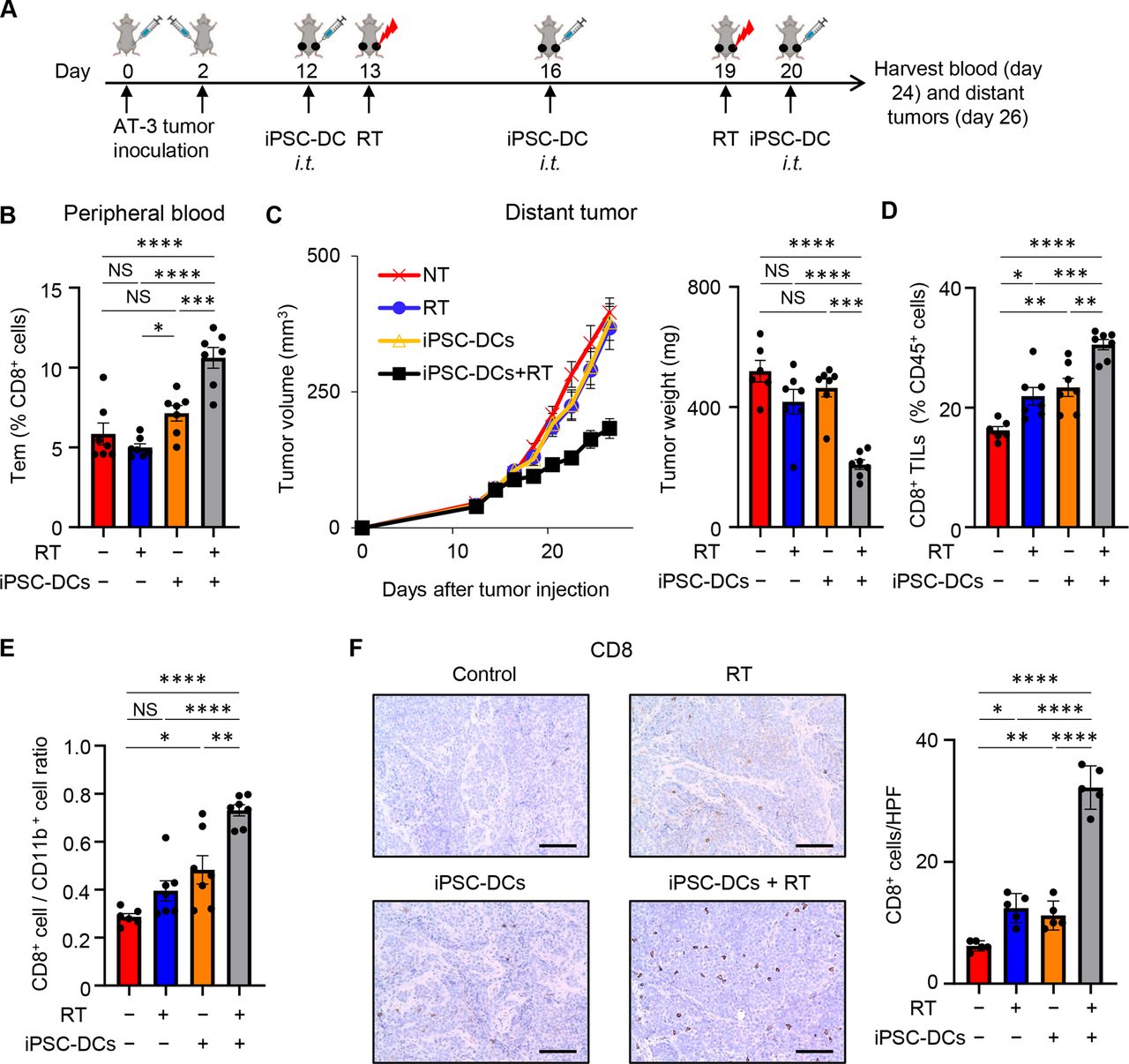
Combining in situ administration of iPSC-DCs with local RT attenuates growth of distant untreated tumors
Given the increased frequency of tumor-specific CD8+ T cells as well as the potent local antitumor response by combination of in situ injection of iPSC-DCs and RT, we next investigated whether this combination therapy would trigger a systemic antitumor immune response. To this end, we used a bilateral mouse tumor model, treated the primary tumor, and monitored the growth of untreated distant tumors (figure 7A). Combination therapy substantially increased the frequency of circulating effector memory CD44+ CD62L− CD8+ T cells (figure 7B). While in situ iPSC-DC administration alone or RT alone had no effect on distant tumor growth, the combination of these attenuated distant tumor growth (figure 7C, online supplemental figure 8). Consistent with these findings, combined therapy synergistically increased CD8+ T cells (figure 7D), and a CD8+/CD11b+ cell ratio (figure 7E), in the untreated distant tumor. Synergistic increase of CD8+ TILs in the distant tumor was confirmed by immunohistochemistry analysis (figure 7F). Together, these results indicate that combined therapy of in situ administration of iPSC-DCs and local RT induces systemic antitumor immunity and is capable of controlling the growth of distant tumors.

