Article Text

Abstract
Background While adoptive transfer of T-cells has been a major medical breakthrough for patients with B cell malignancies, the development of safe and effective T-cell-based immunotherapy for central nervous system (CNS) tumors, such as glioblastoma (GBM), still needs to overcome multiple challenges, including effective homing and persistence of T-cells. Based on previous observations that interleukin (IL)-17-producing T-cells can traffic to the CNS in autoimmune conditions, we evaluated CD8+ T-cells that produce IL-17 and interferon-γ (IFN-γ) (Tc17-1) cells in a preclinical GBM model.
Methods We differentiated Pmel-1 CD8+ T-cells into Tc17-1 cells and compared their phenotypic and functional characteristics with those of IFN-γ-producing CD8+ T (Tc1) and IL-17-producing CD8+ T (Tc17) cells. We also evaluated the therapeutic efficacy, persistence, and tumor-homing of Tc17-1 cells in comparison to Tc1 cells using a mouse GL261 glioma model.
Results In vitro, Tc17-1 cells demonstrated profiles of both Tc1 and Tc17 cells, including production of both IFN-γ and IL-17, although Tc17-1 cells demonstrated lesser degrees of antigen-specific cytotoxic activity compared with Tc1 cells. In mice-bearing intracranial GL261-Quad tumor and treated with temozolomide, Tc1 cells, but not Tc17-1, showed a significant prolongation of survival. However, when the T-cell transfer was combined with poly-ICLC and Pmel-1 peptide vaccine, both Tc1 and Tc17-1 cells exhibited significantly prolonged survival associated with upregulation of very late activation antigen−4 on Tc17-1 cells in vivo. Glioma cells that recurred following the therapy lost the susceptibility to Pmel-1-derived cytotoxic T-cells, indicating that immuno-editing was a mechanism of the acquired resistance.
Conclusions Tc17-1 cells were equally effective as Tc1 cells when combined with poly-ICLC and peptide vaccine treatment.
- brain neoplasms
- central nervous system neoplasms
- immunotherapy
- adoptive
- T-Lymphocytes
- vaccination
Data availability statement
All data relevant to the study are included in the article or uploaded as online supplemental information.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Background
Malignant gliomas, including glioblastoma (GBM), represent a highly unmet clinical need, and their prognosis remains poor.1 Adoptive T-cell transfer therapy, specifically chimeric antigen receptor (CAR) T-cell therapy, has proven effective and led to Food and Drug Administration approvals for patients with B-cell malignancies. However, the development of effective T-cell therapies for GBM remains a challenge due to multiple barriers, including restricted T-cell homing to GBM and the antigenic heterogeneity, as we reviewed recently.2 Especially, the central nervous system (CNS) has anatomical barriers (eg, blood–brain barrier) which tightly regulate the entry of immune cells.3 In the experimental autoimmune encephalomyelitis (EAE) models, interleukin (IL)-17-producing Type17 helper T-cells can migrate to the CNS through their expression of CCR6, thereby mediating the neuroinflammation.4 5 Moreover, CD8+ Type17 T-cells (Tc17 cells) accumulate in the spinal cord through IL-17 production to exacerbate EAE.6 These findings led us to hypothesize that IL-17- and interferon-γ (IFN-γ)-producing CD8+ T (Tc17-1) cells would exhibit enhanced homing to the CNS with potent antitumor activity, leading to improved clinical outcomes in adoptive T-cell therapy for GBM.
Materials and methods
Mice
Pmel-1 T-cell receptor (TCR) transgenic and C57BL/6J mice were purchased from The Jackson Laboratory.
Cell lines
The GL261 cell line was provided by Dr. Robert M. Prins (University of California, Los Angeles). GL261-Quad cells expressing human gp10025–33 (hgp10025-33), chicken OVA257–264 and OVA323–339, and mouse I-Eα52–68 were generated7 and provided by Dr. John R. Ohlfest (University of Minnesota).
Reagents
Detailed information on antibodies and relevant reagents is available in online supplemental table 1.
