Article Text

Abstract
Background Multiple types of immune cells producing IL-17 are found in the tumor microenvironment. However, their roles in tumor progression and exhaustion of CD8+ tumor-infiltrating lymphocytes (TILs) remain unclear.
Methods To determine the role of type 17 immunity in tumor, we investigated the growth of B16F10 melanoma and the exhaustion of CD8+ TILs in Il17a −/− mice, Il17a Cre R26 DTA mice, RORγt inhibitor-treated mice, or their respective control mice. Adoptive transfer of tumor-specific IL-17-producing T cells was performed in B16F10-bearing congenic mice. Anti-CD4 or anti-Ly6G antibodies were used to deplete CD4+ T cells or CD11b+Gr-1hi myeloid cells in vivo, respectively. Correlation between type 17 immunity and T cell exhaustion in human cancer was evaluated by interrogating TCGA dataset.
Results Depletion of CD4+ T cells promotes the exhaustion of CD8+ T cells with a concomitant increase in IL-17-producing CD8+ T (Tc17) cells in the tumor. Unlike IFN-γ-producing CD8+ T (Tc1) cells, tumor-infiltrating Tc17 cells exhibit CD103+KLRG1−IL-7Rαhi tissue resident memory-like phenotypes and are poorly cytolytic. Adoptive transfer of IL-17-producing tumor-specific T cells increases, while depletion of IL-17-producing cells decreases, the frequency of PD-1hiTim3+TOX+ terminally exhausted CD8+ T cells in the tumor. Blockade of IL-17 or RORγt pathway inhibits exhaustion of CD8+ T cells and also delays tumor growth in vivo. Consistent with these results, human TCGA analyses reveal a strong positive correlation between type 17 and CD8+ T cell exhaustion signature gene sets in multiple cancers.
Conclusion IL-17-producing cells promote terminal exhaustion of CD8+ T cells and tumor progression in vivo, which can be reversed by blockade of IL-17 or RORγt pathway. These findings unveil a novel role for IL-17-producing cells as tumor-promoting cells facilitating CD8+ T cell exhaustion, and propose type 17 immunity as a promising target for cancer immunotherapy.
- lymphocytes
- tumor-infiltrating
- cytotoxicity
- immunologic
- CD8-positive T-lymphocytes
- cytokines
- tumor microenvironment
Data availability statement
All data relevant to the study are included in the article or uploaded as online supplemental information.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
- lymphocytes
- tumor-infiltrating
- cytotoxicity
- immunologic
- CD8-positive T-lymphocytes
- cytokines
- tumor microenvironment
Background
The tumor microenvironment (TME) develops a strong immune suppressive network to support tumor growth by limiting the function of immune cells with anti-tumor activities. Formation of the immunosuppressive milieu of the TME is dictated by a complex interplay of cancer cells, organ-specific niche, and various immune cells with immunoregulatory activities.1 Significant advances have unveiled several immune cells, including myeloid-derived suppressor cells (MDSCs) and Foxp3+ regulatory T (Treg) cells, that contribute to the immunosuppressive cloud of the TME.2 3 Nevertheless, many aspects of the immunosuppressive compartments within tumors are not fully understood.
While multiple types of IL-17-producing cells are found in the tumor bed, both in animal models and in humans, their role in tumor progression remains controversial.2 4 A tumor-promoting function of IL-17-producing cells has been proposed based on the observations that mice deficient in Il17a, Il23a, or Rorc show delayed tumor growth in vivo 5–7 and that IL-17 promotes angiogenesis.8 Moreover, increase in IL17A expression or IL-17-producing cell frequencies in tumors has been shown to be associated with poor overall survival in multiple human cancers.9–11 By contrast, the anti-tumor effect of IL-17-producing cells has been supported by the finding that adoptive transfer of IL-17-producing T cells or treatment with RORγt agonists inhibits tumor growth in animal models.12–14 Supporting this notion, IL-17 expression is shown to correlate with a better clinical outcome in patients with various cancers.15–17 These seemingly contradictory results make it difficult to define whether type 17 immunity exerts tumor-promoting or anti-tumor roles.
CD8+ T cells found in the TME exhibit exhausted phenotypes such as expression of multiple immune checkpoint molecules including PD-1, CTLA-4, Tim-3, LAG3 and TIGIT, and are functionally impaired.18 Recent advances revealed that at least two exhaustion states exist among PD-1+ CD8+ tumor-infiltrating lymphocytes (TILs) based on the expression of TCF-1 and TOX; “progenitor exhausted” CD8+ T cells are PD-1intTCF-1+TOX−, and can be expanded and differentiated into PD-1hiTCF-1−TOX+ “terminally exhausted” CD8+ T cells in response to immune checkpoint inhibitors.19–21 T cell intrinsic pathways, such as chronic TcR stimulation and immune checkpoint receptor signaling, are known to contribute to the exhaustion of T cells.22 However, less is known regarding whether T cell extrinsic factor(s) in the TME plays a crucial role in inducing T cell exhaustion. In the present study, we aimed to delineate the role of type 17 immunity in tumor with specific emphasis on the progression of CD8+ T cell exhaustion in vivo.
Methods
Mice
C57BL/6 mice were either home-bred or were purchased from SamTako Ltd (South Korea). B6.SJL, Pmel Tg , Il17a Cre mice were purchased from Jackson Laboratory. Foxp3 DTR mice were kindly provided by Dr. Sin-Hyeog Im (POSTECH) with the permission of Dr. Alexander Rudensky (Memorial Sloan Kettering Cancer Center).23 R26 DTA mice and R26 YFP mice were previously described.24 25 Il17a Cre ×R26 YFP , Il17a Cre ×R26 DTA, and Il17a Cre ×R26 DTA/YFP mouse systems were created by breeding the relevant strains. CCSP Cre ×K-ras G12D mice have been described.7 Male or female mice of 8–12 weeks were used, and each experiment was conducted using age-matched/sex-matched groups. For DTR-mediated depletion experiments, 500 µg of diphtheria toxin (List Biological Laboratories) was intraperitoneally treated every other day after tumor injection in Foxp3 DTR mouse systems. All mice were bred and maintained in a specific pathogen-free facility at either the Seoul National University vivarium or the MD Anderson Cancer Center animal facility.
Tumor induction and antibody treatments
B16F10 and TC-1 mouse tumor cell lines were purchased from ATCC and passaged in DMEM medium containing 10% FBS, 1% penicillin–streptomycin (all from Gibco) in TC-treated Cell Culture Dishes (SPL). At 70%–80% confluency, cancer cells were washed once with PBS (GenDepot) and detached using TrypLE Express (Gibco). B16F10 cells in PBS were either intravenously (1.0–4.0×105 cells), intrasplenically (5.0×105 cells), or subcutaneously (3.0×105 cells) injected into mice. TC-1 cells were intravenously injected into mice (5.0×105 cells). In indicated experiments, mice were intraperitoneally treated with either rat IgG2b (LTF-2; BioXCell) or anti-CD4 depleting antibody (GK1.5; BioXCell) at 100–200 µg/head doses every 4 to 5 days. In addition, anti-mouse CD127 (A7R34; BioXCell), anti-mouse Ly6G (1A8; BioXCell), or anti-mouse PD-L1 (10F.9G2; BioXCell) was intraperitoneally treated at 100 µg/head every 4 days. For RORγt inhibition, ursolic acid (U6753; Sigma Aldrich) dissolved in DMSO was intraperitoneally treated at 150 mg/kg every other day. For Tc17 adoptive transfer experiment, Tc17 cells generated in vitro from naïve CD8+ T cells isolated from CD45.1+ Pmel mice (3.0×106 cells) were adoptively transferred into C57BL/6 mice 1 day before B16F10 tumor intravenous injection. Mice were sacrificed on days 14–19 after tumor cell injection.
