Article Text

Abstract
Background Tertiary lymphoid structures (TLSs) are immune aggregates in peripheral tissues that may support adaptive immune responses. Their presence has been associated with clinical response to checkpoint blockade therapy (CBT), but it is unknown whether TLS have prognostic significance independent of CBT in melanoma. We hypothesized that TLS in melanoma metastases would be associated with increased intratumoral lymphocyte infiltration, but that the intra-TLS immunological milieu would be distinct from the intratumoral immunological milieu. We also hypothesized that the presence of TLS would be associated with improved survival, and that TLS maturation or intra-TLS lymphocyte activity would also correlate with survival.
Methods Cutaneous melanoma metastases (CMM) from 64 patients were evaluated by multiplex immunofluorescence for the presence and maturation status of TLS. Intra-TLS lymphocyte density, proliferation and B-cell Ig somatic hypermutation (AID+) were analyzed, as were markers of T-cell exhaustion and Th1/Tc1 differentiation. Associations between TLS maturation and intra-TLS immunologic activity were assessed, as well as associations with intratumoral immune cell infiltration. Independent associations with overall survival (OS) were assessed using log-rank tests and Cox proportional hazards models.
Results TLS were identified in 30 (47%) of 64 CMM (TLS+) and were associated with increased intratumoral lymphocyte infiltration. However, proliferation of intra-TLS lymphocytes did not correlate with intratumoral lymphocyte proliferation. Most were early TLS; however, subsets of primary or secondary follicle-like TLS were also present. TLS+ lesions were associated with lower risk of tumor recurrence after metastasectomy and with improved OS in multivariate analyses (HR 0.51, p=0.04). OS was longer for TLS with low fractions of CD21+ B-cells (HR 0.29, p=0.02) and shorter for those with low AID+ fraction of B-cells (HR 2.74, p=0.03).
Conclusions The presence of TLS in CMMs is associated with improved OS in patients treated with surgery before CBT, but TLS vary widely in maturation state, in proportions of proliferating T and B cells, and in markers of B cell function, including AID and CD21. Importantly, these features have additional prognostic significance, which suggest that some TLS may have regulatory function, while others functioning to support antigen-driven immune responses, depending on the cellular composition and activation status.
- B-lymphocytes
- immunity
- cellular
- tumor microenvironment
- melanoma
Data availability statement
Data are available on reasonable request. All data available from the authors on reasonable request.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Background
Tertiary lymphoid structures (TLS) are organized lymphoid aggregates that have been identified at sites of chronic antigen exposure, including organ transplants, localized autoimmune reactions and tumors. Characterized by distinct T and B cell areas and lymph-node-like vasculature expressing peripheral node addressin (PNAd),1 2 TLSs have been identified in primary and metastatic melanoma lesions,3–5 and are associated with improved patient survival and response to checkpoint blockade.3 4 6–13 However, these associations have been based on univariate analyses or multivariate models which did not account for some relevant clinical and immunological variables.4 6 13 Another important limitation of prior studies has been a disproportionate reliance on TLS found in metastases to lymph nodes (LNs)4 5 13 which; as secondary lymphoid organs, may not reflect TLS arising de novo at other sites. Thus, uncertainty persists regarding whether TLS are independently associated with clinical outcomes in melanoma, particularly when associated with non-LN metastases.
TLSs vary in composition and expression of functional markers, and such differences may alter their associations with clinical outcomes. For example, TLSs in melanoma have been associated with improved outcomes, while in breast cancer and hepatocellular carcinoma, TLS have been associated with increased tumor grade and decreased overall survival (OS).14–16 Furthermore, in transplanted lungs, TLS are associated with enhanced tolerance to the transplanted organ.17 These contrasting effects of TLS on immune control may also depend on TLS maturation, which progress from immature early TLS (eTLS), to primary follicle-like TLS (pTLS) and subsequently to secondary follicle-like TLS (sTLS). Maturation stage has correlated with clinical outcomes in colorectal and lung cancers, however a similar assessment has not been conducted in melanoma.4 5 7–9 18
Additional questions remain regarding how lymphocyte activation within the TLS (which are usually peritumoral) relates to lymphocyte activation within the tumor parenchyma and to overall tumor control. Among intratumoral immune cells within melanoma metastases, proliferating T-cells and those differentiated towards Th1/Tc1 phenotypes having been associated with improved survival, while exhausted T-cells and regulatory T-cells (Tregs) have been associated with worse clinical outcomes.19–24 Likewise, our group and others have shown that intratumoral B cell density has been associated with prolonged survival of melanoma patients before the checkpoint blockade era25–27 and with higher response rates to checkpoint blockade.4 5 However, it is unknown whether intra-TLS lymphocyte composition or activation correlates with clinical outcomes; likewise, it is unknown whether intra-TLS lymphocyte function mirrors intratumoral lymphocyte function.
To advance understanding of the prognostic significance of TLS, we evaluated TLS and intratumoral regions for immune infiltrates using multiplex immunofluorescence histology (mIFH) in a homogeneous population of cutaneous melanoma metastases (CMM). We hypothesized that mature TLS and would be associated with improved OS in a multivariate model controlling for clinical variables and intratumoral CD8+ T-cell density. We also hypothesized that among patients with TLS, survival would be shorter in those with high proportions of immature TLS, and in those with high proportions of intra-TLS lymphocytes expressing markers of T cell exhaustion (EOMES+), Tregs (FoxP3+), or CD21+CD20+ B-cells. Similarly, we hypothesized that among patients with a TLS, high densities of intra-TLS lymphocytes expressing markers associated with immune activation, such as proliferation (Ki67+), somatic hypermutation by B-cells (AID+), or Th1/Tc1 differentiation by T-cells (Tbet+) would be associated with improved outcomes. Finally, we predicted that lymphocyte activity within the TLS would correlate with lymphocyte activity within the tumor.