Supplemental material
T-cell differentiation
Pmel-1 CD8+CD44- T-cells were sorted using CD8α magnetic MicroBeads system (Miltenyi Biotec) and then purified by MoFlo (Beckman Coulter). These cells were stimulated by mitomycin-C-treated splenocytes loaded with 2 µg/mL hgp10025-33 peptide in the presence of the following cytokines and antibodies from days 0 to 3: Tc1 cells, 100 U/mL IL-2, 2 ng/mL IL-12 and 5 µg/mL anti-IL4 mAb; and Tc17 and Tc17-1 cells, 10 ng/mL TGF-β1, 10 ng/mL IL-6, 10 µg/mL anti-IFN-γ mAb and 5 µg/mL anti-IL4 mAb. After day 4, T-cells were cultured with 100 U/mL IL-2 for Tc1, 10 ng/mL IL-23 for Tc17, and 1 µg/mL IL-12 and 10 ng/mL IL-23 for Tc17-1 cell differentiation.
Flow cytometry
Intracellular cytokine staining Cytofix/Cytoperm Kit was purchased from BD. The data were analyzed using BD Accuri C6 flow cytometer and software.
RNA isolation and quantification of gene expression
Total RNA was extracted and subjected to quantitative real time-PCR analysis as described previously.8 Relative expression of mRNAs compared with control samples was calculated by the ddCt method.
CTL analysis
In vitro cytotoxicity was conducted using a 6-hour 51Cr-release assay as described previously.9 In brief, Tc1, Tc17, and Tc17-1 cells were incubated with GL261 cells loaded with or without hgp10025-33. In some experiments, Tc1 and Tc17-1 cells were sorted from cervical lymph nodes (CLNs) by MoFlo.
ELISA
The amount of IFN-γ and tumor necrosis factor (TNF)-α in the supernatant was measured by BD OptEIA ELISA sets.
Therapeutic mouse models
C57BL/6J mice were intracranially inoculated with GL261-Quad (1×105) cells as described previously.8 Tumor-bearing mice received temozolomide (TMZ; 330 µg/mouse) intraperitoneally on day 9, then intravenously (IV) infusion of Tc1 or Tc17-1 (5×106) cell on day 10 followed by intramuscular injections of polyinosinic-polycytidylic acid stabilized with poly-L-lysine carboxymethylcellulose (poly-ICLC) (2.5 mg/kg) with the peptide vaccine (100 µg/mouse) on day 11 after tumor inoculation. Mice were sacrificed when they showed any of the following signs: hunchback, seizures, hemiparesis, or weight loss of greater than 20%.
Statistical analyses
Log-rank test by GraphPad Prism software (V.8.4.3) was used to determine significant differences in the survival of mice on Kaplan-Meier plots among the groups. Mean values between the two groups were compared using Student’s t-test.
Results
Phenotypic and functional properties of Tc1, Tc17, and Tc17-1 cells in vitro
Using Pmel-1 mouse-derived CD8+CD44- T-cells, we first generated IL-17- and IFN-γ-producing CD8+ T (Tc17-1) cells and compared their characteristics with those of IFN-γ-producing CD8+ T (Tc1) and IL-17-producing CD8+ T (Tc17) cells. Tc1 and Tc17 cells predominantly produced IFN-γ or IL-17, respectively, while Tc17-1 cells represent heterogeneous cell populations, with the majority of cells producing IL-17 and approximately 10% producing both IL-17 and IFN-γ (figure 1A).Consistent with flow cytometry data, Tc17-1 cells expressed lower levels of Ifng mRNA than Tc1 cells (figure 1B). However, Tbx21 mRNA (encoding T-bet) levels were comparable between Tc1 and Tc17-1 cells (figure 1B). On the other hand, mRNA levels of Il17a and Rorc (encoding RORγt) expressed in Tc17-1 cells were at least as high as those in Tc17 cells (figure 1C). Tc17-1 cells expressed CCR6 at higher levels than Tc1 but slightly lower than Tc17, while Tc17-1 and Tc17 cells expressed lower levels of CXCR3 than Tc1 cells (figure 1D and online supplemental figure 1), suggesting that the CCR6 and CXCR3 expression profiles in Tc17-1 cells were closer to those of Tc17 cells than to Tc1 cells.

