Article Text

Abstract
Background There is intense interest in developing novel oncolytic viruses, which can be used in cancer therapies along with immune cells such as natural killer (NK) cells. We have previously developed a particle-based method for in vitro expansion of highly cytotoxic human NK cells (PM21-NK cells). Here, we have tested the hypothesis that oncolytic parainfluenza virus 5 (P/V virus) can combine with PM21-NK cells for targeted killing of lung cancer cells.
Methods PM21-NK cells were assayed for killing of P/V virus-infected A549, H1299 and Calu-1 lung cancer cells in two-dimensional (2D) and three-dimensional (3D) cultures using flow cytometry, luminescence and kinetic imaging-based methods. Blocking antibodies were used to evaluate NK cell activating receptors involved in PM21-NK cell killing of infected target cells. Media transfer experiments tested soluble factors that increase PM21-NK cell killing of both P/V virus-infected and uninfected tumor cells.
Results In 2D cultures, PM21-NK cells efficiently killed P/V virus-infected cancer cells compared with non-infected cells, through involvement of the viral glycoprotein and NK cell receptors NKp30, NKp46 and NKG2D. In 3D spheroid cultures, P/V virus infection was restricted to the outer layer of the spheroid. However, PM21-NK cells were able to more efficiently kill both the outer layer of infected cells in the spheroid and progressing further to kill the uninfected interior cells. Media transfer experiments demonstrated that P/V virus infection produced both type I and type III interferons, which decreased cell growth, which contributed to a reduction in the overall number of uninfected tumor cells in conjunction with PM21-NK cells. Across five cancer cell lines, the contribution of P/V virus infection on PM21-NK cell killing of target cells correlated with interferon induction.
Conclusion Our data support the potential of combining oncolytic parainfluenza virus with PM21-NK cell adoptive therapy against lung cancer.
- killer cells
- natural
- oncolytic virotherapy
- interferon inducers
Data availability statement
Data sharing not applicable as no datasets generated and/or analyzed for this study. Data sharing not applicable as no datasets generated and/or analyzed for this study.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Oncolytic viruses (OVs) are natural or engineered infectious agents that selectively lyse tumor cells while sparing the normal cells.1 There has been an intense interest in developing new OVs to be used as therapeutic agents against a wide range of malignancies.2 3 A number of paramyxoviruses have been developed as oncolytic vectors owing to their intrinsic cytopathic activity and ability to activate immune responses, including mumps virus, Newcastle disease virus and measles virus.4–8 Here, we demonstrate that an oncolytic parainfluenza virus 5 (PIV5) mutant virus can increase NK cell killing of human lung cancer cells through induction of antiviral cytokines.
The wild-type (WT) PIV5 is a non-segmented negative-strand RNA virus, which causes largely non-cytopathic infections, and is a poor inducer of anti-viral host cell responses.9–11 While these properties make WT PIV5 unsuitable as an oncolytic vector, our previous work has shown that introducing substitutions in the PIV5 P/V gene converts the non-cytopathic WT virus into a mutant (P/V virus), which is highly cytopathic and a potent inducer of antiviral cytokines.12 13 Amino acid substitutions in the PIV5 P/V gene render the V protein defective in blocking both type I interferon (IFN) signaling and interferon-beta (IFN-β) synthesis.11–14 In addition, the P/V gene mutant increases cancer cell killing through pathways involving production of double-stranded RNA (dsRNA), activation of caspases, alteration of DNA damage responses and protein kinase R (PKR)-mediated shutoff of protein synthesis.15–19 We have previously shown that the P/V mutant is effective at reducing prostate tumor burden and is also restricted in growth in normal primary prostate tissue culture cells.17 18 Given the natural tropism of PIVs for the respiratory tract, here we have tested the P/V vector in a lung cancer model system.
Natural killer (NK) cells are an integral part of the innate immune system and play pivotal roles in clearance of viral infections as well as tumor cells.20 21 NK cell adoptive therapy is a promising approach to cancer immunotherapy, since NK cells do not require prior sensitization to antigens to kill transformed cells.22 One limitation of NK cell therapy is the ability to obtain sufficient amounts of highly active cells, which can be used in adoptive therapies. We have developed a particle-based method for ex vivo specific expansion of human NK cells that yields highly cytotoxic NK cells.23 24 This involves generation of particles derived from an engineered K562 cell line that expresses the NK cell-stimulating ligands 41-BBL and membrane-bound IL-21.23 These PM21 particles can be used to stimulate specific in vitro expansion of NK cells from unselected PBMCs, and these NK cells can typically multiply >1000-fold and reach >90% of total cell content in ~2 weeks.24 This platform produces vastly superior NK cells, with ~10-fold to 100-fold higher cytotoxicity than NK cells generated with previous methods. We have shown that these NK cells are effective in reducing ovarian tumor load in mice models.25 PM21-NK cells are currently being tested in a clinical trial for treatment of leukemia (NCT04395092) and COVID-19 (NCT04797975).
Although NK cells can be highly cytotoxic toward tumor cells, their killing capacity can be limited due to a number of immune evasion strategies within a tumor microenvironment, including limiting infiltration of solid tumors,26 27 changes in responsiveness28 and elevated expression of inhibitory molecules such as programmed cell death protein 1 (PD-1), programmed death ligand 1 (PDL-1) and cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) on either immune cells or target cells.25 29–31 To address these limitations, there is a need to develop approaches to activate or re-direct NK cells with highly cytotoxic functions and maintain these functions in the context of tumor microenvironment.
Given that NK cells recognize both virus-infected cells and tumor cells, and the need to develop methods to enhance NK cell killing of tumors, we have tested the hypothesis that tumor cell killing will be increased by combined treatment with both the P/V vector and PM21-NK cells. Using 3D spheroid cultures of lung cancer cells, we show that P/V infection is restricted to only the outer layer of cells of the spheroid. Addition of PM21-NK cells resulted in extensive killing of the outer layer infected cells, but unexpectedly, this killing of the cancer cell population also extended to the inner neighboring non-infected cells in the spheroid. Media transfer and reconstitution experiments identified factors released from P/V-infected cells that were capable of increasing NK cell-mediated killing of non-infected cancer cells. These data support the further development of combination therapies of adoptive NK cell therapy with the use of a PIV5 oncolytic vector in cancer treatment.
Methods
Cells and viruses
Cultures of A549 (ATCC) cells were grown in Dulbecco modified Eagle medium supplemented with 10% heat-inactivated fetal bovine serum (HI FBS, Hyclone) at 37°C under humidified, 5% CO2 atmosphere. Nuc Light Red A549 cells expressing a nuclear red fluorescent protein (NLR-A549 cells) were purchased from Incucyte (Incucyte). H1299-NLR, SKOV3-NLR and Calu-1-NLR cells were generated by transduction with NLR lentivirus (Incucyte) and cultured in Roswell Park Memorial Institute (RPMI) with 10% FBS containing 1 µg/mL puromycin. WT PIV5 and the P/V mutant (P/V virus) viruses expressing green fluorescence protein (GFP) were generated and grown as described previously.13 32 Virus titers were determined by plaque assay.33 Multiplicity of infection (MOI) was calculated by counting the number of cells in the culture, multiplying by the desired MOI and dividing by the titer of a virus stock. GFP expression was assessed by flow cytometry.
NK cell preparation
PM21 particles were generated and NK cells were expanded from peripheral blood mononuclear cells (PBMCs) as described previously.23 24 Briefly, PBMCs were depleted of T-cells (EasySep CD3 positive selection kit; STEMCELL Technologies) and then cultured for up to 25 days with 100 U/mL interleukin-2 (IL-2; PeproTech) and 200 µg/mL of PM21 particles in SCGM media (CellGenix) supplemented with 10% non-HI FBS. PM21 particles were derived from CSTX-002 (K562-nmIL21-41BBL) cells, provided by Kiadis Pharma and maintained in RPMI media supplemented with 10% FBS.
Cytotoxicity and cell killing assays
Flow cytometric cytotoxicity assays were carried out as previously described.25 Briefly, mock-infected or virus-infected A549 cells were trypsinized and stained with TFL4 and plated in RPMI containing 10% non-HI FBS (NK cell media) at 30,000 cells per 50 µL in polystyrene U-bottom plates. NK cells were added at different effector:target (E:T) cell ratios in 50 µL and were co-incubated with target cells for 30–45 min in NK cell media containing 100 U/mL IL-2 at 37°C under humidified, 5% CO2 atmosphere. Cells were then washed with phosphate-buffered saline (PBS) and stained with Annexin V (BioLegend). Cytotoxicity was calculated based on the total number of viable target cells (TFL4+/Annexin V-) remaining in each well with effectors (VTC E:T) and referenced to average VTC in ‘target alone’ control wells (VTC T ctrl Cytotoxicity E:T (%) = (1−VTC E:T/AverageVTC Tctrl) × 100.25
Cytotoxity assays were also performed using CytoTox-Glo (Promega) assay. Target cells were infected as described above and at 16 hpi (hours post infection) were re-plated in NK cell media (10,000 cells in 50 µL) in white-walled 96-well plates (Corning). NK cells were added at different E:T ratios in NK cell media containing 100 U/mL IL-2. After 4 hours at 37°C, CytoTox-Glo reagent was added to cultures according to manufacturer’s guidelines. Total max luminescence of target only cells (DCLTmax) was obtained by addition of digitonin. Cytotoxicity was calculated based on dead cell luminescence (DCL) using the following formula Cytotoxicity (%) = [(DCL E:T – DCLE- DCLTmax) / DCLTmax]×100.