Materials and Methods
Patient selection and clinical data collection
With institutional IRB approval (IRB-HSR #10598, #10803), checkpoint-therapy naïve patients with stage IIIB–IV melanoma whose tumors had been included in a tissue microarray (TMA) evaluating for intratumoral immune infiltrates,25 had clinical follow-up after metastasectomy, and available formalin-fixed paraffin-embedded (FFPE) tissue blocks with adequate remaining metastatic tumor tissue (n=130) were selected for analysis. Of these, 100 lesions were identified that were not LN metastases, and these underwent initial screening for presence of TLS by mIFH. To enable a focus on cutaneous metastases, primary tumors and metastases to sites other than skin were excluded, leaving 68 patients with metastases to skin or subcutaneous tissues (hereafter referred to as CMM). Of these, three tumors were excluded due to insufficient tumor on mIFH stains, and one was excluded for have no follow-up after metastasectomy. This left 64 patients for analysis, each with melanoma skin metastases who comprised the study sample. Patients’ clinical histories, disease status and outcomes had been queried.18 Patients were further classified as having undergone metastasectomy with curative intent if they underwent resection of all known melanoma lesions at time of surgery, or metastasectomy with palliative intent if they underwent incomplete resection of their metastatic burden. A participant flow diagram is shown in online supplemental figure 1.
Supplemental material
Tissue microarray
FFPE blocks of CMM were obtained, and TMAs had been constructed from 3 to 4 cores per tumor, each 1.0 mm in diameter, through regions of each selected FFPE block.25 Cores were taken from the tumor center without incorporating surrounding stromal tissue (online supplemental figure 2A). Immune cells had been enumerated through IHC using standard protocols as has been previously reported.28 Cell counts were determined by automated cell counting and averaged across all available cores from a given tumor. Counts were reported as cell density (cell/mm2). Histological patterns of tumorous immune cell infiltration had also been previously described,25 and tumors were subsequently designated immunotype A (no immune infiltrate), immunotype B (immune infiltration adjacent to intratumoral blood vessels) or immunotype C (diffuse immune infiltration).
Multiplex immunohistochemistry and image analysis
Five µm thick sections were cut from FFPE tumor specimens, and human LN samples was used as a positive control. mIFH was performed according to the manufacturer’s protocol using the OPAL Multiplex Manual IHC kit, and antigen retrieval (AR) buffers AR6 and AR9 (Akoya Biosciences, Marlborough, Massachusetts, USA). DIVA Decloaker AR buffer (Biocare Medical, Pacheco, California, USA) was also used. Staining sequence, antibodies and AR buffers were as follows for the 3 panels of 4–6 antibodies:
Panel 1, TLS Identification
AR9, CD8 (1:500, clone C8/144B Agilent Technologies, Santa Clara, California, USA) Opal540; AR6, CD20 (1:1000, clone L26, Agilent Technologies) Opal520; AR6, PNAd (1:1000 clone MECA-79, BioLegend, San Diego, California, USA) Opal620; AR6, Ki67 (1:20, SP6, Abcam, Cambridge, Massachusetts, USA) Opal690; and AR6, spectral DAPI (Akoya Biosciences, Marlborough, Massachusetts, USA). A representative image from panel 1 provided for a TLSpos lesion (online supplemental figure 2) and a TLSneg lesion (online supplemental figure 3).
Panel 2, TLS Maturation
AR9, ILT4/LILRB2 (1:100, Lifespan Biosciences, catalog# LS-B9762 Seattle, WA) Opal620; AR9, AID (1:100, clone ZA001, Thermo Fisher Scientific, Waltham, Massachusetts, USA) Opal570; DIVA, FoxP3 (1:500, clone D2W8E, Cell Signaling Technology, Danvers, Massachusetts, USA) Opal650; AR6, (CD20 1:1000) Opal520; AR6, CD23 (1:100, clone 1B12, Leica Biosystems, Buffalo Grove, Illinois, USA) Opal540; AR9 CD21 (1:500 clone EP3093, Abcam) Opal690; and AR6, spectral DAPI. A representative image from panel 2 is proved in online supplemental figure 4.
Panel 3, TLS T-helper cell lineage
AR9, CD4 (1:100, clone SP35, Cell Marque, Rocklin, CA) Opal520; AR9, CD8 (1:500) Opal540; AR9, EOMES (1:200, Abcam, catalog# ab23345) Opal620; AR6, CD20 (1:4000) Opal570; DIVA, T-bet (1:200, Santa Cruz Biotechnology, Dallas, Texas, USA) Opal650; AR6, Ki67 (1:20) Opal690; and AR6, spectral DAPI. A representative image from panel 2 is proved in online supplemental figure 5.
Stained slides were mounted using prolong diamond antifade (Life Technologies, Carlsbad, California, USA) and scanned using the PerkinElmer Vectra V.3.0 system and Vectra software (Akoya Biosciences, Marlborough, Massachusetts, USA). Regions of interest were selected in Phenochart software, and ×20 magnification images were acquired with the Vectra V.3.0 system. These images were spectrally unmixed using single stain positive control images in the InForm software (Akoya Biosciences, Marlborough, Massachusetts, USA), and immune cells were quantified using HALO software (Indica Labs, Albuquerque, New Mexico, USA). Lymphocyte density was quantified as density of cells expressing a given marker (cells/mm2) within the region of interest. Fractions of lymphocytes expressing a given activation or differentiation marker were calculated by normalizing the density of cells expressing the lymphocyte marker and the activation/differentiation marker by the density of cells expressing the lymphocyte marker (Example: Fraction of Ki67+CD8+ T-cells =  ). Fractions where the denominator was zero were treated as zeros. Normalized densities were used for all comparisons of activation or differentiation marker expression by intra-TLS or intratumoral lymphocytes, and for all assessments of patient outcomes relative to expression of these markers by intra-TLS lymphocytes.
). Fractions where the denominator was zero were treated as zeros. Normalized densities were used for all comparisons of activation or differentiation marker expression by intra-TLS or intratumoral lymphocytes, and for all assessments of patient outcomes relative to expression of these markers by intra-TLS lymphocytes.