Preparation of human tumor samples
Tumor tissue samples were obtained from patients with hepatocellular carcinoma (HCC) (Asan Medical Center, Seoul, Korea) undergoing surgical resection. Lymphocytes were isolated as previously described.26 Briefly, tumor samples were manually cut into small pieces and transferred to a gentle MACS C-Tube containing a mixture of tumor dissociation kit (Milteny Biotec). The samples were homogenized and digested into single cell suspension using the 37C_h_TDK_3 program of the gentle MACS Octo Dissociator (Milteny Biotec). The cells were then washed and cryopreserved. Overall patient profiles and characteristics are summarized in online supplemental table S1.
Supplemental material
Flow cytometry
For mouse sample flow cytometry analysis, lymphoid cells were obtained and stained with PerCP-Cy5.5 or PE-Cy7 anti-mouse CD8α (53-6.7; BioLegend), BUV395 or BUV737 anti-mouse CD8α (53-6.7; BD Biosciences), PerCP-Cy5.5 or APC anti-mouse CD4 (GK1.5; BioLegend), BV510 or PE-Cy7 anti-mouse CD4 (RM4-5; BioLegend), eFluor450 or PE-Cy7 anti-mouse CD90.2 (53-2.1; BioLegend), PerCP-Cy5.5 anti-mouse CD3ε (145-2 C11; BioLegend), BUV395 anti-mouse CD3ε (145-2 C11; BD Biosciences), Pacific Blue anti-mouse TCRβ (H57-597; BioLegend), APC anti-mouse γδTCR (GL-3; eBioscience), PE mouse CD1d tetramer (NIH Tetramer Core facility), APC anti-mouse/human KLRG-1 (2F1/KLRG1; BioLegend), APC-Cy7, FITC, or PE anti-mouse CD103 (2E7; BioLegend), Alexa Fluor 488, PerCP-Cy5.5 or Pacific Blue anti-mouse CD45.1 (A20; BioLegend), PerCP-Cy5.5, Pacific Blue, or BV510 anti-mouse CD45.2 (104; BioLegend), PE-Cy7 anti-mouse PD1 (RMP 1-30; BioLegend), PE or APC anti-mouse Tim3 (RMT3-23; eBioscience), APC or PE-Cy7 anti-mouse/human CD44 (IM7; BioLegend), Alexa Fluor 488 or PerCP-Cy5.5 anti-mouse CD62L (MEL-14; BioLegend), and PE-Cy7 or APC anti-mouse CD127 (A7R34; eBioscience). Intracellular staining was performed using either the IC/fixation buffer or the Foxp3/Transcription factor staining buffer set (ThermoFisher Scientific), and stained with APC, PE, PerCP-Cy5.5, or PE-Cy7 anti-mouse IFN-γ (XMG1.2; BioLegend), Alexa Fluor 488, APC, or PE anti-mouse IL-17A (TC11-18H10.1; BioLegend or eBioscience), Pacific Blue anti-mouse/human granzyme-B (GB11; BioLegend), PE anti-Ki-67 (SolA15; eBioscience), Alexa Fluor 647 anti-mouse/human TCF-1 (C63D9; Cell Signaling), PE anti-mouse/human TOX (REA473; Miltenyi Biotec) and PE anti-active caspase-3 (C92-605; BD Biosciences). The following antibodies were used for the staining of lymphoid cells isolated from human samples: BV421 anti-CD127 (A019D5; BioLegend), BV711 anti-IL-17A (BL168; BioLegend), BV786 anti-CD4 (SK3; BD Biosciences), FITC anti-IFN-γ (4S.B3; eBioscience), PerCP-Cy5.5 anti-CD3 (UCTH1; BD Biosciences), PE-Cy7 anti-KLRG1 (REA261; Miltenyi Biotec), APC anti-CD103 (Ber-ACT8; BioLegend), Alexa Fluor 700 anti-granzyme-B (GB11; BD Biosciences), and APC-Cy7 anti-CD8 (SK1; BD Biosciences). Human samples were stained using the LIVE/DEAD fixable red dead cell stain kit (Invitrogen) to gate out dead cells. Samples were analyzed with either a FACSVerse, FACSLSR II, LSRFortessa, or FACSAria III (BD Biosciences), and data were analyzed using FlowJo software (TreeStar).
Cell isolation and differentiation
Isolation of TILs was previously described.27 For the preparation of TILs from lung tumors, lungs were surgically removed after cardiac perfusion using sterile PBS and were teased into small pieces using a gentle MACS dissociator (Miltenyi Biotec). Samples were digested with RPMI 1640 medium containing 10% FBS (Gibco) and 1% penicillin/streptomycin (Gibco) (RF10), 0.5 mg/mL of Collagenase IV (Gibco), 2 mg/mL of Dispase (Gibco), and 30 µg/mL of DNase I (Bio Basic) for 30 min at 37°C, with gentle agitation. Then, lymphocytes were density-separated using the Lymphocyte Separate Medium (LSM; MP Bio) at 2500 rpm, 20 min at 20°C. Lymphocytes were incubated at 37°C in RF10 medium containing PMA/Ionomycin (Sigma) and Brefeldin A/monensin (ThermoFisher Scientific) for 3–4 hours prior to intracellular staining and flow cytometry analysis. In CCSP Cre ×K-ras G12D experiments, cell isolation was conducted as previously described.7
For in vitro Tc17 or Tc1 differentiation, pooled lymph nodes and spleen cells of CD45.1+ Pmel mice were enriched for the CD8+ population using CD8+ T Cell Isolation Kit (Miltenyi Biotec), followed by naïve CD8+ T cell FACS-sorting using the FACSAria III flow cytometer. In vitro differentiation experiments were conducted by co-culturing sorted naïve CD8+ T cells (5.0 × 104 cells/well) with CD11c+ bone-marrow-derived dendritic cells (1.0 × 104 cells/well) in the presence of soluble anti-CD3ε Ab (145-2 C11; 0.5 µg/mL) in 200 µL of RF10 in 96-well-culture plates (Corning). Co-cultures were treated to Tc17-skewing conditions (LPS (100 ng/mL) plus TGF-β1 (5 ng/mL)) or Tc1-skewing conditions ((LPS (100 ng/mL)) for 3 days. For in vitro stimulation of human TILs, cryopreserved TILs were thawed, resuspended in RF10, and rested overnight at 37°C in a 5% CO2 incubator. Next morning, cells were stimulated with anti-CD3 antibody (1 µg/mL; OKT3, eBioscience). Brefeldin A and monensin were added 1 hour after the initial stimulation and the cells were harvested after additional 5 hours of incubation.
In vitro cytotoxicity assay
Cell Trace Violet (CTV; ThermoFisher Scientific)–labeled TC-1 tumor cells pulsed with mouse gp100 peptides (2 µg/mL; RS Synthesis) for 1 hour at 37°C in a 5% CO2 incubator were used as target cells. In vitro differentiated Pmel Tc17 or Pmel Tc1 cells were co-cultured with target cells at different target:effector ratio. Four hours after the co-culture, frequency of Caspase-3+ cells among gated CTV+ cells were analyzed by flow cytometry.
TCGA analysis
Genomic analyses were performed to explore the associations between Th17-associated genes and exhaustion-related genes using the level 3 data of TCGA which were downloaded on December 18, 2019. For the type 17 signature, genes with positive effect on Th17 development and function were selected from literature curated Th17-relevant gene list.28 For the exhaustion signature, we identified differentially expressed genes (DEGs) between the acute and chronic LCMV infection microarray data29 using limma package (FDR-corrected t test p<0.05; fold change ≥2), and then selected common DEGs on day 15 and day 30, in the same way as previously described.30 Progenitor exhausted and terminally exhausted signature genes were adopted from a previously reported study19 in which differential gene expression analysis was done using single-cell RNA sequencing (RNA-Seq) data using gp33 tetramer+ CD8+ T cells in the LCMV Clone 13 infection model. Scoring of each gene signature was calculated using single sample GSEA. Correlation analysis among gene signatures was done by Pearson and Spearman correlation analysis.