Combining in situ administration of iPSC-DC with RT attenuates growth of distant untreated tumors.(A) Experimental set-up. (B) Frequency of peripheral blood effector memory CD44+ CD62L− CD8+ T cells (Tem) in different treatment groups as indicated (n=6–7). (C) Tumor growth curves (mean) (left) and tumor weight (right) of distant untreated tumors in bilateral AT-3 tumor-bearing mice in different treatment as indicated (n=6–7). Individual tumor volume curves are shown in online supplemental figure 8. (D,E). Frequency of CD8+ T cells among CD45+ cells (D) and CD8+/CD11b+ cell ratio (E) in untreated distant AT-3 tumors in different treatment groups as indicated (n=6–7). (F) Representative images of immunohistochemistry for CD8 in untreated distant AT-3 tumors. Scale bars, 100 µm. Data panels show mean numbers of CD8+ cells per each high-power field (HPF) within five different areas. Each dot represents biologically independent mice (B–E). NS not significant, *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001 by one-way ANOVA with Tukey’s multiple comparisons. Data shown are representative of two independent experiments. Mean±SEM. ANOVA, analysis of variance; iPSC-DC, induced pluripotent stem cell-derived dendritic cells; i.t., intratumorally; RT, radiotherapy; TILs, tumor-infiltrating lymphocytes.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
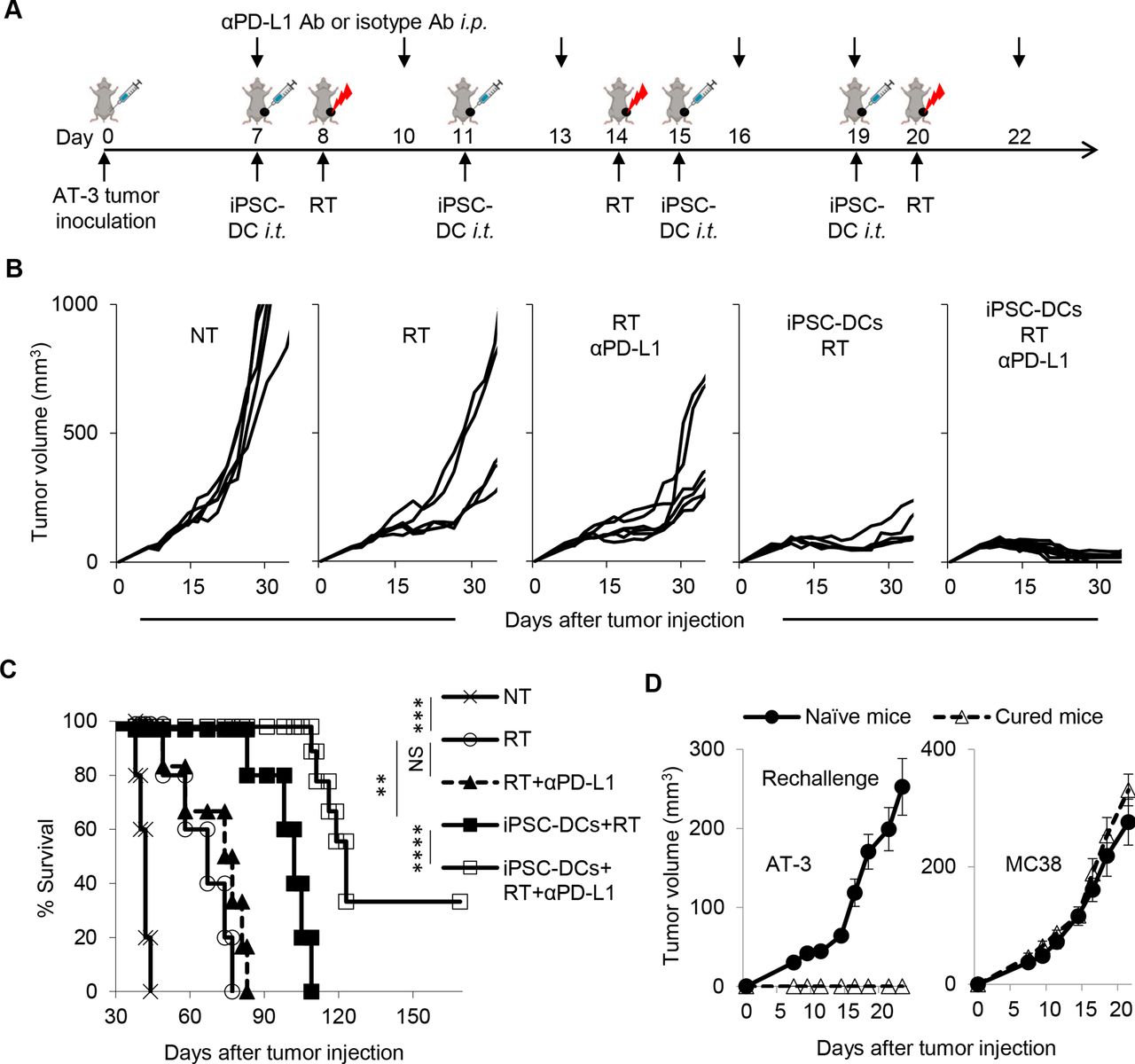
Combined therapy of in situ iPSC-DC injection and RT renders tumors responsive to anti-PD-L1 therapy, has potential to eradicate poorly T cell-inflamed tumors, and establishes immunological memory. (A) Experimental set-up. Mice bearing AT-3 tumors in the left fourth mammary gland were treated with intratumoral iPSC-DC injections, RT, and anti-PD-L1 antibody (αPD-L1 Ab) or isotype Ab. (B) Tumor growth curves (individual) in AT-3 tumor-bearing mice in different treatment groups as indicated (n=5–9). (C) Survival curves in AT-3 tumor-bearing mice in different treatment as indicated. (D) Naïve mice and surviving mice from the experiment (B,C) were rechallenged with AT-3 and MC38 in the right flank at day 123 (AT-3) and on back at day 127 (B16), respectively. NS not significant, **p<0.01, ***p<0.001, ****p=0.0001 by a log-rank test (C). Mean±SEM. Data shown are representative of two independent experiments. i.p., intraperitoneally; iPSC-DC, induced pluripotent stem cell-derived dendritic cells; i.t., intratumorally; PD-L1, PD-1 ligand 1; RT, radiotherapy.
Combined therapy of in situ iPSC-DC injection and irradiation renders tumors responsive to anti-PD-L1 therapy, has potential to eradicate poorly T cell-inflamed tumors, and establishes immunological memory
Our findings of the increased stem-like progenitor exhausted CD8+ TILs and upregulation of PD-L1 expression in tumor-residing macrophages and DCs led us to postulate that antitumor efficacy of the combination therapy could be augmented by PD-1/PD-L1 blockade. To address this, we treated AT-3-bearing mice with combination of in situ injection of iPSC-DCs, RT, and anti-PD-L1 Ab or isotype Ab (figure 8A). Although anti-PD-L1 Ab therapy did not alter the immunogenicity of RT, it substantially improved tumor growth and survival in mice treated with a combination of in situ injection of iPSC-DCs and RT (figure 8B,C). Furthermore, mice with durable regressions rejected the AT-3 tumor rechallenge, but not unrelated MC38 colon adenocarcinoma, demonstrating the establishment of tumor-specific immunological memory (figure 8D). Taken together, these findings suggest in situ induction of iPSC-DCs and RT could overcome primary resistance to anti-PD-L1 therapy.
Discussion
In this study, we demonstrated synergistic antitumor efficacy of in situ iPSC-DC administration and local RT in mouse models of poorly T cell-inflamed tumors. This multimodal intralesional therapy enhanced the priming of tumor-specific CD8+ T cells, generated systemic adaptive T-cell immunity, and delayed growth of untreated distant as well as treated tumors. Furthermore, this combination triggered an influx of stem-like Slamf6+ PD-1int TIM3− CD8+ T cells, activated PD-1/PD-L1 axis in the TME, rendered AT-3 tumors susceptible to anti-PD-L1 therapy, and established tumor-specific immunological memory.