TLS identification and determination of maturation state
CMM-associated lymphoid aggregates were identified in ×20x spectrally unmixed images by visual inspection (IM) and identified as TLS if possessing organized T-cell and B-cell regions in addition to high endothelial venules (HEV)-like vasculature (PNAd+) (figure 1). As TLS mature, they progress from early lymphocyte aggregates (eTLS) to TLS that resemble a pTLS or a sTLS. Accordingly, the maturation state of each TLS was subsequently classified as eTLS, pTLS, or sTLS based on the presence of a CD21+ follicular dendritic cell (FDC) network (pTLS), a CD23+ germinal center (sTLS) or the absence of these features (eTLS), as previously described7 8(figure 2). To estimate the relative abundance of TLS in each maturation state associated with a given tumor, the number of eTLS, pTLS and sTLS identified in each patient’s tumor was divided by the total number of TLS identified in that tumor.
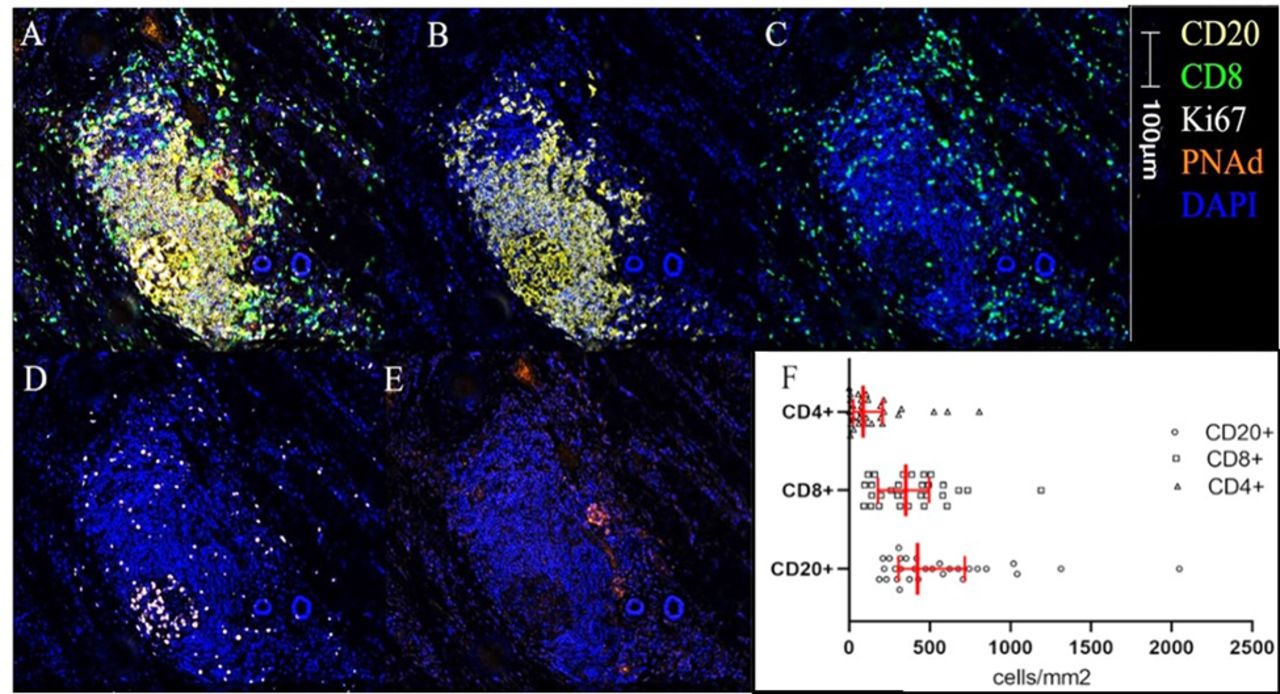
TLS image and immune cell densities. A representative multiplex image of a TLS is shown in (A–E) colors and scare bar are indicated. (A) Five color mIFH staining, (B) CD20+ B-cell aggregate, (C) CD8+ T-cell aggregate, (D) Ki67+ cells, (E) PNAd+ vasculature, (F) histogram of median densities within TLS. IQRs are shown in red bars. Kruskal-Wallis p<0.01. mIFH, multiplex immunofluorescence; PNAd, peripheral node addressin; TLS, tertiary lymphoid structures.

Representative Images of eTLS, pTLS, sTLS (left, middle, right column, respectively). (A) Aggregate CD20+CD21+CD23+ staining (B) CD20+ staining (C) CD21+ staining in a reticular network (pTLS and sTLS) (D) CD23+ germinal center-like structure (sTLS only). eTLS, early tertiary lymphoid structure; pTLS, primary follicle-like TLS; sTLS, secondary follicle-like TLS.
Statistical analysis
T-tests and χ2 tests were used, as appropriate, to test for differences between continuous means or categorical variables, respectively. Where continuous variables were non-parametrically distributed (determined by Shapiro-Wilks test), Kruskal-Wallis tests were used to assess for differences in means. Fraction of TLS in each maturation state (eTLS, pTLS, sTLS) were calculated as described above, and correlations between these fractions and lymphocyte densities or activity markers were assessed with Spearman’s rank correlation coefficient. Survival analysis was also conducted as summarized according to REMARK guidelines in online supplemental table 1. Endpoint was duration of OS as defined by the time from metastasectomy to death. Patients who were alive at last follow-up were censored. Continuous variables were dichotomized into high or low groups using the Contal-O’Quigely method (dichotomization points provided in online supplemental table 2).29 The Kaplan-Meier product limit estimator was used to estimate survival distributions. Multivariate Cox proportional hazards assumptions were tested using Schoenfield residual analysis using a level of significance of 0.01, and where residuals were <0.01, models was stratified accordingly. Four multivariate models were tested, the first on all patients, and the remainder included only patients with a tumor-associated TLS. Covariates of interest for multivariate Cox proportional hazards regression analysis of all patients were selected based on perceived prognostic significance and included disease stage, patient age, patient sex, intratumoral CD8+ T-cell density, and whether the surgical resection was performed with palliative or curative intent. Covariates of interest for multivariate Cox proportional hazards regression analysis of patients with a TLS were selected from variables significant in univariate analyses or of clinical relevance. P values were considered significant when <0.05 and were considered indicative of a trend towards significance when between 0.05 and 0.20. All statistics were conducted using R software V.3.5.3.