Phenotypic and functional properties of Tc1, Tc17, and Tc17-1 cells. Pmel-1-derived Tc1, Tc17, and Tc17-1 cells were prepared as described in materials and methods. (A) Representative flow cytometry data showing the frequency of IFN-γ- and IL-17-producing cells. (B, C) Relative mRNA expression levels of Ifng, Tbx21, Il17a, and Rorc were determined by real-time PCR. Gapdh was used as the internal control. (D) Mean fluorescence intensity (MFI) of CCR6 and CXCR3 expression. (E, F) Tc1, Tc17, and Tc17-1 cells were stimulated with GL261 cells in the presence or absence of hgp10025-33 peptide and evaluated for production of IFN-γ and TNF-α by ELISA (E) and cytotoxic activity by 51Cr-release assay (F). Data are presented as mean±SEM of 2–3 independent experiments performed in duplicate or triplicate. *P<0.05, **p<0.001, ***p<0.0001, ****p<0.00001; one-way ANOVA with interaction followed by Tukey’s multiple comparison test. ANOVA, analysis of variance; IFN-γ, interferon-γ; ND, not detected; TNF, tumor necrosis factor.
To investigate the functional properties of Tc1, Tc17, and Tc17-1 cells, we stimulated them with GL261 glioma cells in the absence or presence of exogenous hgp10025-33 peptide (figure 1E,F). Tc1 cells demonstrated the highest level of IFN-γ production and antigen-specific cytotoxicity compared with those by Tc17-1 and Tc17. While Tc17-1 cells exhibited low but significant antigen-specific IFN-γ production and cytotoxicity, Tc17 showed no cytotoxicity above background levels. On the other hand, Tc17-1 cells produced the highest levels of TNF-α among the three-cell types. Taken together, Tc17-1 cells showed transcriptional characters of both Tc1 and Tc17 cells, while their cytokine and chemokine receptor expression profiles, as well as cytotoxic activity, appear to be closer to those of Tc17 cells in vitro.
Tc17-1 cells did not show effective antitumor activity in glioma-bearing mice treated with TMZ
We next assessed the therapeutic efficacy of intravenously transferred Pmel-1-derived Tc17-1 in mice bearing GL261-Quad gliomas which express the Pmel-1 epitope hgp10025-33.7 Because Tc17 cells showed very low cytotoxic activity compared with Tc17-1 cells (figure 1F), we compared the therapeutic effects of Tc17-1 cells with those of Tc1 but not Tc17 cells (figure 2A). We used TMZ because TMZ is a part of the standard-of-care for GBM therapy,10 and TMZ effectively induced lymphodepletion and expansion of intravenously infused T-cells (online supplemental figure 2). Contrary to our hypothesis, Tc17-1 cells did not significantly prolong the survival of mice compared with the control mice (p=0.1879). In contrast, Tc1 cells showed a modest but significant prolongation of survival compared with TMZ treatment alone (p=0.0180, median survival time: 57 days with TMZ, 70.5 days with TMZ +Tc1 and 60.5 days with TMZ +Tc17-1; n=10 mice/group).

Tc1 cells, but not Tc17-1 cells, prolonged the survival of glioma-bearing mice in vivo. C57BL/6 mice bearing intracranial GL261-Quad glioma received temozolomide (TMZ) on day 9, followed by a single IV infusion of Tc1 or Tc17-1 cells on day 10 after tumor inoculation. (A) Kaplan-Meier survival curve (n=10 per group). Data are pooled from two independent experiments. P=0.0180; log-rank test. (B, C) Representative flow cytometry data showing the frequency of IFN-γ- and IL-17-producing cells (B) and cytotoxic activity against GL261 cells loaded with or without hgp10025-33 peptide (C) in Tc1 and Tc17-1 cells isolated at 7 days after adoptive transfer. Data are representative of two independent experiments and presented as mean±SD. *P<0.05; unpaired two-tailed Student’s t-test. IL-17, interleukin 17; IV, intravenously.
To investigate possible mechanisms underlying the lack of therapeutic efficacy with Tc17-1 cells, 7 days after adoptive transfer of T-cells we isolated the transferred T-cells from glioma-draining CLNs11 and measured cytokine production and cytotoxicity (figure 2B,C). The vast majority of both Tc1 and Tc17-1 cells produced IFN-γ, while IL-17 production in Tc17-1 cells largely diminished (figure 2B). These observations are consistent with a previous report that IL-17-producing CD8+ T-cells convert to IFN-γ-producing phenotype in vivo.12 However, despite the robust IFN-γ production, the antigen-specific cytotoxicity of Tc17-1 cells remained lower than that of Tc1 cells (figure 2C).