For the Incucyte S3 Live-Cell Analysis system (Sartorius), A549-NLR cells were plated in triplicate in 96-well plates (Corning) at 7000 cells/well, and then mock infected or infected with virus at an MOI of 5 and maintained for 16 hours. Virus was removed and cells were washed with PBS before addition of NK cells, which were added at various E:T ratios in NK cell media. Plates were maintained in the Incucyte system at 37°C under humidified, 5% CO2 atmosphere for 3–4 days, while images were captured every 1 hour using 10× objective in red, phase and green channels. Target cell growth/killing was monitored over time and was normalized to initial number of cells present at the time of NK cell addition. For this, the number of viable cells remaining in the well was quantified for each time point based on red object count (ROC) normalized to the value at time 0 (ROC/ROC t(0h)) when NK cells were initially added to co-cultures which corresponds to 16 hpi .
Receptor blocking experiments
Target A549 cells were mock infected or infected at an MOI of 5, and at 16 hpi were incubated for 1 hour with a 1:500 dilution of mouse monoclonal antibodies 1b or 4b against hemagglutinin-neuraminidase (HN) glycoprotein (a kind gift from Dr Randall, University of St Andrews).34 After co-culturing with PM21-NK cells, cytotoxicity was measured by CytoTox-Glo assays as described above.
For experiments blocking NK cell receptors, NK cells were incubated with 10 µg/mL unconjugated antibodies to NK cell receptors NKp30, NKG2D, NKp44 or NKp46 (BioLegend) for 1 hour at 37°C before incubation with NLR-A549 cells that were mock infected or infected at an MOI of 5 with the P/V mutant virus. The cytotoxicity assays were performed on IncuCyte instrument as described above.
Spheroid infections, media transfer experiments, cytokine analysis and RT-qPCR
Cells were plated at 3000 cells/well in a 96 Ultra Low Attachment (ULA) plate (Costar), centrifuged at 130g and cultured for 3 days. Spheroids were infected at an MOI of 50 and maintained in Incucyte incubator for 16 hours. Virus was removed, spheroids were washed with PBS and NK cells were added at different ratios in six replicates in NK cell media containing 200 U/mL IL2. Plates were maintained in the Incucyte for 3–4 days, while images were captured every 4 hours using 4× objective in red, phase and green channels. Target cell growth/killing of three-dimensional (3D) cultures was monitored over time and was normalized to initial number of cells present at the time of NK cell addition. The change in size of spheroids was calculated based on total red integrated intensity (TRII) at each time point normalized to the value at time 0 (TRII/TRIIt0) when NK cells were initially added to the co-cultures (denoted as t=0), which corresponded to 16 hpi. Cytotoxicity at 36 hours was calculated based on the following equation Cytotoxicityt36 (E:T) (%) = [1−(TRIIt36 (E:T)/ Ave. TRIIt36 (Tctrl))] × 100. Respective M42-treated or V42-treated target alone values TRIIt36 (Tctrl) were used to calculate the cytotoxicity for M42-treated or V42-treated conditions.
In transfer experiments, media were collected from A549-NLR cells at 16 hpi (to generate M16 and V16 samples) or 42 hpi (M42 and V42). Virus was inactivated by treatment with UV light as described previously.33 Naive spheroid A549-NLR cultures were treated with M16, V16, M42 or V42 media for the entire experiment, or alternatively, for 16 hours with universal IFN-I, IFN-γ, IFN-III (all from PBL Assay Science) before incubation with PM21-NK cells as described above. For the blocking experiments, spheroids alone were pretreated with 10 µg/mL (saturating concentrations) of Human IFN- λ Receptor 1, Clone MMHLR1 and Human IFN- αβ R2 (IFNAR2) Clone MMHAR-2 (PBL) or isotype controls for 2 hours and then treated with M42 and V42 in the presence of blocking antibodies before adding PM21-NK cells.
Levels of cytokine secretion and RT-qPCR to determine IFN gene expression were performed as previously.33
Fluorescence microscopy
Images of spheroids were captured on 10× objective lens using Keyence microscope (Keyence) over a timecourse of 3 days. The Z-stack images were captured with a 5 µm sections on red, phase and green channels.
Statistical analyses
Statistical analysis was performed using one-way and two-way analysis of variance test and by applying Tukey’s or Dunett’s post hoc test with GraphPad Prism software as detailed in figure legends.
Results
PM21-NK cells effectively lyse lung cancer cells infected with PIV5 P/V oncolytic virus through recognition of viral glycoprotein
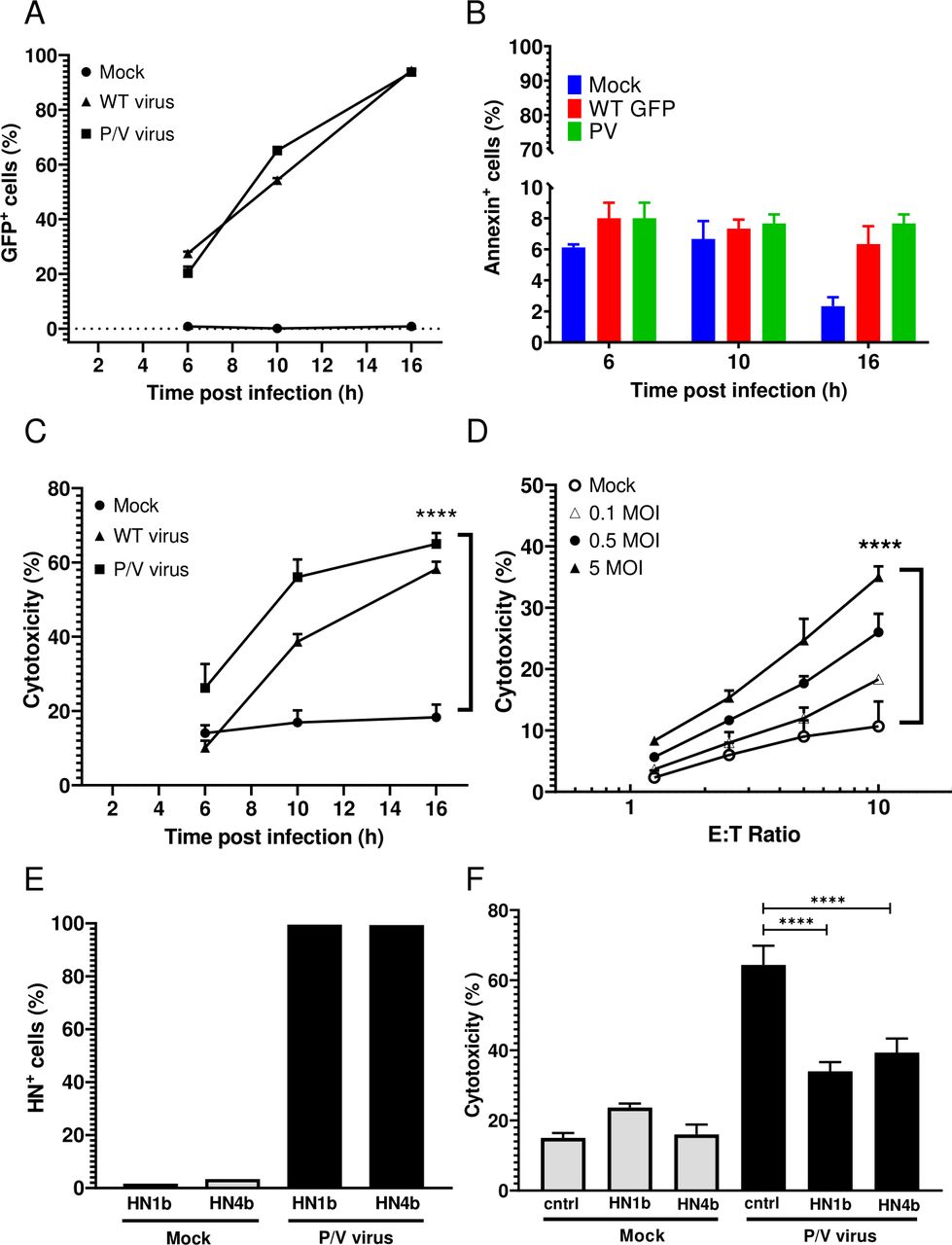
A549 human lung cancer cells were chosen as a target cell line for infection with P/V mutant that encodes GFP as described previously.13 Monolayer two-dimensional (2D) cultures of A549 cells were mock infected or infected with the WT PIV5 or the P/V virus at an MOI of 5 and GFP expression was assayed by flow cytometry at 6, 10 and 16 hpi. As shown in figure 1A, the percentage of GFP+ cells increased from 20% at 6 hpi to >90% by 16 hpi in both the WT PIV5 and P/V virus-infected cultures. Due to limited virus-induced cytotoxicity at 16 hpi (figure 1B), this was used throughout the study as the time postinfection for assays.