Results
Patient demographics
One hundred and thirty CMM from unique patients with clinical stage IIIB–IV disease were selected from a population whose tumors had been previously incorporated in a TMA, with linked clinical data.14 Of these, 66 were excluded for lack of available FFPE blocks, lesion type other than skin metastasis, technical failure or lost to follow-up immediately after metastasectomy, leaving 64 skin metastases which form the basis of the present report (online supplemental figure 1). Each TLS was defined by distinct B and T cell areas and PNAd+ vasculature in the T cell area (figure 1A–C,E). TLS were identified in 30 (47%) of these skin metastases. Most clinical features were similar between TLS+ and TLSneg patients; however, a greater proportion of TLSneg patients suffered disease recurrence after metastasectomy (p=0.02, table 1).
Association of clinical features and intratumoral lymphocyte infiltration with TLS
Enumeration of intra-TLS lymphocyte populations
Densities of CD4+ and CD8+ T-cells and CD20+ B-cells within TLS were enumerated using mIFH. B-cells comprised the largest population of intra-TLS lymphocytes, and CD8+ T cells were more numerous than CD4+ T cells (p<0.01, figure 1F). Hypothesizing that the effects of TLS on tumor control may relate to intra-TLS cellular function rather than intra-TLS cellularity per se, we also determined the fractions of lymphocytes proliferating within the TLS (Ki67+), the fraction of B-cells undergoing somatic hypermutation (AID+), and the fraction of CD4+ T-cells or CD8+ T-cells with a Th1 or Tc1 phenotype (Tbet+), as putative markers to support antitumor immunity. To identify features that may correspond to immune suppression, we also enumerated markers associated with immune regulation, including density of T-regulatory cells (FoxP3+), exhausted T-cells (EOMES+), and CD21+CD20+ B-cells, which have been associated with regulation of autoimmunity.30 Distribution of fractions of intra-TLS and intratumoral lymphocytes expressing a given activation marker are shown in online supplemental figure 6.
TLS maturation does not correlate with Intra-TLS markers of lymphocyte function
TLSs were classified as eTLS, pTLS, or sTLS, using criteria described for cancers of the lung8 and colon7 (figure 2). We hypothesized that TLS maturation state would correlate with proportions of proliferating lymphocytes and intra-TLS B-cells undergoing somatic hypermutation. A total of 334 individual TLS were identified in the 30 TLS+ CMM, of which 63% (212/334) were eTLS, with the remainder consisting of pTLS (51/334, 15%) and sTLS (71/334, 21%). There was significant variation in the fractions of eTLS, pTLS and sTLS across tumors, with nine CMM having only eTLS, while in nine other CMM at least 50% of TLS were pTLS or sTLS. To estimate the relative abundance of TLS in each maturation state for each tumor, the numbers of eTLS, pTLS, and sTLS identified in each CMM was normalized by the total number of TLS identified in that tumor, generating fractions of eTLS, pTLS, and sTLS. There was no significant association between the fraction of proliferating B-cells (CD20+Ki67+ of CD20+) in the TLS and the fraction of eTLS (p=0.68) or sTLS (p=0.30) (online supplemental figure 7). Similarly, the fraction of B-cells expressing AID did not correlate significantly with the sTLS or eTLS fraction (online supplemental figure 8).
TLS neogenesis and maturation is associated with intratumoral lymphocyte density
To determine if intra-TLS lymphocyte activity correlated with intratumoral lymphocyte activity, we first quantified intratumoral immune infiltration using TMAs25 (online supplemental figure 2A). Intratumoral regions of TLS+ tumors were more densely infiltrated by CD8+ T-cells and by CD20+ B-cells and plasma B-cells (CD138+) (table 1). Tumor immunotypes, which define patterns of intratumoral lymphocyte invasion with relation to the intratumoral vasculature14 were also compared between TLS+ and TLSneg legions. A trend was noted towards more Immunotype A (‘cold’) lesions in TLSneg tumors relative to TLS+ lesions, although this did not reach the threshold for statistical significance (44% vs 20%, p=0.13). To determine whether TLS maturation may be associated with intratumoral T-cell infiltration, fractions of eTLS, pTLS, and sTLS were compared with intratumoral CD4 and CD8 T-cell densities. No significant correlations were noted for CD4 T-cell densities with eTLS, pTLS or sTLS fractions (p=0.93, 0.43, 0.20, respectively). With respect to CD8 T-cells, no correlations were noted with eTLS or pTLS fractions (p=0.48, 0.21, respectively), however, a significant correlation was detected between sTLS fraction and density of intratumoral CD8 T-cells (p=0.04, Spearman’s r=0.38).