Statistical analysis
Statistical analysis was performed with GraphPad Prism 6 (GraphPad Software) using unpaired two-tailed Student’s t-test or one-way ANOVA with post hoc Tukey test. Pearson or Spearman correlation analysis was conducted with R software (R Foundation for Statistical Computing) to evaluate correlations between parameters in TCGA analysis. P values are presented within each figure or figure legend.
Results
Depletion of CD4+ T cells induces a concomitant increase in the frequencies of exhausted CD8+ T cells and IL-17-poducing CD8+ T cells in the tumor
As a first step to investigate the regulatory mechanisms governing the exhaustion of CD8+ T subsets in tumor setting in vivo, we sought to determine the contribution of conventional CD4+ T cells, since depletion of CD4+ T cells is known to incapacitate the differentiation of cytotoxic CD8+ T cells in tumor and in infection.31 To this end, we employed a B16F10 lung metastasis model and analyzed TILs after treatment with anti-CD4 depleting antibody or control IgG. Of note, we observed a significant increase in the frequency of PD-1+ cells among CD8+ TILs in the lung of B16F10 melanoma-bearing mice treated with anti-CD4 (figure 1A,B). Further analyses of the PD-1+ cells revealed that anti-CD4 treatment substantially increased the proportion of Tim3+, TOX+, or TCF-1− “terminally exhausted” CD8+ TILs (figure 1A,C‒E), indicating that depletion of CD4+ T cells promotes “terminal exhaustion” of CD8+ T cells in tumor.

Depletion of CD4+ T cells concomitantly increases terminally exhausted CD8+ T cells and IL-17-producing Tc17 cells in tumor bearing mice. (A–E) CD8+ T cells in lung tumor were analyzed 15 days after B16F10 intravenous (i.v.) injection and CD4+ T-cell depletion by anti-CD4 Ab treatment (n=4). Control mice were treated with rat IgG2b isotype control Ab (n=4). (A) Representative contour plots of PD-1, Tim-3, Tox, and TCF-1 expression in CD8+ TILs. (B–E) Frequencies of PD-1+ (B), PD-1+Tim-3+ (C), PD-1+TOX+ (D), and PD-1+TCF-1− (E) cells among CD8+ TILs are shown. (F) IL-17 and IFN-γ production by lung CD8+ TILs of B16F10 tumor-bearing control Ab-treated (n=5) or CD4-depleted mice (n=7) was compared. Representative contour plots and frequencies of IL-17+IFN-γ− cells (Tc17) and IL-17−IFN-γ+ cells (Tc1) are shown. (G) Representative contour plots of IL-17 and IFN-γ production by lung CD8+ TILs of B16F10 tumor-bearing Foxp3 DTR mice after diphtheria toxin (DT) or PBS treatment (n=4 each). Frequencies of Tc17 and Tc1 cells among CD8+ TILs are shown. (H) Composition of IL-17+CD90+ lung TILs in B16F10 tumor-bearing control Ab-treated or CD4-depleted mice. (I–L) Frequencies of tumor-infiltrating Tc17 cells with or without anti-CD4 treatment in B16F10 subcutaneous (s.c.) injection (I), B16F10 intrasplenic (i.s.) injection (J), TC-1 i.v. injection (K), CCSP Cre /K-ras G12D spontaneous tumor models (L). Data are representatives of at least two independent experiments. Data are shown as mean±SD for (B-G), and mean+SD for (I-L). *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. DN17, CD4−CD8−IL-17+; Misc., miscellaneous.
To further investigate the contribution of CD4+ T cells to CD8+ T cell responses in the tumor, we comparatively examined the frequencies of IFN-γ+CD8+ T (Tc1) cells and IL-17+CD8+ T (Tc17) cells in the tumor sites. To our surprise, anti-CD4 treatment significantly increased the frequency of Tc17 cells, while minimally impacting that of Tc1 cells among CD8+ TILs in comparison with control IgG treatment (figure 1F). Unlike anti-CD4 treatment, depletion of Treg cells in B16F10-bearing Foxp3 DTR mice remarkably increased the frequency of Tc1 cells, while minimally impacting Tc17 cell population (figure 1G). Analysis of IL-17-producing lymphoid cells in the TILs revealed that anti-CD4 treatment specifically expanded the Tc17 cell population, while γδT17 cell and NKT17 cell populations were reduced (figure 1H). To determine whether the CD4+ T cell–mediated inhibition of Tc17 cell differentiation observed in the B16F10 lung metastasis model could also be true in other tumor models, we employed four additional well-established tumor models; a subcutaneous injection of B16F10 tumor model, a liver metastasis model induced by intrasplenic injection of B16F10, a TC-1 lung metastasis tumor model, and a spontaneous tumor model driven by Kras G12D mutant expression in lung epithelial cells. In all the tested murine tumor models, anti-CD4 treatment significantly increased the frequencies of Tc17 cells among CD8+ TILs (figure 1I‒L). Thus, conventional CD4+ T cells and Treg cells respectively control the differentiation of Tc17 and Tc1 cells in tumor-bearing mice in vivo in our experimental settings. Together, these results demonstrate that depletion of CD4+ T cells increases the frequency of both terminally exhausted CD8+ T cells and Tc17 cells in the tumor.
Tumor-infiltrating Tc17 cells are phenotypically and functionally distinct from Tc1 cells
Since the nature of tumor-infiltrating Tc17 cells remains incompletely defined, we comparatively analyzed the phenotypes of tumor-infiltrating Tc1 and Tc17 cells. Flow cytometric analysis revealed that, unlike Tc1 cells, the majority of Tc17 cells have CD103+KLRG1− tissue-resident memory (Trm)-like phenotype (figure 2A). The Trm-like features of Tc17 cells were observed in all tested murine models of tumor (online supplemental figure S1A‒D). Intravascular labeling experiments showed that the majority of Tc17 cells in the tumor were protected from intravenous labeling, suggesting that they exhibit tissue-residency property (figure 2B). CD8+ Trm cells have recently emerged as critical players in tumor immunity.32 Of note, Tc17 cells were found to be CD127hi (IL-7Rαhi), while Tc1 cells were mixed populations of CD127+ and CD127− (figure 2C). Moreover, compared with Tc1 cells, Tc17 cells in the tumor were Ki-67hi, suggesting the highly proliferative potential of this unique CD8+ T cell subset (figure 2D). We also found that CD8α− IL-17-producing TILs of CD4-depleted mice as well as Tc17 cells and Th17 cells from tumor-bearing mice treated with control IgG were CD103+KLRG1− and highly expressed CD127, but only Tc17 cells were Ki-67hi (online supplemental figure S1E‒M). Thus, CD103+KLRG1−CD127 hi Trm-like phenotype is likely a common feature of IL-17-producing cells in the tumor regardless of CD4 depletion. When we tested whether the blockade of IL-7R signaling pathway by anti-CD127 Ab treatment impacts Tc17 cell development, we did not see any change in Tc17 cell frequencies, but a significant decrease in Tc1 cell frequencies (online supplemental figure S1N). Cytotoxicity of CD8+ T cells is a hallmark of anti-tumor immunity. We found that the frequency of granzyme B+ cells was significantly lower in Tc17 cells than in Tc1 cells (figure 2E). Consistently, Tc17 cells were significantly less efficient than Tc1 cells in inducing antigen-specific lysis of target cells in in vitro cytotoxicity assay conditions (figure 2F), indicating that Tc17 cells are poorly cytolytic.