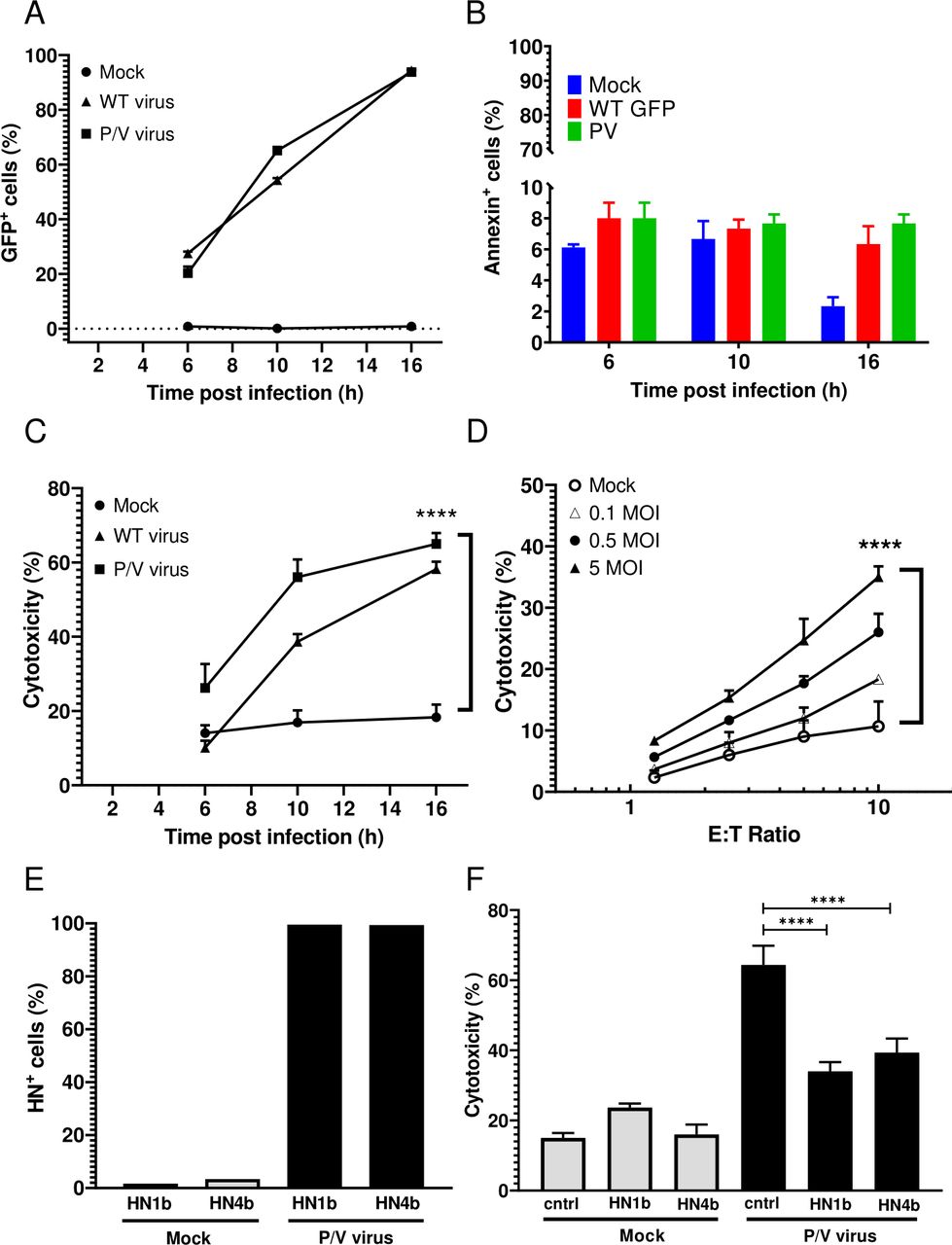
PM21-NK cells effectively lyse lung cancer cells infected with PIV5 P/V oncolytic virus. (A–C) A549 cells in 2D culture were mock infected or infected at an MOI of 5 for the indicated times with either P/V mutant or WT PIV5 virus and cells were assayed by flow cytometry for GFP expression (panel A) or annexin V staining (panel B). Alternatively, cells incubated for 45 min with PM21-NK cells at an E:T ratio of 1:1 before determining the cytotoxicity using a flow cytometric assay (panel C) as described in Methods section. Values are the mean of three replicates and per cent cytotoxicity of mock is compared with virus-infected samples at 16 hpi. Data is representative of four independent experiments conducted with four different NK cell donors with triplicates. (D) A549 cells were infected at the indicated MOIs with the P/V mutant virus for 16 hours, and then incubated for 4 hours with PM21-NK cells at the indicated E:T ratios. Cytotoxicity was assayed using CytoTox-Glo assay. (E) A549 cells were mock infected or infected at an MOI of 5 with P/V virus. At 16 hpi, cells were pretreated with anti-HN monoclonal antibodies 1b or 4b or isotype control antibody and were analyzed for HN surface expression by flow cytometry (panel E) prior to incubation with PM21-NK cells at an E:T of 10:1 (panel F). Per cent cytotoxicity was determined by CytoTox-Glo assay. In all panels, values are the mean of three samples with error bars representing SD. Graphs were analyzed using two-way ANOVA and one-way ANOVA. ****p<0.0001. ANOVA, analysis of variance; E:T, effector:target; GFP, green fluorescence protein; HN, hemagglutinin-neuraminidase; MOI, multiplicity of infection; WT, wild type.
To generate PM21-NK cells, donor PBMCs were depleted of CD3+ cells and then cultured between 12 and 21 days in the presence of IL-2 and PM21 particles to generate NK cells with >90% purity (see details in Methods section). PM21-NK cells were cytotoxic against mock-infected A549 cells with 20% of A549 cells being killed after 1 hour of co-incubation at 1:1 E:T ratio (figure 1C). Killing was increased in the case of virus-infected cells and correlated with the time-dependent increase in number of cells expressing virus-derived GFP. The extent of PM21-NK cell-mediated killing of infected cells was dependent on the E:T ratio and MOI of infection (figure 1C,D), with ~35%–45% killing at an MOI of 5 and at E:T of 10. Since WT PIV5 is not suitable as an oncolytic virus, the remaining studies focused on the P/V virus.
PIV5 expresses two major glycoproteins on the surface of infected cells: the HN protein that serves as the viral attachment protein and the fusion protein (F) that mediates entry into the target cell.35 When analyzed with either of two monoclonal anti-HN antibodies 1b or 4b,34 ~95% of cells were positive for HN surface expression at 16 hpi (figure 1E). When infected A549 target cells were pretreated with either anti-HN 1b or 4b antibody, cell lysis by PM21-NK cells was significantly reduced compared with pretreatment with a control antibody (figure 1F).
A real-time assay for PM21-NK cell killing of P/V virus-infected cancer cells identifies a role for NK cell receptors NKp30, NKG2D and NKp46
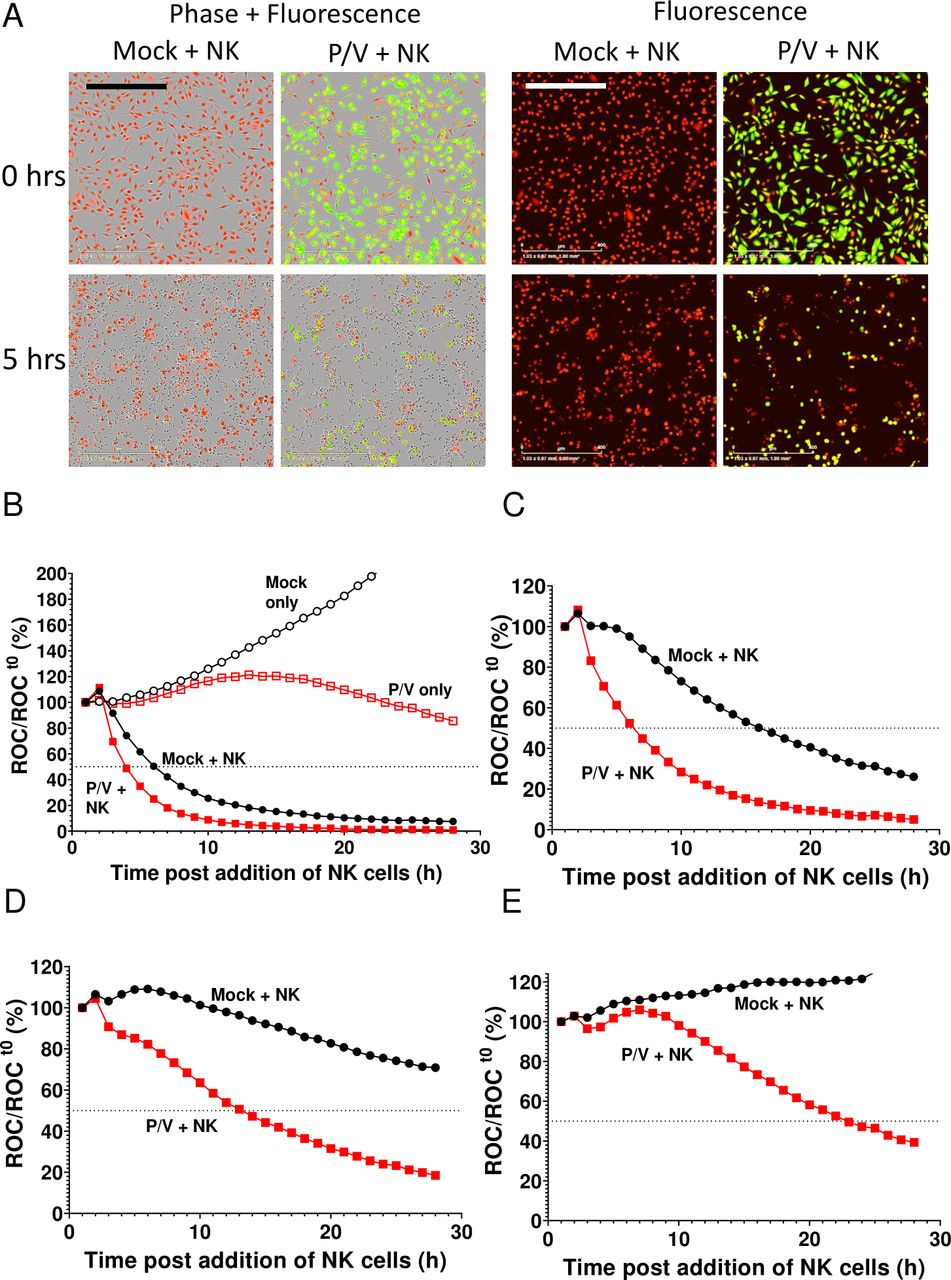
Monolayer cultures of A549 cells that express a nuclear red fluorescence protein (A549-NLR cells) were used in a kinetic assay for NK cell-mediated killing of infected cells. PM21-NK cells were incubated with A549-NLR cells that had been mock infected or infected at an MOI of 5 with P/V virus. Brightfield and fluorescence images were recorded at 1-hour intervals over time. Figure 2A shows A549-NLR target cells in red, infected cells in green (due to GFP expression) and PM21-NK cells as small dark cells visible in brightfield. Comparison of the number of red cells at time 0 to 5 hours of incubation with PM21-NK cells shows a loss of target cells for mock-infected cultures, but a much greater loss in the case of P/V virus-infected cultures. When plotted as a per cent of starting fluorescent cells when adding PM21-NK cells, real-time quantitative analysis of the images showed that mock-infected A549-NLR cells without NK cells increased in number due to cell growth (figure 2B, mock only curve), while infected A549-NLR cells without NK cells showed a plateau of growth over time (P/V only curve). By contrast, when co-cultured with PM21-NK cells at an E:T of 10, both mock-infected and P/V virus-infected cells showed a rapid loss of red fluorescence due to cell death. Increased killing of virus-infected A549-NLR cells compared with killing of mock-infected cells was even more evident at lower E:T ratios of 5 (panel C), 2.5 (panel D) and 1.25 (panel E).