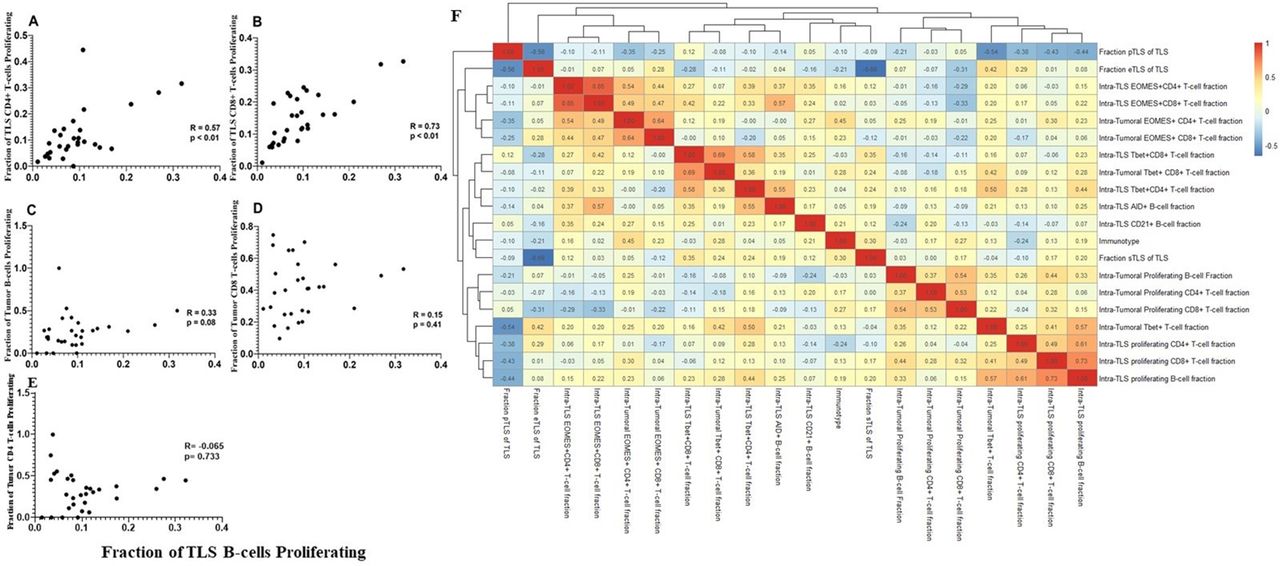
We then assessed whether proliferation of T and B cells correlated in TLS and tumor compartments. Significant correlations were noted between intra-TLS B-cell proliferation and intra-TLS T-cell proliferation rates (p<0.01) (figure 3A,B). On the other hand, intra-TLS B-cell proliferation and intratumoral B-cell or CD8+ T-cell proliferation were not significantly correlated (figure 3C–E). A heatmap was also generated comparing intratumoral lymphocyte activity markers with intra-TLS activity markers; again, no correlations were evident between activation of tumorous lymphocytes and intra-TLS lymphocytes (figure 3F). These data suggest that lymphocyte activity within the intratumoral and intra-TLS niches is independently regulated.

T cell proliferation at TLS sites is correlated with B cell proliferation. Correlation graphs comparing the relationship between the fraction of proliferating B cells at the TLS site, to the fraction of proliferating CD4+ (A) or CD8 +T cells (C) at the TLS site, and to the fraction of proliferating intratumoral B (B), CD8+ cell (D) or CD4+ (E) infiltrates. R, p values calculated by Spearman rank correlation test. (F) Non-hierarchical clustering of correlations between fraction of lymphocytes expressing a given activity marker in TLS and tumor, as well as fractions of TLS in each maturation state (eTLS, pTLS, sTLS). eTLS, early tertiary lymphoid structure; pTLS, primary follicle-like TLS; sTLS, secondary follicle-like TLS.
Features of TLS correlate with OS
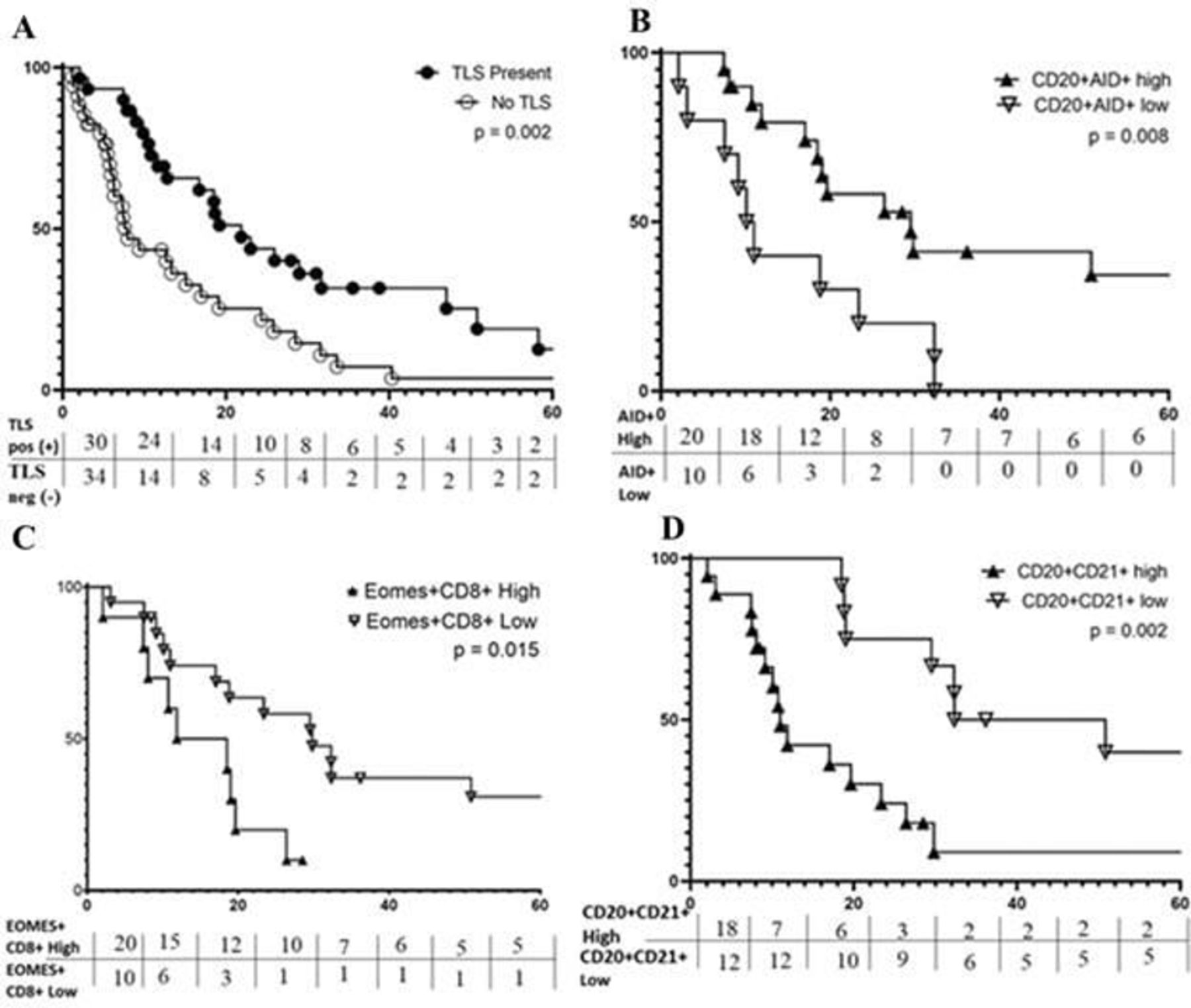
We hypothesized that OS, would be longer for patients with TLS+ CMM. In univariate analysis, TLS+ patients had significantly prolonged OS (Median OS 21.9 vs 8.0 months, p=0.002, figure 4A). This association was validated in a multivariate Cox model incorporating disease stage, patient age, sex and intratumoral CD8+ T-cell density; presence of TLS remained significantly associated with improved OS (HR=0.51, p=0.04) (table 2).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Features of TLS are Associated with OS. Kaplan-Meier curves for the indicated immune markers are shown in A–D. (A) Prolonged OS is associated with TLSpos metastases (median 21.9 months) vs TLSneg (median 8.0 months); (B) prolonged OS is associated with a high fraction of CD20+ B-cells expressing aid, median 29.5 vs 10.5 months; (C) prolonged os is associated with low fraction of CD8+ T-cells expressing EOMES, median 29.8 vs 15.2 months; (D) prolonged OS correlates with low fraction of CD20+ B-cells coexpressing CD21, median OS=29.5 vs 10.5 months. OS, overall survival; TLS, tertiary lymphoid structure.
Multivariate Cox proportional hazards model of all patients
We next tested the hypothesis that TLS maturation or markers of a pro-inflammatory immunologic milieu within the TLS would be associated with OS among patients with TLS+ CMM. TLS maturation fraction was not significantly correlated with OS (p=0.30, 0.33, 0.73 for eTLS, pTLS and sTLS, respectively). High fractions of proliferating lymphocytes, B-cells undergoing somatic hypermutation, and high proportions of CD4+ or CD8+ T-cells differentiating towards a Th1 or Tc1 phenotypes, were used as markers of a pro-inflammatory immunologic milieu within the TLS. Among TLS+ tumors, median OS did not differ based on fractions of proliferating lymphocytes, or fraction of intra-TLS CD4+ or CD8+ T-cells differentiating toward a Th1/Tc1 phenotype (Tbet+) (online supplemental table 3). However, as hypothesized, a high fraction of B-cells expressing AID+ was correlated with significantly improved OS (figure 4B, online supplemental figure 9). Further, AID+ fraction was independently correlated with improved OS in a multivariate Cox proportional Hazards model controlling for surgical-intent at metastasectomy and stratified by sex (table 3A).