Tc17-1 and Tc1 cells equally inhibited glioma growth when combined with poly-ICLC and peptide vaccine
Very late activation antigen (VLA)-4 expression on T-cells mediates T-cell homing into the CNS,11 13 and we have reported that poly-ICLC enhances VLA-4 expression and glioma-homing of vaccine-reactive T-cells, thereby extending the survival of GL261 glioma-bearing mice.14 We, therefore, evaluated the addition of poly-ICLC and the hgp10025-33 peptide vaccine on adoptive transfer of Pmel-1 Tc1 or Tc17-1 cells in the GL261-Quad glioma model. While the combination of poly-ICLC and peptide vaccine did not prolong the survival, mice receiving Tc17-1 or Tc1 cells showed equally prolonged survival when they also received poly-ICLC and the peptide vaccine (median survival time: 57.5 days with TMZ, 61 days with TMZ+poly+vaccine, 92.5 days with TMZ+poly+vaccine+Tc1 and 91 days with TMZ+poly+vaccine+Tc17-1) (figure 3A). There was no significant difference in median survival time between mice receiving Tc17-1 or Tc1 cells (p=0.6751). We then evaluated the effects of poly-ICLC and the peptide vaccine on Pmel-1-derived (CD90.1+) T-cells. Poly-ICLC+vaccine increased the percentage of VLA-4+ and IFN-γ+ cells in mice that received Tc17-1 cells (figure 3B,C), although the frequency of VLA-4+ cells was lower in Tc17-1 than in Tc1 cells even after the poly-ICLC+vaccine (figure 3B). The percentage of VLA-4+ cells among Tc1 cells slightly decreased with the poly-ICLC+vaccine. In the spleen on day 3 following the T-cell transfer, Tc17-1 cells showed a trend for higher frequency among all CD8+ cells compared with Tc1 cells regardless of the additional treatment (figure 3D). The migration of transferred T-cells in brain tumor tissues was detectable at variable levels on day 25 (figure 3E) in all treatment groups but not on day 3 (data not shown).

Tc17-1 and Tc1 cells are equally efficacious when combined with TMZ, poly-ICLC, and the peptide vaccine. C57BL/6 mice bearing intracranial GL261-Quad glioma received TMZ treatment on day 9, then a single IV infusion of Tc1 or Tc17-1 cells on day 10, followed by poly-ICLC and the peptide vaccine on day 11 after tumor inoculation. (A) Kaplan-Meier survival curve (n=10 per group). Data are pooled from two independent experiments. ***p<0.0001; log-rank test. (B–D) GL261-Quad-bearing mice in each treatment group were analyzed on day 13 (n=3 per group). Frequency of VLA-4- (B) and IFN-γ-expressing cells (C) on Pmel-1-derived infused Tc1 and Tc17-1 cells in cervical lymph nodes (CLNs) and spleen, respectively. Frequency of CD90.1+ cells on CD8+ T-cells in the spleen (D). Data are representative of 2–3 independent experiments and presented as mean±SD. *P<0.05, **p<0.001, ****p<0.00001; one-way ANOVA with interaction followed by Tukey’s multiple comparison test. ns, not significant. (E) Representative flow cytometry data showing the frequency of CD8+CD90.1+ cells in BIL at around day 25 after adoptive transfer (n=2 per group). ANOVA, analysis of variance; BIL, brain-infiltrating lymphocytes; TMZ, temozolomide.