Real-time assay for PM21-NK cell killing of P/V virus-infected lung cancer cells. Monolayers of A549-NLR cells were mock infected or infected with the P/V mutant virus. At 16 hpi, cells were incubated with PM21-NK cells at different E:T ratios. Red fluorescence (red object count (ROC)) in the cultures was recorded at 1-hour intervals using the IncuCyte instrument and is expressed as per cent of time 0 when PM21-NK cells were added to culture. (A) Phase and fluorescence microscopy (10× magnification) of cultures at 0 and 5 hours postaddition of NK cells at an E:T of 5. The scale bar represents 400 µm. (B–E) Time-dependent red intensity curves for cultures of A549-NLR cells plus PM21-NK cells at E:T ratios of 10 (B), 5 (C), 2.5 (D) and 1.25 (E). Each timepoint represents the mean of three samples, with error bars representing SD. Dotted line indicated 50% mark. Note the y-axis scale is larger in (panel B) to accommodate the data from mock-infected cells alone and P/V virus-infected cells as controls. E:T, effector:target; NK, natural killer.
To test the role of NK cell receptors in PM21-NK cell killing, monolayer 2D cultures of A549-NLR cells were mock infected or infected with P/V virus for 16 hours prior to addition of PM21-NK cells that had been pretreated with blocking antibodies to surface receptors NKp44, NKp30, NKG2D or NKp46 and analyzed by Incucyte assay. As shown in online supplemental figure S1, binding of antibodies to NKp30, NKG2D and NKp46 resulted in significant reduction in NK cell-mediated killing, whereas antibody to NKp44 had little effect. The combination of antibodies to NKp46, NKG2D and NKp30 had a greater effect than single antibody treatment (online supplemental figure S1E).
Supplemental material
P/V virus infection increases NK cell killing of both infected outer layers and uninfected core of 3D spheroid lung cancer cells
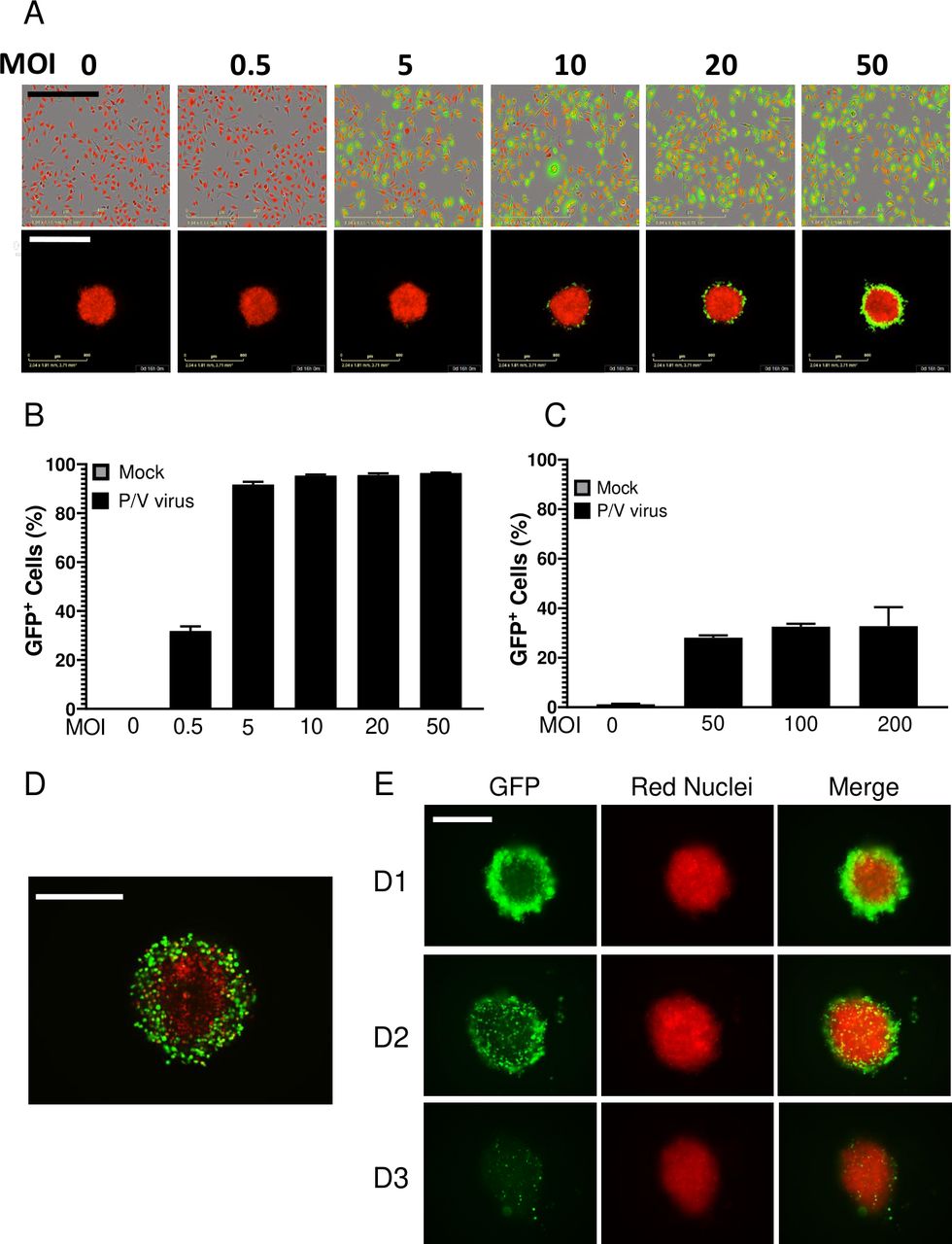
As shown in our prior work13 36 and in the images on the top row in figure 3A, A549-NLR cells in 2D monolayers were highly sensitive to P/V virus infection, with >90% GFP+ cells at MOI of 5 or higher (quantified in figure 3B). By sharp contrast, P/V virus infection of 3D spheroids resulted in only a maximum of ~30% GFP+ cells even at MOI as high as 200 (figure 3A, bottom row and C). High-resolution microscopy (figure 3D) revealed that the P/V virus had only infected the outermost shell of the 3D spheroid, with the remaining inner core cells showing no GFP expression. As shown in figure 3E by the representative micrographs at D1, D2 and D3 pi, the infected GFP-positive outer cells in the spheroids disappeared over time due to virus-induced cytopathic effects, leaving the GFP-negative A549-NLRs in the core intact. P/V virus infection of 3D spheroids is more clearly shown in the time-lapse movie provided in online supplemental figure 2.
Supplementary video

Comparison of P/V virus infection of two-dimensional (2D) monolayer versus three-dimensional (3D) spheroid cultures of A549 lung cancer cells. (A–C) A549-NLR cells grown as 2D monolayers or 3D spheroids were mock infected or infected with the P/V virus at the indicated MOIs and cultured on the IncuCyte instrument. (Panel A) shows an example 10× (2D) or 4× (3D spheroids) micrograph of A549-NLR cells (red) and infected cells (green) at 16 hpi. The percentage of GFP+ cells in the culture at 16 hpi was quantified by flow cytometry for 2D monolayers (panel B) or 3D spheroid cultures (panel C). For quantification with 3D spheroids, each sample comprised of three spheroids pooled together and trypsinized. Data are the mean of three samples (nine spheroids total) with bars representing SD. Scale bars for 2D monolayers and 3D spheroids correspond to 400 µm and 800 µm, respectively. (D) High-resolution Z-stack 10× imaging of a 3D spheroid at D1 Pi with P/V virus. (E) Spheroid 3D cultures of A549-NLR cells were infected with P/V virus at an MOI of 50. Microscopy 10× images of GFP and red fluorescence were captured at D1, D2 and D3 pi. The scale bars in D and E represent 500 µm. GFP, green fluorescence protein; MOI, multiplicity of infection.