Multivariate Cox proportional hazard models of TLS+ patients
To identify TLS which could inhibit immunologic tumor control, we assessed the intra-TLS density of (Tregs, CD4+FoxP3+), the fraction of T-cells expressing the exhaustion marker, EOMES,19 31 32 and the fraction of B-cells expressing CD21+CD20+. Intra-TLS density of Tregs was not associated with significant changes in OS. High fractions of exhausted CD8+ T-cells (representative mIFH images shown in online supplemental figure 10), were significantly correlated with worse OS (15.2 vs 29.8 months, p=0.02, figure 4C); however, this association between fraction of exhausted CD8+ T-cells and OS at the univariate level was not significant in a multivariate model incorporating sex and surgical-intent at metastasectomy (online supplemental table 4) (p=0.38). On the other hand, high fraction of CD21+ CD20+ B-cells was strongly correlated with worse OS in both univariate analyses (11.0 vs 41.6 months, p=0.002) (figure 4D), and multivariate Cox model analyses controlling for patient sex and surgical intent (table 3B).
Discussion
In this sample of CMM from 64 checkpoint-blockade naïve patients with stage IIIB–IV melanoma, 47% had tumor-associated TLS. This prevalence rate is concordant with a prior report in which TLS were identified in 14 out of 29 (48%) CMM.18 Prior studies of TLS in melanoma have predominantly studied LN metastases4 5; however, the present study excluded LN metastases in the interest of a more homogeneous sample population that avoids potential biases in studying TLS within pre-existing secondary lymphoid structures. Although a prior report did not identify mature TLS in CMM,5 we found that 21% of all TLS were mature (sTLS). TLSs were associated with higher densities of intratumoral CD8+ T-cells, B-cells and plasma B-cells, and while there were strong correlations between T and B cell proliferation within TLS, intra-TLS B cell proliferation did not correlate with intratumoral B cells or CD8+ T cells proliferation. Taken together, these data suggest that overlap may exist between the factors that promote TLS formation and those that drive intratumoral lymphocyte invasion, as TLSpos lesions were more densely infiltrated by lymphocytes at the tumor center. Nonetheless, lymphocyte activity within the TLS was not correlated with lymphocyte function within tumor parenchyma, implying that intratumoral lymphocyte activity is regulated by factors distinct from those which regulate intra-TLS lymphocyte activity. With respect to clinical outcomes, TLS+ CMM were independently associated with improved OS in a multivariate model incorporating key clinical variables and intratumoral CD8+ T-cell infiltration (HR=0.51, p=0.04). In a subgroup analysis of only TLS+ lesions, higher fractions of intra-TLS B-cells undergoing somatic hypermutation and decreased fractions of CD21+CD20+ B-cells were associated with improved OS in separate multivariable hazard models, strongly suggesting that TLS may exert either immunogenic or immunoregulatory effects on tumor control, which are driven by intra-TLS B-cell phenotype.