Supplemental material

Tumor-infiltrating Tc17 cells exhibit tissue resident memory-like phenotypes in mice and humans. (A–E) Lung CD8+ tumor-infiltrating lymphocytes (TILs) in CD4-depleted B16F10 tumor-bearing C57BL/6 mice were analyzed. (A) Representative contour plots of cell surface expression of CD103 and KLRG1 on Tc17 and Tc1 cells. (B) PerCP-Cy5.5-labeled anti-mouse CD8α Ab was intravenously (i.v.) injected into CD4-depleted B16F10 tumor-bearing mice 5 min before sacrifice for analysis. Representative contour plots of CD8 α-PerCP-Cy5.5+ cells among total CD8+ T cells in spleen and lung tumor are shown. (C) Representative histogram of CD127 expression and frequencies of CD127+ cells of Tc17 and Tc1 cells (n=4). (D) Representative histogram of Ki-67 expression and frequencies of Ki-67+ cells of Tc17 and Tc1 cells (n=7). (E) Frequencies of granzyme B(GzmB)+ cells of Tc17 and Tc1 cells (n=4). (F) Cytotoxicity of in vitro generated gp100-specific Tc1 cells or Tc17 cells (effector cells) against gp100-pulsed TC-1 tumor cells (target cells). Frequencies of Caspase-3(Casp3)+ among CTV-labeled target cells were analyzed 4 hours after co-culture with effector cells. (G–J) CD8+ TILs isolated from tumor tissues of patients with hepatocellular carcinoma (HCC) were analyzed by flow cytometry. (G) Representative contour plots of IL-17 and IFN-γ production by Tc17 and Tc1 cells in TILs of patients with HCC and frequencies of Tc17 and Tc1 cells among CD8+ TILs (n=11). (H) Cell surface expression of CD103 and KLRG1 on each CD8+ T cell subset. (I) Representative histogram of cell surface expression of CD127 on each CD8+ TIL subset and frequencies of CD127hi cells (n=6). (J) Representative histogram of GzmB expression in each CD8+ TIL subset and frequencies of GzmB+ cells in each CD8+ TIL subset (n=6). Data are representatives of at least two independent experiments for (A–F) and (H–J) and combined results of two independent experiments for (G). Data are shown as mean±SD for (C-E, G, I, J) and mean±SEM for (F). *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.
To determine whether Tc17 cells found in human tumors also exhibit similar phenotypes as those observed in mice, we analyzed the phenotypes of CD8+ TILs isolated from patients with hepatocellular carcinoma (HCC) (online supplemental table S1). In addition to Tc1 cells, we observed a small, but distinct population of Tc17 cells (figure 2G). The Tc17 cells from patients with HCC uniformly exhibited CD103+KLRG1− Trm-like phenotype, while Tc1 cells were found to be mixed populations of CD103+KLRG1− and CD103−KLRG1+ cells (figure 2H). Moreover, more than 80% of Tc17 cells were CD127hi, which was in sharp contrast to the CD127lo/int-dominant phenotype of Tc1 cells, bearing less than 50% of CD127hi cells on average (figure 2I). Unlike the high frequency of granzyme B+ cells in Tc1 (about 70%), tumor-infiltrating Tc17 cells were mostly granzyme B− (figure 2J). Taken together, these results demonstrate that, unlike Tc1 cells, tumor-infiltrating Tc17 cells exhibit a Trm-like CD103+KLRG1−CD127hi surface phenotype and poorly cytotoxic, which seems to be conserved between mouse and human.
IL-17-producing cells promote the exhaustion of tumor-infiltrating CD8+ T cells in vivo
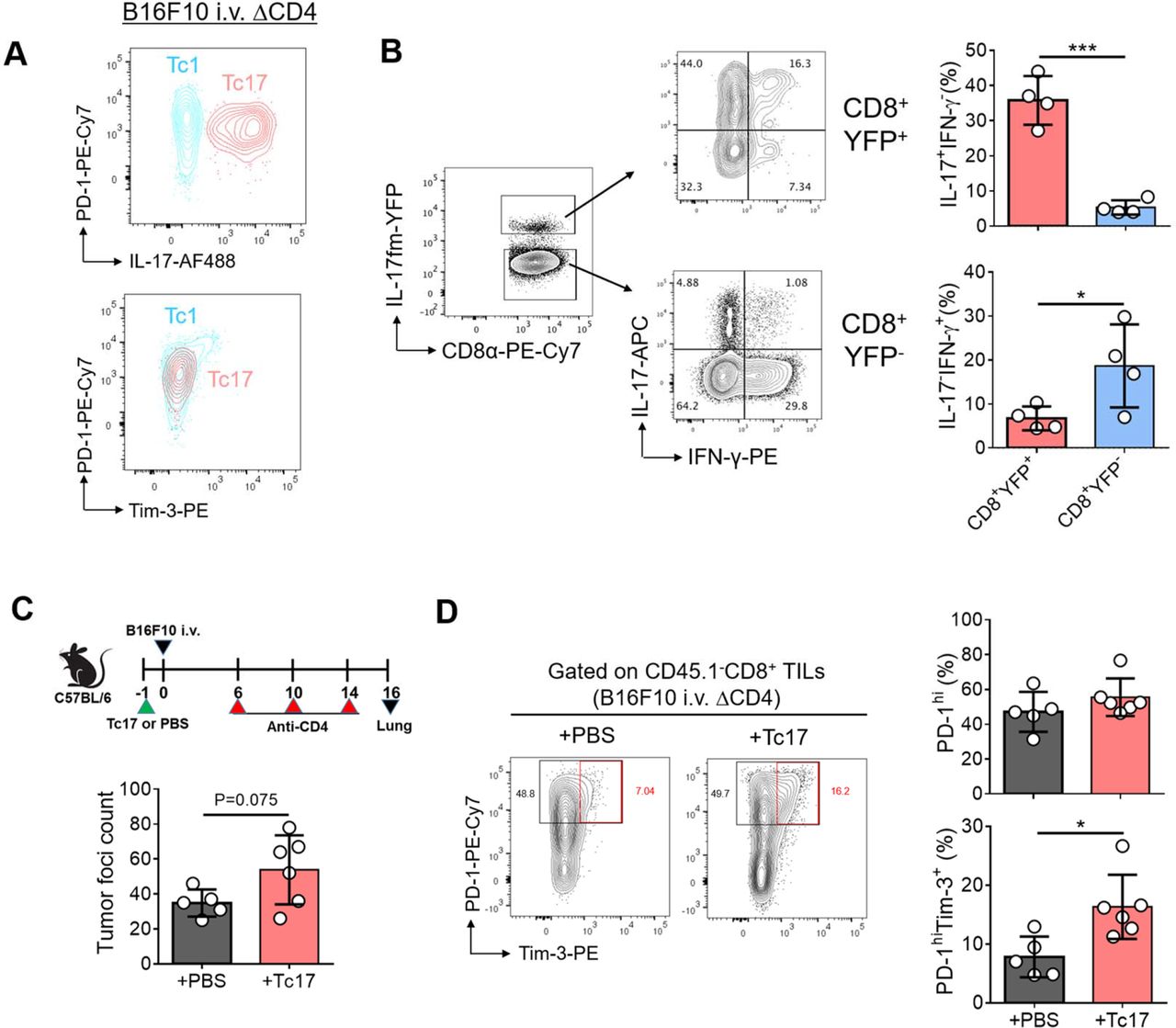
Concomitant increase in the frequencies of Tc17 cells and PD-1hiTim3+ cells by anti-CD4 treatment prompted us to hypothesize that IL-17-producing cells might exhibit “terminally exhausted” phenotypes. However, flow cytometric analysis of cell surface PD-1 and Tim-3 expression showed that Tc17 cells were PD-1intTim3−, while PD-1hiTim3+ “terminally exhausted” CD8+ TILs were confined to Tc1 population (figure 3A). Th17 cells are known to transdifferentiate into Th1 cells on repeated stimulation with IL-23 or IL-12.33 Moreover, in vitro–generated Tc17 cells have been shown to become Tc1 cells when transferred in vivo.34 To determine if in vivo–generated tumor-infiltrating Tc17 cells transdifferentiate into Tc1 cells in response to environmental stimuli in the TME, we employed Il17a Cre R26 YFP fate mapping (fm) mice in which IL-17-producing cells are permanently labeled with YFP. We injected anti-CD4 into B16F10 bearing Il17a Cre R26 YFP mice, and found that only a small fraction of CD8+YFP+ T cells (<10%) was IL-17−IFN-γ+ Tc1 cells, while the majority was still IL-17+ in the TME (figure 3B). These results suggest that in vivo–generated tumor-infiltrating Tc17 cells are stable in the TME in our experimental setting.