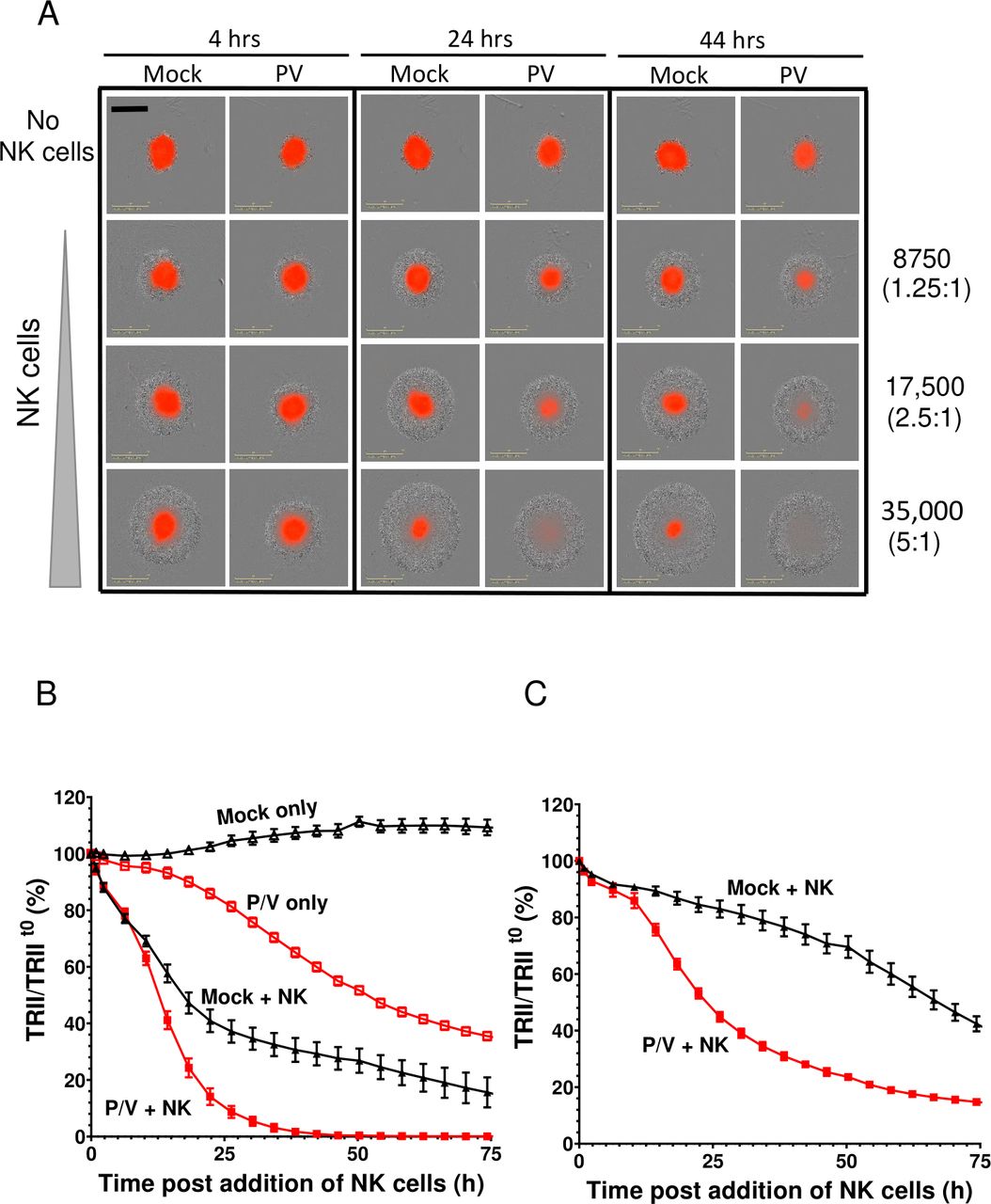
We tested the hypothesis that killing of 3D spheroid cultures of lung cancer cells could be increased by combining P/V virus infection with PM21-NK cells. A549-NLR spheroids were either mock infected or infected at MOI of 50 with P/V virus. At 16 hpi, cells were incubated with PM21-NK cells at E:T ratios of 1.25, 2.5 or 5. Levels of red fluorescence were recorded at 4-hour intervals. A representative snapshot of co-cultured cells is shown in figure 4A, with A549-NLR cells shown in red and PM21-NK cells shown as a gray halo visible on the brightfield image. PM21 cells were found to be very effective at killing P/V-infected target spheroid cells, with almost all red fluorescence gone by 24 hours postaddition of NK cells at an E:T of 5 or by 44 hours with an E:T of 2.5. This contrasts with mock-infected target cells, where NK cells were less effective at killing, as evidenced by the residual red fluorescence even at E:T of 5 and 44 hours of co-culture (bottom right corner, figure 4A). These differences in NK cell killing are also evident in the timecourses of target cell counts figure 4B,C for E:T ratios of 5 and 1.25, respectively. Compared with mock-infected cultures, the rate of PM21-NK cell killing of P/V virus-infected cells was greatly accelerated at early times (figure 4B) and at lower E:T ratio (figure 4C). This is seen in the time-lapse movies in the online supplemental figures S3 and S4.
Supplementary video
Supplementary video

PM21-NK cells combine with P/V virus infection to increase killing of three-dimensional (3D) spheroid cultures of A549 lung cancer cells. Spheroid 3D cultures of A549-NLR cells were mock infected or infected with P/V virus at a multiplicity of infection of 50. At 16 hpi, PM21-NK cells were added to the cultures at E:T ratios of 1.25, 2.5 or 5 and red image fluorescence (total red integrated intensity (TRII)) was calculated from images recorded on the IncuCyte instrument at 4-hour intervals. (A) Representative spheroids pictures recorded at 4, 24 and 44 hours postaddition of NK cells at E:T ratios of 1.25, 2.5 and 5. (B and C) Time-dependent changes in red image fluorescence (TRII) expressed as per cent of time 0 when PM21-NK cells were added to culture are shown for E:T ratios of 2.5 (panel B) and 1.25 (panel C). Data are from six replicate cultures. E:T, effector:target; NK, natural killer.
Soluble factors released from P/V virus-infected 3D spheroid cultures slow target cell growth and increase NK cell killing of uninfected cancer cells
Media transfer experiments were used to test the hypothesis that cellular factors released during P/V virus infection acted on naive uninfected 3D spheroids to sensitize them to killing by PM21-NK cells. As outlined in figure 5A, media was collected from mock-infected or P/V virus-infected A549-NLR spheroids at 16 and 42 hpi and were designated as M or V for mock or virus and the time of collection (eg, M16 for mock media at 16 hpi). Samples were treated with UV light to inactivate infectious virus33 and then used to treat naive 3D cultures of uninfected A549-NLR spheroids. PM21-NK cells were added at the same time at an E:T ratio of 1.25 and red fluorescence images were recorded over time by IncuCyte instrument and quantified. Figure 5B shows a representative experiment using M42 and V42 media, but similar results were also seen with M16 and V16. As quantified in the timecourse in figure 5B, target cell cultures without added PM21-NK cells showed a time-dependent increase in fluorescence due to cell growth. Treatment of naive target cells with M42 did not alter either cell growth or the efficiency of PM21-NK cell killing (compare control and M42 samples±NK cells). Importantly however, treatment of naive uninfected 3D spheroids with the V42 media from P/V virus-infected cells had two apparent effects. First, in the absence of added PM21-NK cells, the cells treated with V42 media did not show the increase in fluorescence due to cell growth as seen with M42 treatment. Thus, as quantified in figure 5B, the V42 media appears to have a cytostatic effect on growth of naive 3D A549 culture. Second, treatment of naive cells with V42 media resulted in an increase in target cell killing when cultured with PM21-NK cells (compare M42 plus NK with V42 plus NK; figure 5B). To determine if this treatment increased NK cell killing, cytotoxicity was calculated as described in Methods section using respective M42-treated or V42-treated target alone controls to account for cytostatic effects of V42. When normalized for cytostatic effect, treatment of naive target cells with V42 media showed that the combination of conditioned media and NK cells leads to a stronger reduction in tumor cells compared with M42-treated target cells (figure 5C). Taken together, these results support the contention that media from P/V virus-infected A549 cells can alter a 3D spheroid population of cancer cells through mechanisms that include an inhibition of cell growth, which contributes to increased overall killing when incubated with PM21-NK cells.