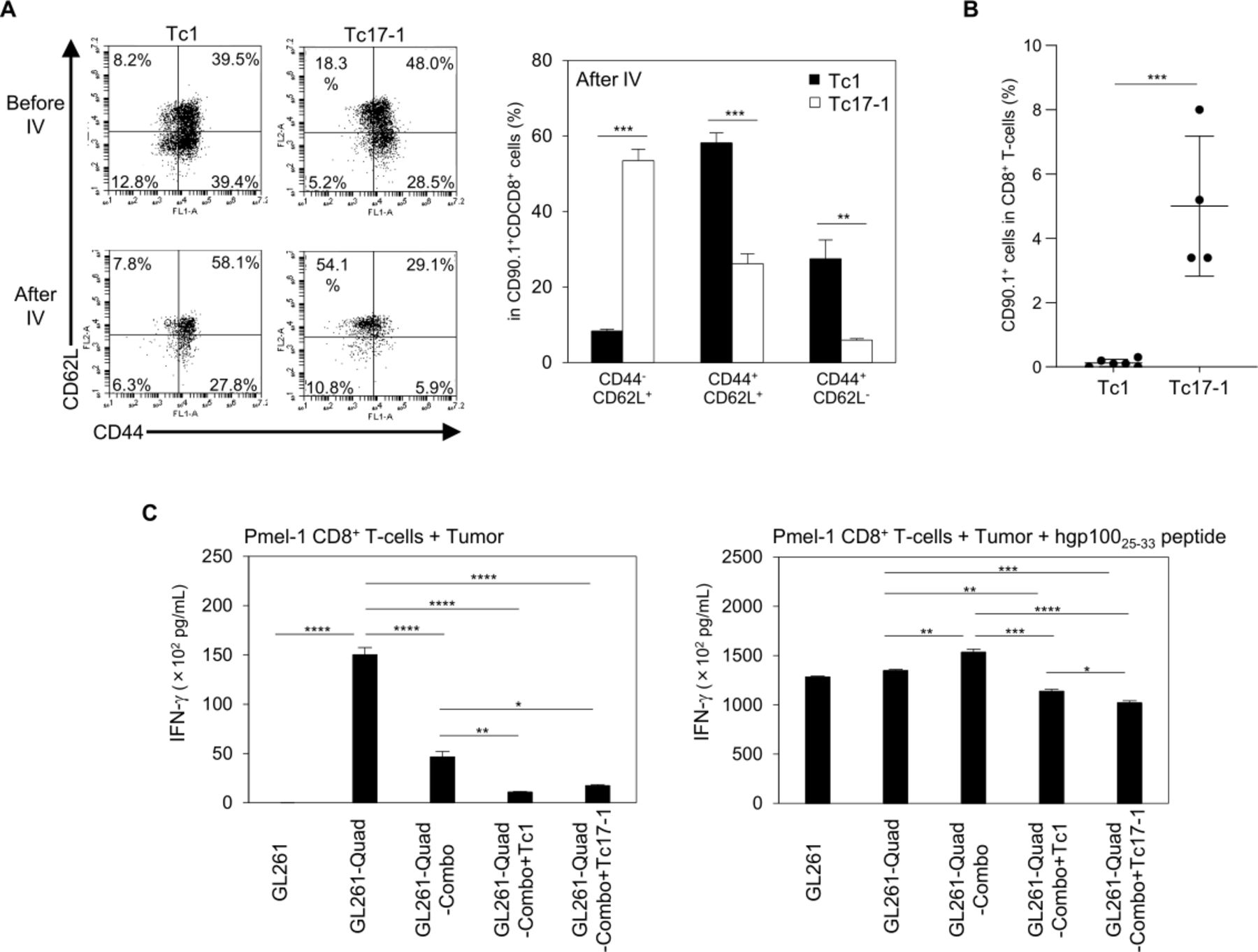
While we observed a trend toward better persistence of Tc17-1 cells over Tc1 cells in the spleen (figure 3D), this character of Tc17-1 cells did not translate to improved therapeutic effects in mice bearing intracranial glioma (figures 2A and 3A). To address the underlying mechanisms of these observations, we further evaluated the phenotype and pharmacodynamics of adoptively transferred Tc17-1 and Tc1 cells. As shown in figure 4A, before infusion, the majority of both Tc1 and Tc17-1 cells were CD44+ including CD62L+ central memory and CD62L− effector memory subsets. However, on day 7 following intravenously infusion, a majority of the Tc17-1 cells in CLNs acquired a CD44-CD62L+ stem-like phenotype while Tc1 cells showed similar profiles as observed before the intravenously infusion (figure 4A). Furthermore, on day 50 following the intravenously transfer (figure 4B), more Tc17-1 cells were present in peripheral blood compared with Tc1 cells. In vitro, Tc17-1 cells showed higher levels of Rorc (figure 1C), Gata3, and Bcl2 (online supplemental figure 3) but lower CXCR3 (figure 1D) when compared with Tc1 cells. These are consistent with a stem-like phenotype as reported recently.15–17

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Tc17-1 cells acquire a stem-like phenotype and show long-term persistence after in vivo transfer. (A, B) GL261-Quad-bearing mice received a single IV infusion of Tc1 or Tc17-1 cells on the day after TMZ treatment, and their splenocytes were evaluated on day 7 (A) and day 50 (B) after T-cell transfer. Representative flow cytometry data of Tc1 and Tc17-1 cells before and after IV transfer, and frequency of CD44-CD62L+, CD44+CD62L+ and CD44+CD62L- cells in CD90.1+CD8+ T-cells (A) and CD90.1+ cells in CD8+ T-cells (B). Data are representative of 2 independent experiments and presented as mean±SD (n=4–5 per group). **P<0.001, ***p<0.0001; unpaired two-tailed Student’s t-test. (C) GL261-Quad cells were isolated from mice treated with Combo, Combo+Tc1, or Combo+Tc17-1 cells and cocultured with Pmel-1-derived CD8+ T-cells. IFN-γ production in the supernatant was determined by ELISA. GL261 and GL261-Quad cells were used as negative and positive controls, respectively. Data are presented as mean±SEM of 2 independent experiments performed in triplicate. *P<0.05, **p<0.001, ***p<0.0001, ****p<0.00001; one-way ANOVA with interaction followed by Tukey’s multiple comparison test. ANOVA, analysis of variance; IFN-γ, interferon-γ; IV, intravenously; TMZ, temozolomide.
Finally, to investigate mechanisms underlying the recurrence of the GL261-Quad tumors following the adoptive transfer therapy, we established cell lines from each of the recurrent GL261-Quad tumors in the following treatment arms: TMZ, poly-ICLC, and hgp10025-33 peptide vaccine (referred as Combo); Combo and Tc1 cells; Combo and Tc17-1 cells. We used in vitro-cultured GL261 and GL261-Quad cells as negative and positive controls, respectively. Pmel-1 CD8+ T-cells produced IFN-γ in response to GL261-Quad-Combo, but significantly lower levels to GL261-Quad-Combo+Tc1 or GL261-Quad-Combo+Tc17-1 cells (figure 4C; left). The IFN-γ production was restored by exogenous hgp100 peptide (figure 4C; right), indicating that both Tc1 and Tc17-1 cells induced immune-editing in GL261-Quad cells in vivo.