A major advantage of using iPSC-DCs is the ability to generate unlimited numbers of phenotypically defined, functional, and autologous professional antigen-presenting cells (APCs)12 for repeated vaccinations. Previous studies demonstrated that mouse PSC-derived DCs exhibited comparable expression levels of surface DC markers, and antigen-presenting capacity with BM-DCs,7 9–12 and that vaccination with antigen-loaded or transduced mouse PSC-derived DCs had protective and therapeutic efficacy in preclinical models.9–13 Our results agreed with these, and further revealed equivalent antitumor efficacy of intratumorally injected iPSC-DCs and BM-DCs in combination with local RT. It is likely, however, that unstimulated iPSC-derived cells in GM-CSF cultures have APCs with various differentiation status. Therefore, iPSC-derived CD11c+ MHC class II+ cells may comprise a heterogeneous population similar to BM-derived CD11c+ MHC class II+ cells,41 and more work is needed to elucidate the cellular heterogeneity of iPSC-derived cells by bioinformatics and systems biology.
Our findings described herein may provide novel implications for the therapeutic application of iPSC-DCs in the clinical setting. There seems to be a strong rationale for combining in situ delivery of iPSC-DCs with RT based on compelling evidence that tumor irradiation not only induces immunogenic tumor cell death leading to release of TAA, but also augments DC maturation, and increases the ability of DCs to cross-present antigens and prime T cells.16 19 42–44 In this study, we demonstrated that RT could enhance the migratory capacity of intratumorally injected iPSC-DCs to TdLN, which is necessary to initiate the process of the adaptive immune response. Moreover, imaging flow cytometry displayed increased frequencies of GFP+ iPSC-DC/CD8+ T cell and GFP+ iPSC-DC/XCR1+ DC/CD8+ T cell aggregates, representing TAA-loaded DC-T cell cross-talk in TdLN. Poor trafficking of DCs from injection sites to the LN has been recognized for many years, and increasing migratory capacity of DCs after immunization is paramount for effective cancer immunotherapy.1 Of note, we chose two to three fractions of 9 Gy based on our recent study suggesting that this dose could cause immunogenic tumor cell death and promote maturation of tumor-residing DCs.26 However, optimal dose, fractionation and interval between doses in combination with in situ iPSC-DC administration remain to be determined. Given the negligible apoptotic iPSC-DCs in the TME after local RT (figure 4C), increasing dose or fractionation might further improve antitumor efficacy of intratumorally injected iPSC-DCs. Future studies are necessary to optimize several parameters including dose, fractionation and timing of RT in relation to the in situ iPSC-DC administration.
One advantage of the in situ vaccination strategy is to elicit tumor-specific adaptive response without the need for identification of patient-specific and tumor-specific antigens. Our findings of the increased frequency of tumor antigen-specific CD8+ T cells after combined intratumoral iPSC-DC administration and RT confirmed this scenario, and are in line with our previous study using BM-DCs and RT.22 Our work further demonstrated that antitumor T cells generated at treated tumor sites were capable of controlling distant tumor growth. Another advantage of this approach is to generate diverse repertoires of T cells targeting heterogeneous tumors. Therefore, it was reasonable to observe that the antitumor efficacy of intratumoral iPSC-DC administration and RT was further potentiated by anti-PD-L1 therapy, which also targets diverse repertoires of intratumoral T cells. One caveat from the current study is that response to anti-PD-L1 therapy might be associated with the type (Slamf6+ TIM3−) of infiltrating CD8+ T cells rather than the frequency of CD8+ T cells in the TME.
In our experiments, the beneficial effect of anti-PD-L1 therapy was not seen in mice treated with RT alone whereas other preclinical studies showed synergistic antitumor efficacy of RT and PD-1/PD-L1 blockade.44–47 The reason for this discrepancy remains unclear although it may be explained by the size of the tumor at the initiation of the treatment, poor immunogenicity of AT-3 tumors or different radiation dose-fractionation regimens. In our study, the profound therapeutic efficacy of anti-PD-L1 therapy was identified in tumors treated with combined in situ iPSC-DCs and RT that are enriched with Slamf6+ PD-1int TIM3− CD8+ T cells. Our findings are in line with the emerging evidence that progenitor exhausted T cells that express Slamf6 and intermediate-level of PD-1 found in tumors are critical for a durable response to PD-1/PD-L1 blockade therapy.26 39
Although the frequency of CD8+ T cells and CD8+/CD11b+ cell ratios were increased in distant untreated tumors, these were not seen in tumors treated by intratumoral injection of iPSC-DCs and RT. This is likely because the majority of injected iPSC-DCs remained in situ after administration. Even though RT increased the trafficking of iPSC-DCs to TdLN, only approximately 0.5% of injected iPSC-DCs were identified in the TdLN (online supplemental figure 6A). The reason for this remains unclear, but might be associated with the maturation status of DCs. We used non-activated (immature) DCs due to their ability to efficiently capture antigens,2 which was shown to be effective against established poorly immunogenic tumors in combination with local RT.22 However, immature DCs could induce immune tolerance,2 3 48 and would require activation after in vivo capture of dead tumor cells to be able to migrate to TdLN and prime T cell responses. Regarding this issue, we have recently shown that in situ dual toll-like receptor 3/CD40 stimulation facilitates trafficking of tumor-residing DCs to TdLNs.26 A CD40 agonist would be particularly attractive in this regard because it could provide maturation and anti-apoptotic signals to DCs,48 49 which might activate iPSC-DCs expressing CD40 (figure 4E) and decrease the frequency of apoptotic iPSC-DCs in TdLN (figure 4C). Therefore, a future area of investigation will be to add immunomodulators to stimulate iPSC-DCs to the combined therapy. We observed upregulation of CD40 in migrated iPSC-DCs after RT (figure 4E).