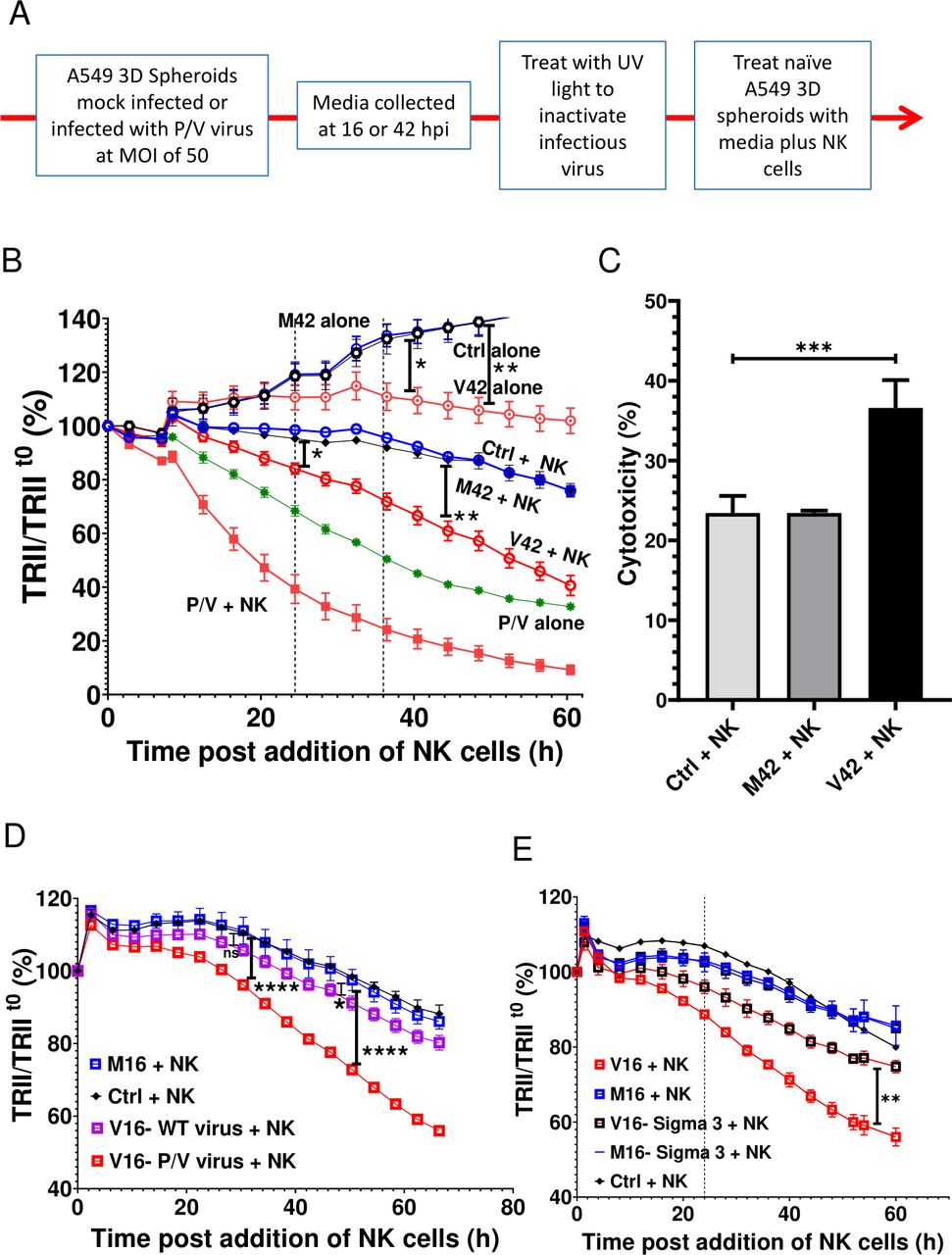
Media from P/V virus-infected cells confers an increase in killing of naive uninfected lung cancer cells by PM21-NK cells. (A) Experimental approach for transfer of media from uninfected and virus-infected spheroids. (B and C) Media was collected from mock-infected (M) and P/V virus-infected (V) cells at 42 hpi (M42, V42) as described in (panel A) and used to treat spheroid 3D cultures of naive-uninfected A549-NLR cells. At the same time, spheroids were either incubated alone or incubated with PM21-NK cells at an E:T ratio of 1.25 and red image fluorescence (TRII) was recorded on the IncuCyte instrument at 4-hour intervals (panel B). Each data point represents values from three individual spheroid culture, with bars representing SD. The data is representative of multiple independent experiments performed in triplicates with two to three different NK cell donors. Panel C shows per cent cytotoxicity at 36 hours postaddition of NK cells with error bars representing the SD. (D and E) Spheroid 3D cultures of naive-uninfected A549-NLR cells were treated with UV-inactivated media from mock-infected cells (M16) or cells (V16) infected with either WT PIV5 or P/V mutant (panel D) or P/V-infected normal A549 or Sigma3 expression A549 cells (panel E). Cells were in incubation with PM21-NK cells as described for (panel C). For all panels, Ctrl is a control of uninfected untreated A549-NLR cells without addition of PM21-NK cells. Data was analyzed using two-way ANOVA test and Tukey’s post hoc test for the indicated time points with comparison between indicated groups. For all graphs, the adjusted p values were *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. Panel C was analyzed using one-way ANOVA test. ANOVA, analysis of variance; E:T, effector:target; MOI, multiplicity of infection; NK, natural killer; TRII, total red integrated intensity.
Given that WT PIV5 is a poor inducer of antiviral cytokines14 37 compared with the P/V mutant virus,13 38 we tested the hypothesis that media from WT PIV5-infected cells would be poor at sensitizing target cells for PM21-NK cell killing. As shown in figure 5D, PM21-NK cells effectively killed uninfected 3D spheroids that had been treated with V16 media from P/V virus infection, but treatment with V16 media from WT PIV5 infection did not significantly change killing above that seen with mock-infected control media (M16).
We previously described an A549 cell line engineered to express the reovirus sigma3 dsRNA-binding protein (A549-sigma3), which can reduce P/V virus-induced cytokine release by sequestering viral dsRNA.36 As shown in figure 5E, treatment of target naive A549 spheroids with V16 media from P/V virus-infected unmodified A549 cells (solid red line) increased PM21-NK cell killing as seen in prior experiments above. By contrast, cell killing in the population in the presence of PM21-NK was greatly reduced when target uninfected cells were treated with V16 media generated from infected A549-sigma3 cells—to levels similar to that seen with treatment of target cells with M16 or M16 from A549-sigma3 cells. These data support the hypothesis that P/V virus infection activates dsRNA-sensing pathways, which in turn results in secretion of factors that can modify uninfected target cells to increase the overall PM21-NK cell-mediated killing of the population of cells.
Media from P/V virus-infected cells increases PM21-NK cell killing of non-infected lung cancer cells through type I and type III IFN receptors
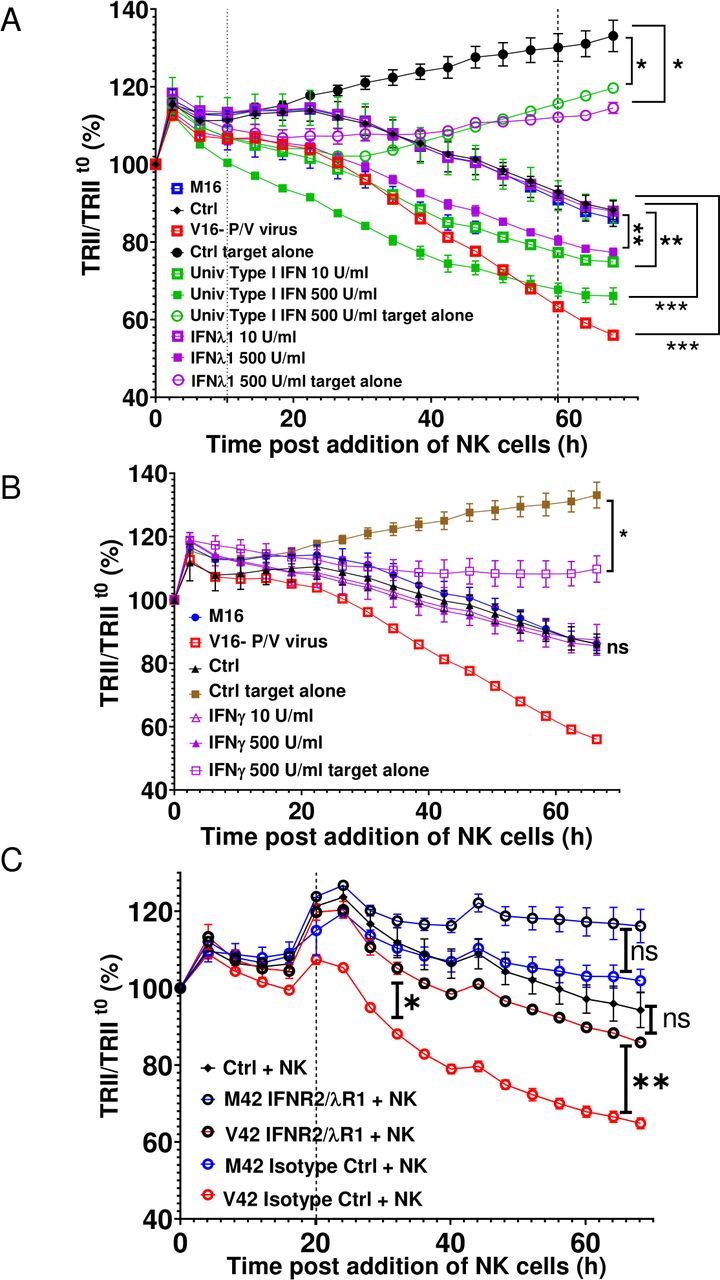
To determine if there is increased secretion of IFNs from virus-treated spheroids, the induction of IFN genes was analyzed by qRT-PCR while media from the mock or PV-treated spheroids were analyzed for presence of IFN-I and IFN-III proteins. As shown in online supplemental figure 5, P/V virus infection of 3D A549-NLR spheroids induced high level expression of IFN-β and IFN-λ at the protein and RNA levels. To test whether cytokine pretreatment of uninfected target cancer cells altered NK cell killing, naive 3D A549-NLR spheroids were treated for 16 hours with two concentrations of IFN-I or IFN-III, washed and then incubated with PM21-NK cells at an E:T ratio of 1.25. TRII was recorded on the IncuCyte instrument at 4-hour intervals. When compared with untreated or M16-treated spheroids, pretreatment of uninfected target cells with IFN-I showed a dose-dependent increase in PM21-NK cell-mediated killing (figure 6A). Similarly, pretreatment of target cells with IFN-III also increased killing, but only at the highest concentration of cytokine. Pretreatment of naive A549 spheroids with type II IFN-γ did not alter PM21-NK cell killing (figure 6B).