Tc17 cells are phenotypically not exhausted but can promote the exhaustion of CD8+ T cells in the tumor. (A) Representative contour plots of PD-1, IL-17, and Tim-3 expression of lung tumor-infiltrating Tc17 and Tc1 cells in CD4-depleted B16F10 tumor-bearing mice. (B) Fate-mapping (fm) of Tc17 cells within the tumor microenvironment. Il17a Cre R26 YFP mice were intravenously inoculated with B16F10 tumor cells and were treated with anti-CD4 Ab. Representative contour plots of IFN-γ and IL-17 expression in IL-17fm-YFP+ cells and IL-17fm-YFP− cells within the CD8+ tumor-infiltrating lymphocytes (TILs) and the frequencies of IL-17+IFN-γ− and IL-17−IFN-γ+ cells among IL-17fm-YFP+ or IL-17fm-YFP− CD8+ TILs. (C,D) C57BL/6 mice were adoptively transferred with in vitro differentiated CD45.1+ Pmel Tc17 cells 1 day before B16F10 intravenous (i.v.) injection. Control mice were injected with PBS and CD4+ T cells were depleted by anti-CD4. (C) Tumor foci count in the lung. (D) Representative contour plots of PD-1 and Tim-3 expression of CD45.1− lung CD8+ TILs and frequencies of PD-1hi and PD-1hiTim-3+ cells among CD45.1− lung CD8+ TILs are shown. Data are representatives of at least two independent experiments. Data are shown as mean±SD. *p<0.05 and ***p<0.001.
These results together raised a possibility that Tc17 cells contribute to the exhaustion of Tc1 cells in the TME. To test this possibility, we stimulated naïve CD8+ T cells isolated from CD45.1+ Pmel mice under Tc17-skewing conditions in vitro and adoptively transferred them into B16F10 tumor-bearing mice (CD45.1−) before analyzing host CD8+ TILs. The number of tumor foci was slightly increased in the recipients of Tc17 cells (figure 3C). Compared with those from the PBS-injected mice, host CD8+ TILs from the tumor-specific Tc17 cell-recipient mice showed a significant increase in Tim-3+ or TCF-1−TOX+ population. The increase in TCF-1−TOX+ population was associated with a concomitant decrease in TCF-1+TOX− cell frequencies among the PD-1hi population, although the frequency of PD-1hi cells remained comparable (figure 3D and online supplemental figure S2A,B). Thus, Tc17 cells can accelerate the terminal exhaustion of tumor-infiltrating CD8+ T cells in vivo.
Based on the observed exhaustion-promoting function of Tc17 cells, we speculated that depletion of IL-17-producing cells could reverse the exhaustion of CD8+ TILs, thereby suppressing tumor growth in vivo. To prove this hypothesis, we established Il17a Cre R26 DTA mice in which all IL-17-producing cells can be depleted by expressing diphtheria toxin. We confirmed the depletion of IL-17-producing cells in the TILs by comparing YFP+CD90+ cell frequency between Il17a Cre R26 WT/YFP mice and Il17a Cre R26 DTA/YFP mice, both of which selectively express eYFP reporter protein in IL-17-producing cells (figure 4A). Indeed, compared with Il17a Cre R26 WT mice, Il17a Cre R26 DTA mice exhibited significantly lower tumor burdens as evidenced by tumor foci and lung weight in the B16F10 lung metastasis model (figure 4B). The frequency of PD-1hiTim-3+ as well as PD-1hiTCF1− “terminally exhausted” population within CD8+ TILs was significantly lower in the latter group of mice (figure 4C,D). Similar reduction of PD-1hiTim-3+ CD8+ TILs was observed in Il17a Cre R26 DTA mice bearing subcutaneous B16F10 without anti-CD4 treatment (figure 4E) as well as in a TC-1 lung metastasis tumor model (online supplemental figure 2C,D). These results collectively demonstrate that IL-17-producing cells promote the terminal exhaustion of CD8+ T cells in the tumor per se, regardless of tumor types, tumor injection routes, and CD4 depletion in our experimental settings in vivo.

IL-17-producing cells promote CD8+ T cell exhaustion and tumor progression in vivo. (A) Il17a Cre R26 WT/YFP (n=8) or Il17a Cre R26 DTA/YFP mice (n=5) were intravenously (i.v.) injected with TC-1 tumor cells and were treated with anti-CD4 Ab. Representative contour plots of IL-17fm-YFP and CD90 expression in lymphocytes of lung tumor and frequencies of YFP+CD90+ cells among lymphocytes. (B) Il17a Cre R26 WT (n=7) or Il17a Cre R26 DTA mice (n=7) were i.v. injected with B16F10 tumor cells and were treated with anti-CD4. Representative photograph of lung tumor burdens and summary of lung weights at day 19 after tumor inoculation. (C,D) PD-1, Tim-3, and TCF-1 expression of lung CD8+ tumor-inflitrating lymphocytes (TILs) of CD4-depleted Il17a Cre R26 WT (n=6) or Il17a Cre R26 DTA mice (n=5) were analyzed at day 16 after B16F10 tumor injection. Frequencies of PD-1hiTim-3+ cells (C) or PD-1hiTCF-1− cells (D) among CD8+ TILs. (E) Il17a Cre R26 WT mice (n=4) and Il17a Cre R26 DTA mice (n=5) were subcutaneously injected with B16F10 tumor cells. Representative contour plots of PD-1 and Tim-3 expression in CD8+ TILs of B16F10 tumor and the frequencies of PD-1hi and PD-1hiTim-3+ cells among CD8+ TILs at day 14 after tumor injection. Data are representatives of at least two independent experiments. Data are shown as mean±SD. *p<0.05, **p<0.01, ***p<0.001.
IL-17 deficiency delays the exhaustion of tumor-infiltrating CD8+ T cells
Since IL-17 plays pivotal roles in type 17 cell-mediated tissue inflammation and host defense,35 we wondered if blockade of IL-17 pathway could recapitulate the phenotypes observed in the tumor-bearing Il17a Cre R26 DTA mice. We compared the tumor growth and the exhaustion progression of CD8+ TILs between tumor-bearing wild-type and IL-17-deficient mice after anti-CD4 treatment. Indeed, a significantly lower tumor burden was observed in IL-17-deficient mice than wild-type mice, associated with a lower frequency of PD-1hiTim3+ CD8+ TILs (figure 5A,B). Consistently, we also observed a significant reduction in PD-1hiTim3+ CD8+ TIL frequencies of IL-17-deficient mice without CD4 depletion in the B16F10 subcutaneous model (figure 5C), indicating that the IL-17 deficiency attenuated the terminal exhaustion of tumor-infiltrating CD8+ T cells in vivo.