Discussion
In this study, in mice that had received TMZ pre-treatment, Tc17-1 cells demonstrated a superior level of persistence compared with that of Tc1 cells. On the other hand, Tc-1 cells, but not Tc-17 cells, demonstrated the antitumor effects associated with higher expression levels of the CNS-homing receptor VLA-4. When combined with poly-ICLC+vaccine treatment, however, Tc-1 and Tc17-1 cells equally improved survival in mice with glioma. These data may imply that Tc1 cells, which may be more feasible for manufacturing owing to fewer cytokines, are the choice for clinical trials. However, additional studies (eg, the use of BALB/c mice) are warranted to drive a more definitive conclusion about the relative efficacy of Tc1 versus Tc17-1 cells. Nevertheless, our data from the current study may provide some guidance for data-driven designing of clinical trials
Regarding the clinical relevance of Pmel-1-derived TCR-transgenic T-cell models, we identified the HLA-A*02.01-restricted, H3.3K27M-derived neoantigen epitope in diffuse midline glioma (DMG) and isolated an H3.3K27M-specific TCR.18 We recently reported the safety and efficacy of an H3.3K27M-peptide vaccine in patients with H3.3K27M+ DMG,19 and we are currently developing a phase I study of H3.3K27M-TCR-transduced adoptive T-cell therapy. Although the efficacy of peptide-based cancer vaccines in clinical trials has been modest to date, optimization of the effector phenotype and selection of adjuvants, as we addressed in the current study, may lead to the development of a more effective vaccine and adoptive cell therapy regimens. When we develop more potent strategies, although we did not observe any obvious toxicity in the current study, we should address close attention to possible CNS toxicities associated with peritumoral inflammation or CNS autoimmunity as we discussed recently.20
Although Tc17-1 cells expressed CCR6 at higher levels than Tc1 cells, based on our data, CCR6 may not contribute significantly to antiglioma activity. EAE models demonstrated that Th17 cells cross the choroid plexus epithelial barrier in CCR6/CCL20- and LFA-1/ICAM-1-dependent manners, while Th1 cells cross the blood–brain barrier using VLA-4.21 Additionally, patients with CNS autoimmunity present high levels of CXCL10 and increased CXCR3+ T-cells in the cerebrospinal fluid,22 and we previously reported that poly-ICLC with a peptide vaccine-induced CXCL10 and increased T-cell migration in gliomas.23 These results, together with our findings that Tc1 cells expressed high levels of VLA-4 even in the absence of poly-ICLC+vaccine, suggest that expression of VLA-4 and/or CXCR3 on effecter T-cells may improve infiltration in gliomas.