Supplemental material
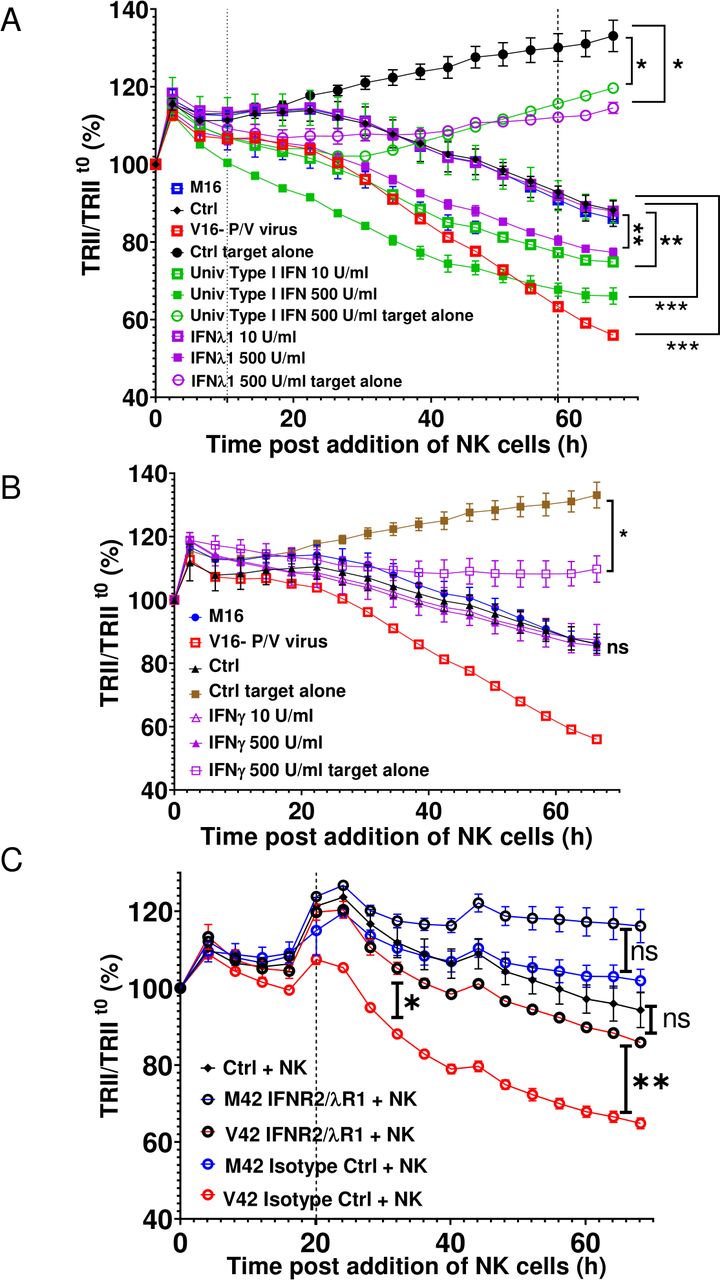
Media from P/V virus-infected cells enhance PM21-NK cell killing of lung cancer cells through type I and type III IFN receptors. (A and B) Spheroid 3D cultures of naive uninfected A549-NLR cells were left untreated (Ctrl) or treated with the indicated concentrations of cytokines, M16 or V16 for 16 hours. Cells were washed before incubation with PM21-NK cells at an E:T ratio of 1.25. Red image fluorescence (TRII) was recorded on the IncuCyte instrument at 4-hour intervals. (C) Naive uninfected A549-NLR cells were treated with the indicated anti-IFN receptor antibodies or isotype control antibodies before addition of M42 or V42 media. Samples were then incubated with PM21-NK cells at an E:T ratio of 1.25 along with antibodies. Red image fluorescence (TRII) was quantified based on images recorded on the IncuCyte instrument at 4-hour intervals. For all graphs, each data point represents values from three individual spheroid cultures, with bars representing SDs. Data was analyzed using two-way ANOVA test and applying Tukey’s post hoc test when comparing between multiple groups or Dunett’s post hoc test when comparing with a single control group. Comparisons were made at the indicated time points for the groups as shown. For all graphs, the adjust p values after applying post hoc tests were *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. ANOVA, analysis of variance; E:T, effector:target; IFN, interferon; NK, natural killer; TRII, total red integrated intensity.
To determine if IFN-I and IFN-III contained within the bone fide V42 media played a role in enhancing PM21-NK cell killing, naive 3D A549-NLR spheroids were treated with neutralizing antibodies, which block both the IFN-I and IFN-III receptors on the target cells or with isotype control antibodies. As shown in figure 6C, V42 media treatment of naive uninfected spheroids in presence of isotype control antibodies (open red symbols), followed by incubation with PM21-NK cells led to a stronger reduction in tumor cell numbers than treatment with M42 media. Most importantly however, PM21-NK cell killing of V42-treated target cells was significantly reduced when antibodies to the IFN-I and IFN-III receptors were present (open black circles). As shown in online supplemental figure 6, addition of blocking IFN antibodies did not alter the growth of the A549 cells or the kinetics of PM21-NK cell-mediated killing of target cells.
Supplemental material
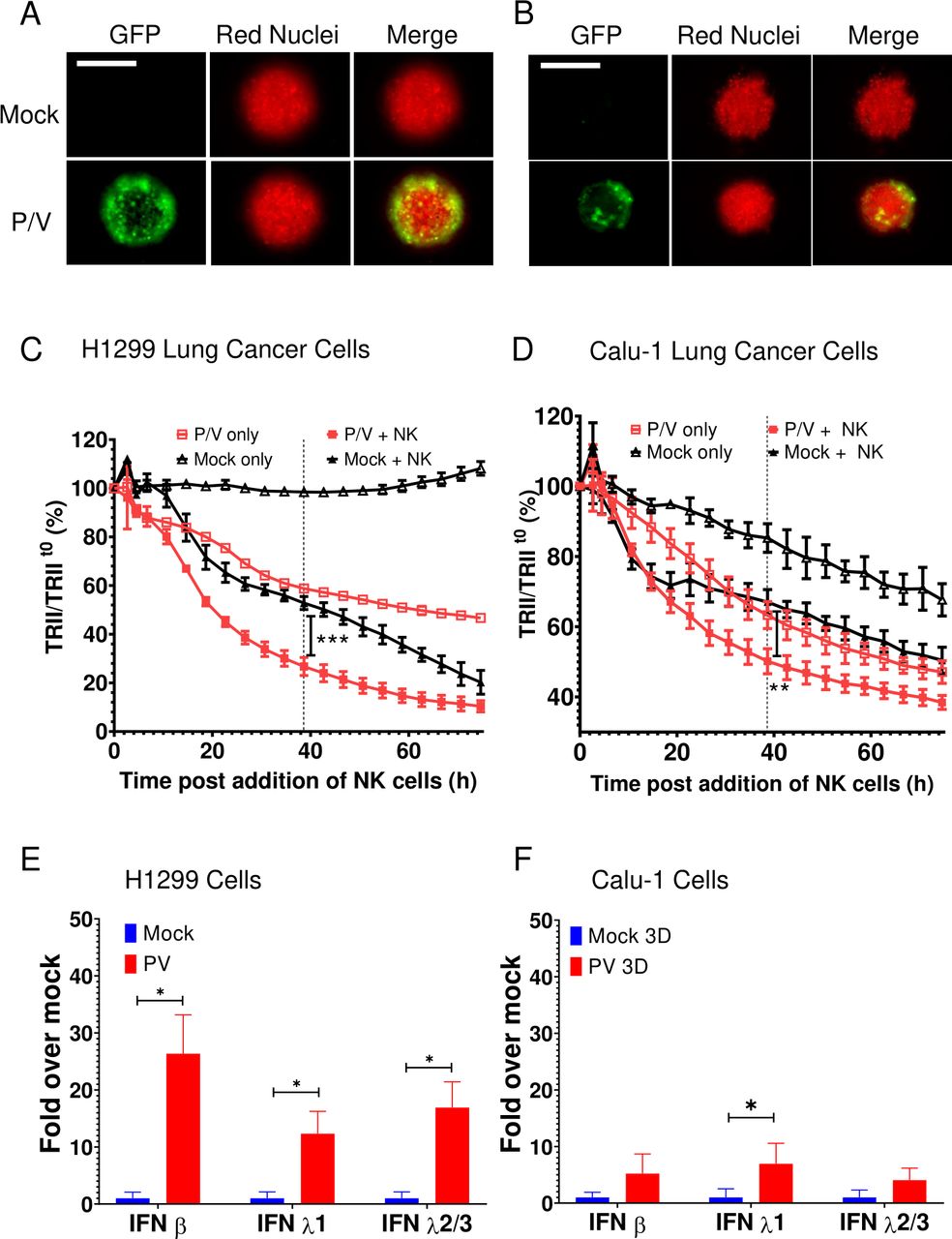
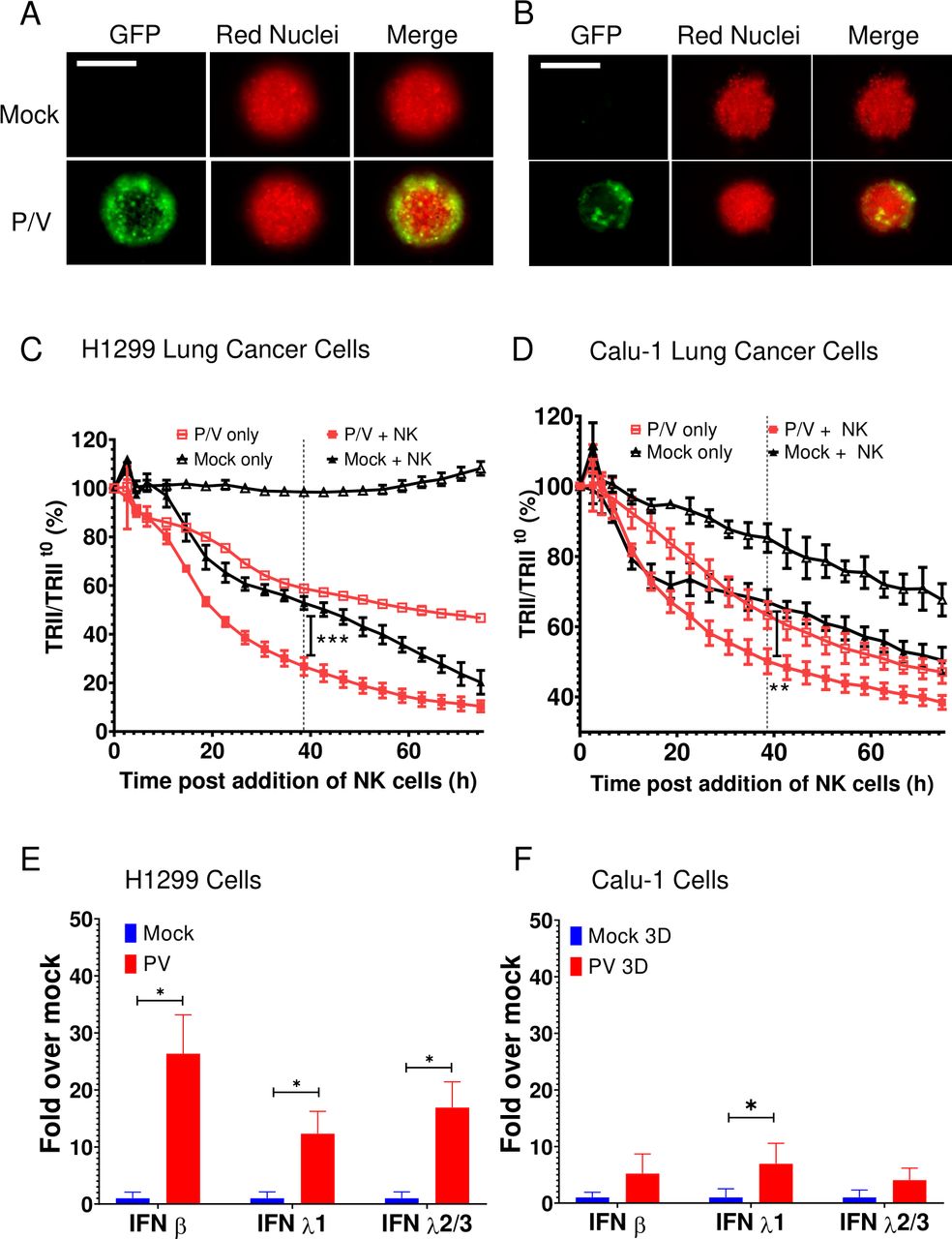
To determine if the above results also apply to other lung cancer cell lines, 3D spheroid cultures of H1299-NLR cells or Calu-1-NLR cells were infected with P/V virus at an MOI of 50 and analyzed for time-dependent killing by PM21-NK cells. Similar to A549 cells, P/V virus infection of these cells was limited to the outer shell of the spheroid (figure 7A,B) and P/V infection increased the extent of killing by PM21-NK cells (figure 7C,D). Likewise, P/V virus infection led to an increase in IFN-β and IFN-λ1 and 2/3 gene expression. Furthermore, very similar results were seen with two other non-lung cancer cell lines—SKOV3 ovarian cancer cells and A375 melanoma cells (online supplemental figure 7). Taken together, results from five tumor cell lines suggest that the ability of P/V virus infection to increase cell killing by PM21-NK cells correlated directly with levels of IFN induction by virus infection.