TLSs are defined as lymphoid aggregates that are segregated into B- and T-cell compartments and are associated with vascular endothelial cells expressing PNAd, a ligand that is also expressed on HEV in secondary lymphoid organs.1 TLS neogenesis has been linked to secretion of pro-inflammatory cytokines, notably CXCL13. CXCL13 is a proinflammatory chemokine which recruits B cells to peripheral tissues and is upregulated in TLS-associated colorectal cancers and expressed by perivascular stromal cells in TLS-associated lung squamous cell carcinoma.2 8 33 In separate studies, our group is investigating the mechanistic role of fibroblasts in TLS generation both in murine models and in pilot studies of CXCL13 expression by fibroblasts in human TLS. These pilot data suggest that CXCL13 positive cells are more abundant in TLS than the tumor parenchyma.34
As described previously, TLS may be further classified as eTLS, consisting of B-cell and T-cell aggregates, pTLS, which are characterized by networks of FDC, or sTLS, which demonstration germinal center formation, and mimic the architecture of a germinal center within a true secondary lymphoid follicle.4 7 8 10 Previously, TLS maturation was correlated with reduced tumor recurrence in colorectal cancer and with improved survival in chemotherapy-naïve lung cancer patients.7 8 In melanoma, TLS with germinal centers (sTLS) were previously identified almost exclusively in TLS associated with LN metastases, which is likely reflective of the fact that earlier works were performed using samples from heterogeneous tissue types and included only a very small number of CMM, limiting TLS characterization in these lesions.4 5 Thus, these data represent the largest characterization of melanoma-associated TLS maturation outside the LN. Most TLS were eTLS; however, a significant fraction (21%) of TLS in these CMM were sTLS.
Patients with TLS+ metastases had longer OS in a multivariate analysis controlling for clinical variables and intratumoral CD8+ T-cell density. These patients were all treated prior to the checkpoint blockade era; thus, patient outcomes reflect spontaneous antitumor immune responses. This extends an earlier report wherein TLS+ lesions were associated with improved OS in a multivariate analysis only when tumors were also densely infiltrated by intratumoral CD8+ T-cells.5 As intratumoral CD8+ T-cell density is a well-recognized marker of improved prognosis in melanoma and other cancers,28 35–38 it would be valid to question whether differences in survival associated with TLS+ tumors that were also densely infiltrated by CD8+ T-cells can be attributed to the TLS themselves or are simply a reflection of intratumoral CD8+ T-cell density.
Importantly, our multivariate models controlled for CD8+ T-cell tumor infiltration and for surgical intent, which was not incorporated into the model used in earlier reports. In patients with metastatic disease, surgical intent can be either curative, in which all known metastatic lesions are removed at time of surgery, or palliative, in which known metastatic disease is left unresected during metastasectomy. It has been well-documented that complete resection, reflective of curative surgical intent, correlates with patient outcomes after metastasectomy39 40; thus, our finding that TLS+ lesions tended to be more commonly resected with curative intent (70% vs 50%, p=0.17) suggests that multivariate models assessing associations between TLS and patients outcomes should control for surgical intent in patients with metastatic disease. After controlling for surgical intent, patient age, patient sex and disease stage and intratumoral CD8+ T-cell infiltration, we confirmed that the presence of TLS was independently associated with improved OS.
It has been suggested that TLS may improve outcomes by enhancing the activity of intratumoral CD8+ T-cells.5 In the present study, TLS+ CMM were more densely infiltrated by intratumoral CD8+ T-cells, B-cells and plasma B-cells, which have been previously associated with improved outcomes in melanoma.17 This may indicate that TLS support antigen-driven proliferation of the T- and B-cells that infiltrate the tumors associated with them. Intriguingly, the density of intratumoral CD8+ T-cells correlated with increasing fraction of sTLS but not with fraction of eTLS or pTLS, suggesting that TLS maturation may be correlated with cytotoxic T-cell infiltration into the tumor center. However, it is notable that proliferation rates of intra-TLS lymphocytes were not correlated with proliferation rates of intratumoral lymphocytes, suggesting that factors driving proliferation of T-cells and B-cells within TLS differ from those infiltrating among tumor cells.
We hypothesized that among TLS+ metastases, markers of immune activation or regulation within the TLS would be associated with better and worse patient outcomes, respectively. Specifically, we hypothesized that increased intra-TLS lymphocyte proliferation, T-cell differentiation to a CD4+ Th1 or CD8+ Tc1 phenotype and increased somatic hypermutation by B-cells would be associated with improved OS. Contrary to these hypotheses, the fractions of B-cells and T-cells proliferating within the TLS were not associated with changes in OS, which is consistent with prior findings.5 Also, intra-TLS Th1-dominance of CD4+ T-cells populations and Tc1-dominance of CD8+ T-cell populations were not associated with associated with improved OS. The fraction of B-cells undergoing somatic hypermutation, however, was significantly associated with improved OS.
Others have demonstrated that B-cells in CMM-associated TLS express AID and undergo both antibody class switching and clonal amplification, which are the hallmarks of antibody affinity maturation.18 Affinity maturation occurs in B-cells undergoing somatic hypermutation (AID+) and is dependent on duration of antigen presentation by local antigen presenting cells in the presence of helper T-cell stimulation.41 B-cells within the tumor-adjacent TLS are colocalized with FDC and CD4+ T-cells and are likely exposed to higher concentration of tumor antigen than B-cells in tumor draining LNs. Consequently, there is reason to speculate that antibody affinity maturation within CMM associated TLS could produce more effective antitumor antibodies than antibodies produced by B-cells in tumor draining LNs. Interestingly, even in a small study, wide variation in the percent of intra-TLS B-cells expressing AID (0%–60%) has been observed.18 In the present larger study of TLS-associated CMM, we confirm that intra-TLS B-cells express markers of somatic hypermutation and that expression is highly variable across TLS. Furthermore, we expand on this prior work, demonstrating that TLS+ patients with a high proportion of intra-TLS B cells undergoing somatic hypermutation had improved OS relative to other TLS+ patients, and that this association persisted in a multivariate model controlling for patient sex and surgical intent.