In situ induction and activation of Batf3-dependent conventional type 1 dendritic cells (cDC1s) has been shown to enhance immunogenicity of RT in preclinical models and humans.26 50 In this study, we have used OP9-DL1 feeder cells at step 2 of an established differentiation protocol12 in an attempt to differentiate mouse iPSC-derived hematopoietic progenitor cells (HPCs) on OP9 cells to cDC1s based on recent evidence that notch signaling facilitates in vitro generation of mouse and human cDC1s.28 29 However, we found that generated iPSC-DCs expressed CD24, CD11b, F4/80, CD80 and IRF4, similar to cDC2 not cDC131 32 even in the presence of GM-CSF and Fms-like tyrosine kinase 3 ligand (data not shown). The exact reason for this remains unclear, but can be explained by the difference between hematopoietic stem cells and iPSC-derived HPCs.51 Further investigation is needed to delineate the culture conditions for the generation of cDC1s from iPSCs.
Despite potent antitumor efficacy and synergy with anti-PD-L1 therapy, this combinatorial strategy has some drawbacks. Generation, characterization, maintenance, and differentiation of iPSCs remain a labor and time-consuming task while part of this limitation can be alleviated by the use of iPSC banks to provide human leukocyte antigen-matched iPSC products.52 Similar to other vaccination regimens, development of adaptive T cell immunity requires repeated administration of iPSC-DCs and RT, which might not be suitable for rapidly growing tumors. Genetic modification of iPSCs or iPSC-derived myeloid cells for generation of myeloid lineage cells with proliferation capacity9 53 may mitigate these limitations. A recent study showed that local administration of IFNα-producing iPSC-derived proliferating myeloid cells (iPSC-pMCs) could recruit cDC1s to the TME, and yield superior antitumor efficacy compared with untransduced iPSC-pMCs.14
In summary, this work highlights the antitumor efficacy of in situ administration of iPSC-DCs in combination with RT against poorly immunogenic tumors. The combined treatment leads to the favorable TME for anti-PD-L1 therapy to be effective, and triggers antitumor reactivity in untreated distant tumors. Our data suggest the therapeutic potential for a novel vaccination strategy with the use of iPSC-derived products rationally integrated with traditional cancer treatment.
Supplemental material
Data availability statement
Data are available upon reasonable request.
Acknowledgments
We acknowledge Prometheus Laboratories for kindly providing rhIL-2 and BioRender.com for illustrations. We thank Drs Scott Abrams, Sharon Evans (Roswell Park Comprehensive Cancer Center), and Weiping Zou (University of Michigan) for AT-3, B16-OVA, and MC38 cells, respectively, and Ms Cheryl Eppolito and Drs Takayoshi Yamauchi and Kumiko Iwabuchi (Roswell Park Comprehensive Cancer Center) for technical assistance.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors TO designed and performed experiments, analyzed data, and wrote the manuscript. KM, RK, and TY designed and performed experiments, and revised the article. RA and MA provided mouse iPSCs, and revised the article. HM performed imaging flow cytometry analysis and revised the article. AEC provided intellectual input on the project and revised the article. AO provided resources (OT-I TCR transgenic mice), and revised the article. FI developed the concept, designed experiments, analyzed data, revised the article, coordinated author activities, and provided final approval of the version to be submitted.
Funding This work was supported by National Cancer Institute (NCI) grant P30CA016056 involving the use of Roswell Park’s Flow and Image Cytometry, and Mouse Tumor Model Shared Resources. This work was supported by the Melanoma Research Alliance and the Sarcoma Foundation of America (FI), Uehara Memorial Foundation (TO), National Cancer Institute (NCI) grant, K08CA197966 (FI) and R50CA211108 (HM).
Competing interests AO was a co-founder of Tactiva Therapeutics and receives research support from AstraZeneca and Tessaro.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.