Supplemental material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
P/V virus infection of other lung cancer cells induces type I and type III IFN gene expression and leads to increased killing by PM21-NK cells. (A and B) Spheroid 3D cultures of H1299-NLR (A) and Calu-1-NLR (B) were mock infected or infected with P/V virus at an MOI of 50. Microscopy images at 10× were captured at D1 pi. The scale bar represents 200 μm. (C and D) At 16 hpi, PM21-NK cells were added to the H1299-NLR (panel C) and Calu-1-NLR (panel D) cultures at E:T ratios of 2.5 and red image fluorescence (TRII) was recorded on the IncuCyte instrument at 4-hour intervals. Each data point represents values from three individual spheroid cultures with bars representing the SD. (E and F) Spheroid cultures of H1299-NLR (panel E) or Calu-1-NLR (panel F) were either mock infected or infected with P/V virus at an MOI of 50. At 16 hpi, 32 spheroids were pooled and total cellular RNA was evaluated for the expression of IFN-β, IFN-λ1 and IFN-λ2/3 by RT-qPCR. Values are the mean of three biological replicates with error bars representing SD. For panels C and D, data was analyzed by two-way ANOVA test and by applying Tukey’s post hoc test when comparing the indicated data. For all graphs, the adjusted p values were *p<0.5; **p<0.01; ***p<0.001. ANOVA, analysis of variance; E:T, effector:target; IFN, interferon; MOI, multiplicity of infection; NK, natural killer; TRII, total red integrated intensity.
Discussion
NK cells are powerful innate immune cells that recognize and lyse both virus-infected cells and cancer cells,22 raising the hypothesis that oncolytic virus infection of cancer cells could be combined with adoptive NK cell immunotherapy. Oncolytic virus infection can alter cell surfaces for enhanced NK cell recognition, through providing a new foreign surface ligands such as viral glycoproteins,39 or by altering levels of cellular proteins as seen with the induction of NKG2D ligands in the case of adenovirus,40 or downregulation of MHC-I molecules in case of Myxoma virus infection.21 41 42 Here, we show that infection of lung cancer cells with an oncolytic PIV5 P/V vector results in surface expression of the viral glycoprotein HN, which is recognized by PM21-NK cells for increased cell lysis. Most importantly however, the P/V oncolytic virus has two additional properties, which were evident in 3D spheroid cultures—induction of cytokines, which have the dual effect of reducing tumor cell growth as well as increasing the susceptibility of uninfected lung cancer cells in the population to PM21-NK cell killing.
Recent data have shown that 3D cultures of lung cancer cells are more appropriate models for research on cancer therapeutic compared with conventional 2D cultures.43 It has been shown that 3D cultures of tumor cells can mimic the microenvironment found in tumors in vivo with respect to cell morphology, cell architecture, cell–cell interactions and cellular metabolism. In our studies, the efficiency of P/V infection differed dramatically when comparing 2D versus 3D cultures of lung cancer cells. While 2D cultures were very susceptible to infection with P/V virus at traditional MOIs (eg, 5), maximal infection of 3D lung cancer spheroids required a high MOI of 50 and even then, was limited to cells in the outermost layer of the 3D cell population. This selective infection of subpopulations of cells within the spheroid could reflect differences in different layers of cells with regard to nutrient availability, metabolic profiles or landscape of IFN-stimulated gene (ISG) products. In addition, there could be differential expression of the sialic acid receptor utilized for P/V entry at different layers of spheroids as shown for other viral receptors in 3D ovarian cancer spheroids.44
Our most striking results came from media transfer experiments coupled with real-time assays for NK cell-mediated killing of 3D A549 lung cancer spheroids. Our results support a working model that P/V virus infection is limited to the outer most layer of cells in the 3D structure, resulting in release of soluble factors through cellular pathways that are activated by viral dsRNA. When PM21-NK cells are present, there is rapid killing of the outermost P/V virus-infected cells, followed by continued killing of uninfected cells located within the inner core of the spheroid. Support for a role of type I and type III IFNs in increased NK cell-mediated killing of the cancer cell population comes from our findings that: (1) media from P/V virus-infected cells increased NK cell killing of naive cancer cells in the absence of virus infection, (2) there was no increase with media from cells infected with WT PIV5, which is a poor inducer of IFN or from P/V virus-infected cells expressing sigma3, which decreases IFN induction by sequestering dsRNA, (3) direct treatment of naive target cells with purified type I and type III IFN increased their overall killing by PM21-NK cells and (4) blocking type I and type III IFN receptors on target cells reduced the stimulating activity of media from P/V virus infection.
We found that factors released from P/V-infected cells have the effect of decreasing growth of naive A549 lung cancer cells. It is well established that IFNs can be cytostatic, and while the mechanisms for this may be incompletely understood,45 it is nevertheless a desirable property of any antitumor agent. Future work will be focused on the signaling pathways and epigenetic changes in 3D spheroids, which are altered by IFN-I and IFN-III to reduce cell proliferation.
NK cells secrete IFN-γ when activated by exposure to virus-infected cells or tumor cells.21 In this regard, it is noteworthy that unlike type I and type III IFNs, treatment of naive target A549 spheroids with type II IFN-γ did not increase overall killing of cancer cells by PM21-NK cells. This suggests that the ISGs that are activated by type I and type III IFN differ substantially from those induced by IFN-γ.46 We have not determined whether the landscape of ISG expression that results in sensitization of target cells by IFN-I is the same or distinct from that induced by IFN-III; that is, distinct signatures induced by IFN-I and IFN-III that both result in sensitization to NK cell killing or a common signature that is shared through IFN-I and IFN-III signaling. Future work will investigate these questions as well as which other antiviral cytokines can alter naive lung cancer cells to make them more susceptible to NK cell killing.
Prior work has shown that type I IFN can act on NK cells to enhance effector functions such as antibody-dependent cytotoxicity.47 While our results do not rule out a role for type I or type III IFN acting on PM21-NK cells to augment effector functions, our reconstitution experiments allowed us to demonstrate a role for these cytokines acting on the target cells to increase their susceptibility to killing. Whether the cytostatic effect of IFNs on the target cell is sufficient to account for increased NK cell killing of the cancer cell population or if there are additional consequences to IFN signaling is not clear at this point. Future work will focus on determining the IFN-inducted signatures on target cells and the factors that increase their recognition and activation of NK cell functions.
Our results suggest novel contributions that OVs can make in combination with NK cells toward the overall killing of a cancer cell population, through: (1) direct virus killing, (2) providing viral glycoprotein as a signal for NK cell recognition and (3) inducing secretion of cytostatic IFNs, which decrease cancer cell growth to shift the growth versus killing balance in favor of NK cell killing. There is great potential to engineer viral vectors for expression of novel immune-modulatory molecules,48 such as immunoglobulins, cytokines or stimulating co-factors, and to determine how this can be used to further harness the activity and specificity of NK cells for tumors.
Data availability statement
Data sharing not applicable as no datasets generated and/or analyzed for this study. Data sharing not applicable as no datasets generated and/or analyzed for this study.
Ethics statements
Acknowledgments
We thank members of the Parks lab for input and Kritika Kedarinath for excellent technical assistance. We are grateful to Dr Candace Fox for her generous help with making NLR cell lines.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors NV performed experimental work, data analyses and wrote the manuscript. JLO contributed to experimental procedures. AC conceived the ideas, designed the study, supervised the project, analyzed and interpreted data and revised the manuscript. GDP conceived the idea, designed the study, supervised the project, analyzed and interpreted data and wrote and revised the manuscript.
Funding This work was supported by American Lung Association grant LCD-619391.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.