IL-17 deficiency delays the exhaustion of tumor-infiltrating CD8+ T cells and tumor growth via suppressing the recruitment of CD11b+Gr-1hi myeloid cells. (A,B) C57BL/6 (WT) or Il17a −/− (IL-17 KO) mice were intravenously (i.v.) injected with B16F10 tumor cells and were treated with anti-CD4 Ab. (A) Summary of lung weights of WT (n=6) and IL-17 KO (n=7) at day 17 after tumor inoculation. (B) Frequencies of PD-1hiTim-3+ cells of lung CD8+ tumor-infiltrating lymphocytes (TILs) of WT (n=12) and IL-17 KO mice (n=13). (C) WT and IL-17 KO mice were subcutaneously injected with B16F10 tumor cells. Representative contour plots of PD-1 and Tim-3 expression in CD8+ TILs and frequencies of PD-1hiTim-3+ cells in CD8+ TILs of WT (n=5) and IL-17 KO mice (n=5). (D) Frequencies of CD11b+Gr-1hi cells among CD45+ cells in lung tumor of WT (n=12) and IL-17 KO mice (n=13) in (B). (E) Frequencies of CD11b+Gr-1hi cells among CD45+ cells of lung tumor in CD4-depleted B16F10 tumor bearing Il17a Cre R26 WT mice (n=7) and Il17a Cre R26 DTA mice (n=7). (F) B16F10 tumor-bearing C57BL/6 mice were treated with anti-CD4 Ab alone (n=7) or anti-CD4 Ab plus anti-Ly6G Ab (n=7). PD-1hiTim-3+ cells or PD-1hiTOX+ cells among lung CD8+ TILs were analyzed. Data are representatives of at least two independent experiments for (A,C,E,F) and combined results of two independent experiments for (B,D). Data are shown as mean±SD. *p<0.05, **p<0.01.
In addition to the profound decrease of PD-1hiTim3+ CD8+ T cells, we observed a remarkable reduction of CD11b+Gr-1hi(Ly6Ghi) myeloid cells among the TILs of IL-17-deficient mice as well as in Il17a Cre R26 DTA mice in comparison with their respective controls (figure 5D,E). Conversely, anti-CD4 treatment increased the accumulation of CD11b+Gr-1hi myeloid cells into the tumor, likely due to increase of Tc17 cells (online supplemental figure S3A,B). As we repeatedly observed a concomitant decrease in the frequencies of exhausted CD8+ TILs and CD11b+Gr-1hi myeloid cells in Il17a Cre R26 DTA mice and IL-17-deficient mice, we sought to determine if CD11b+Gr-1hi myeloid cells are involved in the IL-17-mediated exhaustion of CD8+ TILs. Depletion of the CD11b+Gr-1hi myeloid cells by anti-Ly6G treatment partially reduced the frequency of PD-1hiTim3+ or PD-1hiTOX+ CD8+ TILs in anti-CD4-treated mice (figure 5F). Thus, IL-17-producing cells in the TME facilitate the terminal exhaustion of CD8+ TILs, at least in part, via the recruitment of CD11b+Gr-1hi myeloid cells into the tumor.
Blockade of RORγt suppresses the exhaustion of CD8+ TILs induced by anti-CD4 or anti-PD-L1
Type 17 immunity exerts its effector functions via IL-17 as well as other pro-inflammatory cytokines such as IL-17F, IL-22, and GM-CSF, whose production largely depends on the activity of a transcription factor RORγt.36–38 To determine if targeting RORγt could reinvigorate the exhaustion of tumor-infiltrating CD8+ T cells in vivo, we examined the expression of PD-1, Tim-3, and TOX in CD8+ TILs in mice bearing B16F10 treated with vehicle or ursolic acid (UA), a small molecule inhibitor of RORγt.39 Treatment with UA remarkably reduced the tumor burden as well as the frequency of Tc17 cells among CD8+ TILs, while that of Tc1 cells was minimally affected (figure 6A‒C). Similar to the results observed in Il17a Cre R26 DTA mice and IL-17-deficient mice, UA treatment significantly decreased the frequency of PD-1hiTim-3+ or PD-1hiTOX+ terminally exhausted CD8+ TILs (figure 6D‒F).
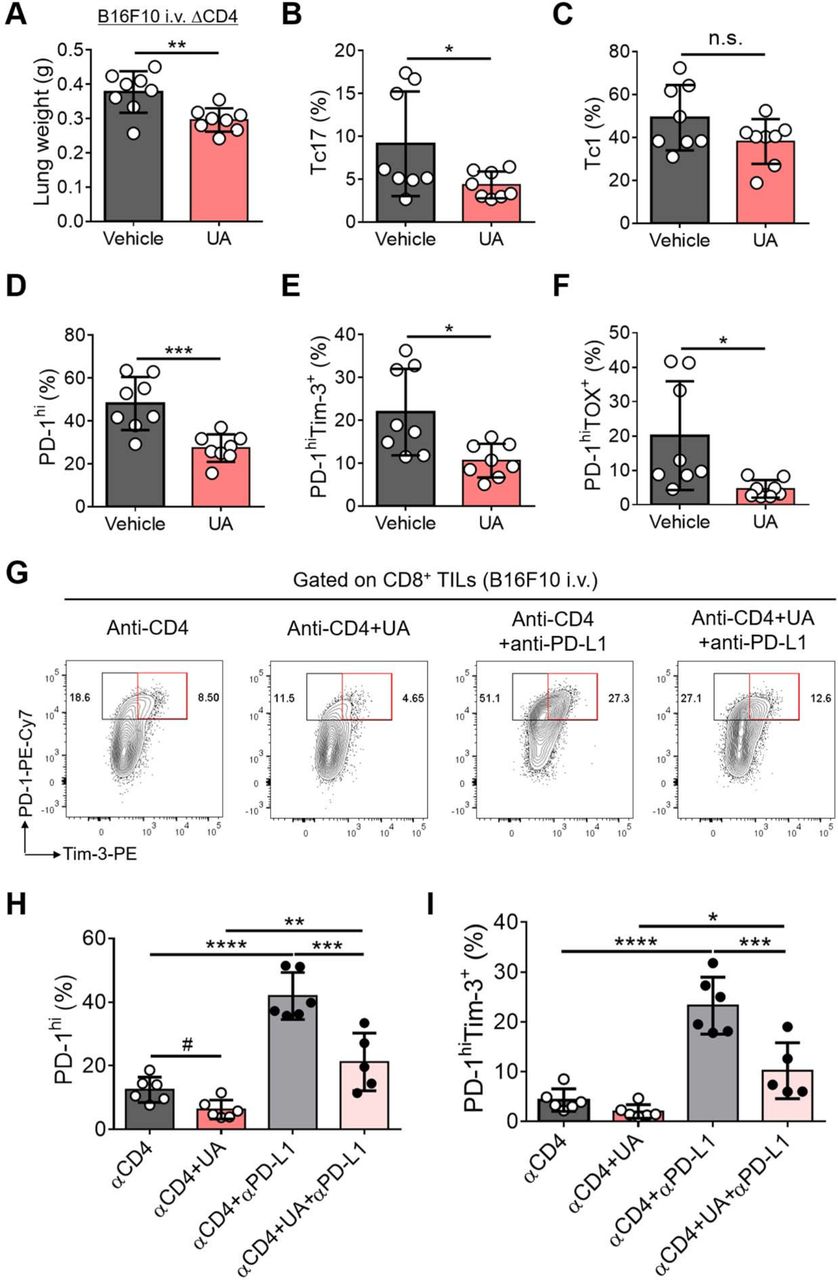
Inhibition of RORγt represses the exhaustion of CD8+ T cells induced by anti-CD4 or anti-PD-L1 in vivo. (A–F) C57BL/6 mice were intravenously (i.v.) injected with B16F10 tumor cells and were treated with anti-CD4 Ab every 4 days starting from day 6 after tumor injection. The CD4-depleted tumor-bearing mice were treated with ursolic acid (UA) or vehicle every other day starting from day 6 (n=8 each). Lung weight (A) and the frequencies of Tc17 (B), Tc1 (C), PD-1hi (D), PD-1hiTim-3+ (E), and PD-1hiTOX+ cells (F) among lung CD8+ TILs were analyzed at day 15 after tumor injection. (G–I) CD4-depleted B16F10 tumor-bearing mice were either untreated or treated with UA or anti-PD-L1 or both. (G) Representative contour plots of PD-1 and Tim-3 expression in lung CD8+ TILs. (H,I) Frequencies of PD-1hi (H) or PD-1hiTim-3+ cells (I) among lung CD8+ TILs. Data are representatives of at least two independent experiments. Data are shown as mean±SD. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001 (unpaired t-test for (A–F) and one-way ANOVA for (H,I)). #p<0.05 (unpaired t-test). n.s. not significant.