We also investigated the possibility that some TLS may have immune suppressive or regulatory features, as others have also theorized that some TLS may serve as immunotolerant niches harboring malignant cells, particularly if the TLS is enriched with cell types that promote immune tolerance or exhaustion.9 12 42 We found broad variance among patients in the intra-TLS proportions of several putative immunoregulatory cell types, including Tregs, CD21+CD20+ B cells, and CD8+EOMES+ T cells, which can be associated with T-cell exhaustion.32 43 We hypothesized that high proportions of these cell types would be associated with worse patient outcomes. In contrast to our hypothesis, density of intra-TLS Treg cells was not associated with patient outcomes. High proportions of EOMES+CD8+ cells were correlated with worse OS in a univariate analysis, but this was not significant in multivariate analysis. However, CD21+CD20+ B-cell fraction was strongly correlated with worse OS and was significant in a multivariate proportional hazards model controlling for patient sex and curative-intent resection.
The significance of CD21 as a B-cell marker in cancer is poorly understood. In melanoma patients, others have noted that intratumoral B-cells exhibited lower CD21 expression than peripheral blood B-cells; however, no specific assessment was made of the function or prognostic significance of these cells.4 Intriguingly, it has recently been demonstrated that circulating CD21low B-cells are upregulated in melanoma patients after the initiation of checkpoint blockade, and that these B-cells were a more clonal population than circulating CD21+ B-cells.44 In autoimmune disease, low CD21 expression by B-cells is associated with decreased fractions of T-reg cells in patients with common variable immune deficiency (CVID).45 Decreasing fractions of B-cells expressing CD21 correlated with severity of autoimmune reactions in patients with rheumatoid arthritis, systemic lupus erythematosus, CVID and Sjogren’s syndrome.30 46 47 These B-cells have been found to home preferentially to peripheral tissues that are targeted in autoimmune conditions, such as the synovial fluid of patients with rheumatoid arthritis.3 Given the established role of CD21+ B-cells in regulating humoral autoimmune disease, and the increase in circulating CD21low B-cells after checkpoint blockade, it is tempting to speculate that CD21+ B-cells may also play a role in attenuating the antitumor immune response in TLS, and our findings would be consistent with this hypothesis.
Many questions remained unanswered regarding how TLS enhance or inhibit tumor control in CMM. TLS have been found to predict response to checkpoint blockade and were associated with improved survival in patients who received checkpoint blockade therapy.4 5 As samples from our study predated the checkpoint blockade era, it would be worthwhile to investigate whether the associations between intra-TLS B-cell phenotypes and patient survival identified herein also correlate with patient response to checkpoint blockade. Likewise, the possibility that TLS maturation may influence response to checkpoint blockade, or the activation states of these intratumoral lymphocytes, have yet to be explored.
We observed a strong association between patient outcomes and fraction of B-cells expressing CD21+ within TLS, identifying a potential subset of TLS which may contribute to immune regulation and inhibition of tumor control. Similarly, the favorable prognostic association of AID expression by B cells in TLS suggests that antibodies produced by B cells in TLS may enhance immunologic tumor control. For all of these, further work is needed to elucidate the mechanisms underlying these associations. It is tempting to speculate that novel therapies targeting these B-cell populations, in conjunction with checkpoint blockade, may improve outcomes in patients with TLS-associated advanced melanoma.
In summary, this study provides the first detailed assessment of a population of patients with non-LN metastases evaluated for TLS and their clinical associations. Practically speaking, CMMs are common in melanoma and are readily accessible for biopsy, and therefore could provide useful windows to the host-tumor relationship. TLS were identified in 47% of lesions, and were peritumoral in location, but were associated with increased intratumoral lymphocyte infiltration. However intra-TLS lymphocyte function was distinct from intratumoral lymphocyte activity. A significant fraction of the identified TLS had matured into pTLS or sTLS. The presence of TLS was associated with improved OS in a multivariate hazard models controlling for surgical intent and intratumoral CD8+ T-cell activity. Intra-TLS B-cell activity was also strongly correlated with OS in multivariate hazard models, with high fractions of B-cells undergoing somatic hypermutation or expressing CD21 associated with better and worse patient outcomes, respectively. These data suggest that each TLS may exist in a unique immunological niche which is distinct from the intratumoral niche, and influence of this niche on B-cell activity within the TLS is a strong predictor of systemic tumor control.
Data availability statement
Data are available on reasonable request. All data available from the authors on reasonable request.
Ethics statements
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors Conception and design: IM, CLS. Development and execution of methodology: IM. Acquisition of data: IM. Analysis and interpretation of data (eg, statistical analysis, computational analysis): KL, MM, NW, IM and CLS. Writing, review and/or revision of the manuscript: KL, MM, IM, CLS, VE. Administrative, technical or material support (assisting in experiments): SY
Funding Support provided by the US Public Health Services Training Grants T32HL007849 (KL) and T32CA163177 (MM), as well as the Rebecca Clary Harris Memorial Fellowship from the University of Virginia (KL). Additional support was received from the Biostatistics Shared Resource, University of Virginia Cancer Center (NW) and the Cancer Center Support Grant P30CA044579 (CLS) This work was supported in part by the University of Virginia Cancer Center Support Grant (NIH P30 CA044579: Molecular and Immunologic Translational Studies Core, Biorepository and Tissue Research Facility, Biostatistics Core).
Competing interests CLS has the following disclosures, none of which are felt to represent conflicts of interests regarding the present manuscript: Research support to the University of Virginia from Celldex (funding, drug), Glaxo-Smith Kline (funding), Merck (funding, drug), 3M (drug), Theraclion (device staff support); Funding to the University of Virginia from Polynoma for PI role on the MAVIS Clinical Trial; Funding to the University of Virginia for roles on Scientific Advisory Boards for Immatics and CureVac. Also CLS receives licensing fee payments through the UVA Licensing and Ventures Group for patents for peptides used in cancer vaccines.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.