Previous reports have indicated improved in vivo persistence and antitumor responses of TCR-transgenic Th17 cells24 and ICOS-based CAR T-cells expressing Th17 profile.25 Consistent with these results, we found long-term persistence and a stem-like phenotype of Tc17-1 cells after infusion. A recent study showed that stem-like CD8+ T-cells in tumors contribute to the antitumor immune responses by differentiating into effector T-cells.26 Although the association of Th17 cell infiltration with prognosis in cancer remains controversial,27 our data suggest adoptive Tc17-1 or Tc1 cell therapy combined with poly-ICLC+vaccine may confer anti-tumor immunity against brain tumors.
We did not evaluate the effect of either poly-ICLC or peptide vaccine alone in this study because we have previously reported that the combination poly-ICLC and peptide vaccine, but not either of those as monotherapy, significantly enhanced antiglioma immunity.14 23
Our data evaluating recurrent gliomas following therapy indicated that those tumors had escaped from the Pmel-1-specific T cell responses, as observed in a variety of cancer immunotherapy trials.28 We still need to determine whether the escape was caused by the loss of Pmel-1 antigen or other antigen-presentation mechanisms. Such future studies may allow us to develop strategies to mitigate the issue, such as the use of epigenetic modulators DNA methyltransferase and histone deacetylase inhibitors to upregulate tumor antigens.29 Another way to reduce the risk of tumor escape is to use TCR- or CAR-transduced T-cells that recognize truncal/driver mutations as neoantigens or multiple antigens.30
It is critically important to understand the factors that promote the persistence of therapeutic T-cells. Tc17-1 cells expressed higher levels of Rorc and Gata3 and lower levels of CXCR3 and CD44 than Tc1 cells, which is consistent with a recent report that stem-like T-cells express high levels of RORC and GATA3 and low levels of CXCR3 and CD44.15 Furthermore, we found Tc17-1 cells express higher levels of Bcl2 than Tc1 cells. Chen et al16 demonstrated that Bcl2-expressing CD8 T cells compose a cell cluster that retains capacities to proliferate, regenerate, and produce differentiated cells. Our data showing the lack of therapeutic response by Tc17-1 cells (figure 2) suggest that the stem-like phenotype itself is not sufficient to mediate therapeutic response against solid cancer. Further investigations are warranted in multiple models and mouse strains to determine the most suitable T-cell types for immunotherapy of glioma.
In conclusion, our findings have important implications for the development of more effective strategies to treat brain tumors and indicate equal efficacy of Tc17-1 and Tc1 cells in combination with poly-ICLC and peptide vaccine.
Data availability statement
All data relevant to the study are included in the article or uploaded as online supplemental information.
Ethics statements
Ethics approval
All mice were maintained and handled in accordance with the Animal Facility at the University of Pittsburgh per an Institutional Animal Care and Use Committee-approved protocol.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
TO and AK contributed equally.
Contributors HO conceptualized and supervised this study. TO, AK, and HO designed the study. TO and AK performed the experiments and acquired and analyzed the data. TO, AK, MI, AMS, and HO interpreted data and wrote the manuscript. AMS provided the poly-ICLC. MI provided administrative and technical supports.
Funding This work was supported by Grants to HO from The National Institutes of Health (NIH)/National Institute for Neurological Disorders and Stroke (NINDS) (R35 NS105068, R21 NS083171), the National Cancer Institute (NCI R01) (CA222965) and Musella Foundation for Brain Tumor Research and Information. This project used University of Pittsburgh Cancer Institute (UPCI) shared resources (Animal Facility, In Vivo Imaging Facility and Cytometry Facility) that are supported in part by NIH P30 CA047904.
Competing interests AMS is Chairman, CEO, Scientific Director and cofounder of Oncovir.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.