Blockade of PD-1/PD-L1 pathway is known to induce the expansion of PD-1intTCF-1+ progenitor exhausted CD8+ TILs and also the terminal exhaustion of the progenitor exhausted CD8+ TILs.19 20 Indeed, we observed that blockade of PD-1/PD-L1 pathway by anti-PD-L1 treatment further increased the frequency of PD-1hi population, particularly PD-1hiTim-3+ terminally exhausted population among CD8+ TILs in anti-CD4-treated mice bearing B16F10 tumor (figure 6G‒I). UA treatment, however, significantly reduced the frequency of PD-1hi or PD-1hiTim-3+ CD8+ TILs induced by anti-PD-L1 in anti-CD4-treated mice (figure 6G‒I). These findings together demonstrate that blockade of RORγt efficiently represses the exhaustion of CD8+ TILs induced by anti-CD4 or anti-PD-L1 in tumor-bearing mice in vivo.
Type 17 gene set positively correlates with terminal exhaustion gene set in multiple human cancers
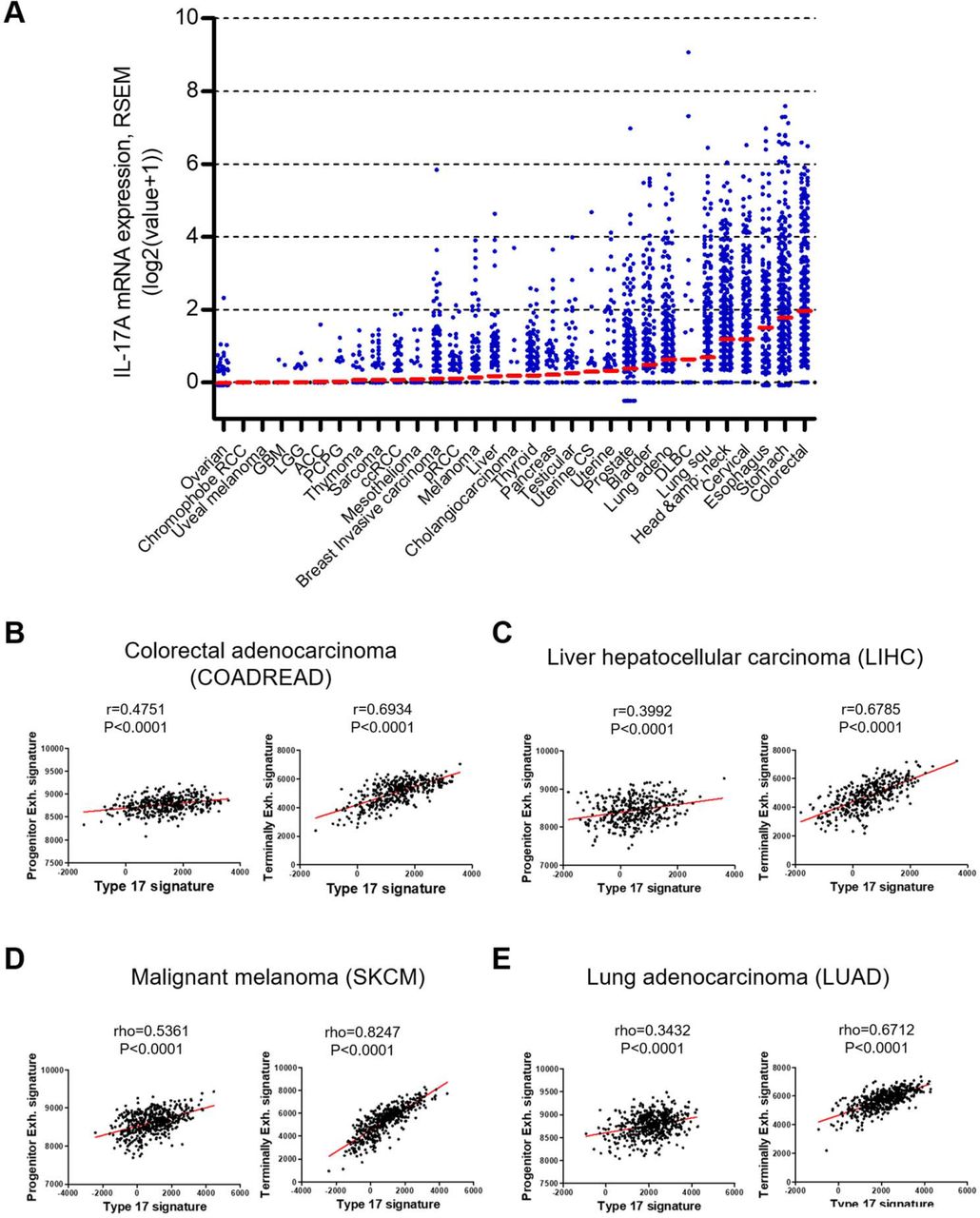
To investigate the clinical relevance of the type 17 immunity-mediated exhaustion of CD8+ TILs observed in animal models, we examined the expression of type 17 and exhaustion genes in human tumors by analyzing data from The Cancer Genome Atlas (TCGA). Significant heterogeneity in IL17A transcript expression was observed between individuals of the same cancer. In addition, we also noticed wide variability across various cancer types (figure 7A). We selected four cancers—colorectal cancer, melanoma, lung adenocarcinoma and liver cancer—for further analysis based on IL17A expression and whether immune checkpoint blockers were being used in the clinic for a given indication. We conducted correlation tests between type 17 signature and exhaustion signature gene sets, with each signature being defined by previously reported signature gene sets.28–30 Interrogation of the TCGA dataset revealed that type 17 gene expression was closely associated with exhaustion gene expression in three out of four tumor types examined: colorectal adenocarcinoma, liver hepatocellular carcinoma, and malignant melanoma (online supplemental figure S4A‒D). The highest correlation coefficient was scored by malignant melanoma (rho=0.4617, p<0.0001), while the lowest was observed in lung adenocarcinoma (rho=0.1026, p=0.0861).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Expression of type 17 signature genes positively correlates with exhaustion signature genes in multiple human cancers. (A) IL-17A mRNA expression in tumor was compared across 31 cancer types. Red bars indicate mean of log2(value+1). All data were obtained from TCGA using cbioportal website (www.cbioportal.org). (B–E) Correlation between type 17 signature genes expression and either progenitor exhausted T cell (Progenitor Exh.) signature genes expression (left) or terminally exhausted T cell (Terminally Exh.) signature genes expression (right) in colorectal adenocarcinoma (B), liver hepatocellular carcinoma (C), malignant melanoma (D), and lung adenocarcinoma (E). All data were obtained from TCGA. Statistical significance is shown with a Pearson correlation coefficient (r) or a Spearman correlation coefficient (rho).
By interrogating the correlation of type 17 gene set with either the progenitor exhausted (Progenitor Exh.) T cell gene set or the terminally exhausted (Terminally Exh.) T cell gene set,19 28 we found that type 17 gene expression level was tightly associated with expression level of Progenitor Exh. gene set and the correlation coefficient was higher than that of exhaustion signature gene set in all four tumor types (figure 7B‒E and online supplemental figure S4A‒D). Of note, the correlation of type 17 gene set with Terminally Exh. gene set was much stronger than that with Progenitor Exh. gene set, in which the correlation coefficient was over 0.6 in all four cancer types. While the highest correlation coefficient was scored by malignant melanoma (rho=0.8247, p<0.0001), the correlation coefficients for other three cancer types were also comparably high (rho=0.6712~0.6934, p<0.0001) (figure 7B‒E). Thus, type 17 gene expression positively correlates with exhaustion gene expression, particularly “terminal exhaustion” genes in multiple human cancers, which is consistent with the role of IL-17-producing cells in CD8+ TIL exhaustion in animal models of tumor in the present study. Together, these results strongly suggest a role for type 17 immunity in promoting terminal exhaustion of CD8+ T cells within the tumor in humans.
Discussion
Dissecting the mechanisms governing the exhaustion of CD8+ T cells is important for our understanding on the fate-decision program of T cells and also critical for the development of effective therapeutic interventions for cancer. By employing multiple animal models of tumor and genetic tools and human TCGA analyses, the present study convincingly demonstrates that type 17 immunity promotes the exhaustion of CD8+ T cells in tumor based on the following observations: (1) the enhanced progression of CD8+ T cell exhaustion in anti-CD4-treated mice is associated with a sharp increase of tumor-infiltrating Tc17 cells, (2) adoptive transfer of IL-17-producing tumor-specific T cells facilitates the terminal exhaustion of CD8+ T cells in tumor-bearing mice, (3) both the tumor growth and CD8+ T cell exhaustion progression are significantly delayed in Il17a Cre R26 DTA mice and in IL-17-deficient mice, (4) blockade of RORγt pathway represses the exhaustion of CD8+ TILs induced by anti-CD4 or anti-PD-L1 treatment, and (5) expression of type 17 gene set shows a strong positive correlation with terminal exhaustion gene set in multiple human cancers. Hence, we propose that type 17 immunity exerts tumor-promoting activity in vivo by promoting the terminal exhaustion of CD8+ T cells within the TME.
The role of IL-17-producing cells in cancer has been controversial. By using animal models lacking IL-17-producing cells, we demonstrate that IL-17-producing cells generated in vivo exert tumor-promoting activities. The tumor-promoting activities of type 17 cells are likely mediated in part by the induction of terminally exhausted CD8+ T cells in the TME, based on the finding that PD-1hiTim-3+ (or PD-1hiTCF-1−TOX+) fractions of CD8+ TILs were significantly lower in IL-17-deficient mice as well as in type 17 cell-deficient Il17a Cre R26 DTA mice. Adoptive transfer studies with tumor-specific Tc17 cells confirmed that type 17 cells facilitate the terminal exhaustion of CD8+ TILs. In line with these findings, recent studies demonstrated upregulation of intratumoral type 17 gene expression in non-responders of immune checkpoint blockade in patients with colorectal cancer and melanoma,40 41 suggesting that type 17 gene expression may preclude responses to immune checkpoint blockade. In this regard, a recent preclinical study demonstrated that neutralization of IL-17 enhanced anti-PD-1-mediated recruitment of neoantigen-specific CD8+ T cells in the TME.42 Thus, inhibition of type 17 immunity might improve the clinical efficacy of anti-cancer immunotherapies by blocking terminal exhaustion of CD8+ TILs.43
How do IL-17 and IL-17-producing cells promote the exhaustion of CD8+ TILs? The diminished recruitment of CD11b+Gr-1hi myeloid cells into the TMEs of Il17a Cre R26 DTA and IL-17-deficient mice and the delay in the progression of CD8+ TIL exhaustion by anti-Ly6G treatment strongly suggest that IL-17 promotes the exhaustion of CD8+ T cells indirectly via the recruitment of CD11b+Gr-1hi myeloid cells. Supporting this notion, it has been reported that IL-17 triggers the recruitment of CD11b+Gr-1+ MDSCs to mediate CD8+ T cell exhaustion in hepatitis B virus infection44 and the CD11b+Gr-1+ MDSCs have been suggested to promote CD8+ T cell exhaustion through Tim-3/Galectin-9 pathway in myelodysplastic syndromes.45 Given the role of IL-17RA signaling in CD8+ T cell survival and dysfunction in parasite infection; however, we do not rule out the direct effect of IL-17 on CD8+ TIL exhaustion.46 Further studies are needed to elucidate the detailed cellular and molecular mechanisms by which IL-17-producing cells exert CD8+ T cell exhaustion.
Previous studies proposed that depletion of CD4+ T cells exacerbated the exhaustion of CD8+ T cells in tumor as well as in chronic viral infection31 47 48; however, the mechanism whereby absence of CD4+ T cells promotes CD8+ T cell exhaustion has been elusive. In the present study, conventional CD4+ T cells appeared to suppress the differentiation of Tc17 cells, but not Tc1 cells. Given the exhaustion-promoting function of Tc17 cells observed in the present study, we propose that depletion of CD4+ T cells facilitates the exhaustion of CD8+ T cells, at least in part, via inducing Tc17 cell differentiation. Thus, Tc17 cells serve as one of the major suppressive immune cells in CD4-depleted mice. Based on these findings, we propose that targeting type 17 immunity might be effective in converting the immunosuppressive niche of the TME into a more inflammatory nature, particularly in patients with defective CD4+ T cells such as patients with cancer with HIV infection or those who are receiving CD4-depletion treatment as an immunotherapy.49
Conclusion
Our findings identify a novel role for IL-17-producing cells in regulating anti-cancer immunity via induction of CD8+ TIL exhaustion. The present study adds another dimension of complexity to the immunosuppressive network of the TME. It also proposes that blockade of IL-17 or RORγt pathway by themselves or in combination with other current immunotherapies might be a promising strategy for enhancing anti-tumor immunity.
Supplemental material
Data availability statement
All data relevant to the study are included in the article or uploaded as online supplemental information.
Ethics statements
Ethics approval
All experiments were performed according to protocols approved by the Institutional Animal Care and Use Committees of Seoul National University (protocol No. SNU-170210-3, SNU-141203-1, SNU-181030-5) or the MD Anderson Cancer Center animal facility (protocol No. 00000849-RN01). Human patient samples were obtained via protocols approved by the Institutional Review Boards at Asan Medical Center (IRB No. 2016-0845). All patients provided informed consent.
Acknowledgments
We thank Dr. Kyu-Won Kim (Seoul National University) for support in flow cytometry, Dr. Sin-Hyeog Im (POSTECH) and Dr. Alexander Rudensky (Memorial Sloan Kettering Cancer Center) for Foxp3 DTR mice, Dr. Eric Vivier (Aix-Marseille University) for R26 YFP mice, and the entire Chung laboratory for suggestions and discussion. The following reagent was obtained through the NIH Tetramer Core Facility: PE-conjugated mouse CD1d tetramer.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors B-SK designed and performed experiments, analyzed and interpreted the data, and wrote the manuscript. D-SK and C-HK performed experiments, analyzed and interpreted the data, and wrote the manuscript. H-DK analyzed human samples and interpreted the data. SH collected the human samples. E-CS interpreted the human sample data and gave suggestions for the manuscript. SHC performed CCSP Cre×K-ras G12D mice experiment and analyzed the data. SK and YKJ analyzed and interpreted the TCGA data. Y-JP, GC and JK performed some in vivo tumor experiments. KWK assisted with hepatic metastasis experiments. S-JK provided R26 DTA mice and gave suggestions for the manuscript. C-YK provided R26 YFP mice and mouse CD1d tetramer. HYK provided reagents and gave suggestions for the manuscript. CD gave suggestions for the study and the manuscript. YC designed the experiments, interpreted the data, and wrote the manuscript.
Funding This work was supported by the National Research Foundation of Korea. (NRF-2020R1A3B2078890 and NRF-2021R1C1C101279111).
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.