Article Text

Abstract
Background Therapeutic regimens designed to augment the immunological response of a patient with breast cancer (BC) to tumor tissue are critically informed by tumor mutational burden and the antigenicity of expressed neoepitopes. Herein we describe a neoepitope and cognate neoepitope-reactive T-cell identification and validation program that supports the development of next-generation immunotherapies.
Methods Using GPS Cancer, NantOmics research, and The Cancer Genome Atlas databases, we developed a novel bioinformatic-based approach which assesses mutational load, neoepitope expression, human leukocyte antigen (HLA)-binding prediction, and in vitro confirmation of T-cell recognition to preferentially identify targetable neoepitopes. This program was validated by application to a BC cell line and confirmed using tumor biopsies from two patients with BC enrolled in the Tumor-Infiltrating Lymphocytes and Genomics (TILGen) study.
Results The antigenicity and HLA-A2 restriction of the BC cell line predicted neoepitopes were determined by reactivity of T cells from HLA-A2-expressing healthy donors. For the TILGen subjects, tumor-infiltrating lymphocytes (TILs) recognized the predicted neoepitopes both as peptides and on retroviral expression in HLA-matched Epstein-Barr virus–lymphoblastoid cell line and BC cell line MCF-7 cells; PCR clonotyping revealed the presence of T cells in the periphery with T-cell receptors for the predicted neoepitopes. These high-avidity immune responses were polyclonal, mutation-specific and restricted to either HLA class I or II. Interestingly, we observed the persistence and expansion of polyclonal T-cell responses following neoadjuvant chemotherapy.
Conclusions We demonstrate our neoepitope prediction program allows for the successful identification of neoepitopes targeted by TILs in patients with BC, providing a means to identify tumor-specific immunogenic targets for individualized treatment, including vaccines or adoptively transferred cellular therapies.
- breast neoplasms
- antigens
- neoplasm
Data availability statement
Data are available upon reasonable request. Data have largely been presented in the text; other data are available upon request.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Background
Cancer therapies based on immunological approaches are rapidly becoming standard for many cancer types.1–4 In particular, immunological checkpoint inhibitors such as cytotoxic T-lymphocyte associated protein 4 (CTLA-4), programmed death ligand 1 (PD-L1) and programmed death protein 1 (PD-1) antibodies are gaining approval for the treatment of a growing number of cancer types; however, they have shown little clinical efficacy in breast cancer (BC), with response rates of 20%–25% in previously untreated cases of advanced triple-negative breast cancer (TNBC) and 5%–10% in pretreated patients; nonetheless, when efficacy is seen, it is often durable. Larger phase III trials for checkpoint inhibitors in BC are under way.5 6 Results have been more promising for combination chemotherapy and immune checkpoint therapy as shown by the recent Food and Drug Administration and European Medicines Agency approval of use of the PD-L1 inhibitor atezolizumab in combination with nab-paclitaxel as first-line therapy for metastasized or unresectable locally advanced TNBC.7 However, there continues to be a large unmet clinical need for the development of effective immunotherapies for the treatment of BC.
Expansion and subsequent adoptive transfer of large numbers of tumor-infiltrating lymphocytes (TILs)8 9 have been shown to be effective in treating some cancers, demonstrating the clinical potential for individualized immune therapies. However, current strategies largely rely on non-specific expansion and reinfusion of TILs without identification and isolation of tumor-specific T cells. Neoepitopes are epitopes created by non-synonymous mutations in the tumor genome. As these mutations are restricted to the tumor, neoepitopes can induce high-avidity mutation-specific T-cell responses. In contrast, tumor-associated antigens, which are purely overexpressed in the tumor as compared with healthy tissue, often fail to induce high-avidity T-cell responses as these T cells are largely depleted during thymic development. Mutation-specific targeting of the tumor is also less likely to induce autoimmunity. Here we establish a feasible cancer neoepitope discovery and validation program that can inform the development of patient-specific neoepitope-targeted therapies using either adoptively transferred neoepitope-specific TILs or neoepitope-targeted therapeutic vaccines.
Previously, to establish our method, we used GPS Cancer,10 NantOmics research (including several breast cancer samples), and The Cancer Genome Atlas (TCGA) databases (dbs) by whole-genome sequencing (WGS) data and RNA sequencing (RNA-Seq) data for 750 patients with cancer across 23 cancer classifications, to identify neoepitopes, to determine whether recurrent neoepitopes were prevalent in invasive BC, and to identify those neoepitopes that may be capable of binding to the HLA. We have previously used this program to identify neoepitopes in the clinical trial QUILT 2.025 (NCT03552718).
These techniques were refined and tested experimentally using the well-characterized human breast carcinoma basal-type triple-negative cell line, MDA-MB-231.11 12 Because MDA-MB-231 cells express the HLA-A2 restriction element, we were able to use established assays to compare the in silico predicted HLA-A2 binding of potential neoepitopes to in vitro binding assays. This allowed us to directly assess the efficacy by which binding algorithms can accurately predict peptides capable of being presented by a patient’s HLA. Using the cell line, we confirmed the feasibility of the pipeline. We then demonstrated its clinical feasibility in two patients with BC enrolled on the Tumor-Infiltrating Lymphocytes and Genomics (TILGen) study.13 14
Patient samples and methods
Neoepitope prediction and prioritization by successive filtering using genomics, transcriptomics and HLA typing
To predict specific neoepitopes, we established an analysis workflow pipeline. It starts with genomic analysis to identify possible neoepitopes, followed by expression analysis and further HLA-binding prediction to narrow an initial large set of possible neoepitopes down to those most likely to be actionable. As an example of this, for 26 patients with TNBC, we identified single-nucleotide variants (SNVs) and insertions/deletions (INDELs) as previously described15 using TCGA WGS. Tumor mutational burden (TMB) was determined as part of these analyses, and the results reported here are based, in part, on data generated by the TCGA Research Network (https://www.cancer.gov/tcga).
Genomic analysis was followed by RNA-Seq-based expression determination. For these analyses, tumor tissue data were compared with germline data. This filtering step eliminates all non-expressed possible neoantigens.
After filtering by WGS and RNA-Seq data, potential neoepitopes underwent HLA-binding prediction to further reduce the pool of potentially actionable neoepitopes.
Since HLA class I alleles predominantly bind to 9-mer peptide fragments,16 17 we chose to focus on the identification of 9-mer neoepitopes. Neoepitopes were identified by windowing around all possible 9-mer amino acid sequences derived from an identified non-silent SNV or INDEL. As a means to reduce possible off-target effects of a particular neoepitope, we filtered all identified neoepitopes against all possible 9-mer (HLA class I epitopes) and 15-mer peptide (HLA class II epitopes) sequences created from every known human gene using the reference human genome. These epitopes represent the normal epitopes that the immune system should ‘ignore’; that is, they should not provoke an immune response. In addition, we added single-nucleotide polymorphisms (SNPs) from the db for SNPs (http://www.ncbi.nlm.nih.gov/SNP/) to account for rare protein sequences that we may have missed within the sequencing data. Neoepitopes were ranked by RNA expression based on RNA-Seq data as well as the allele frequency of the observed coding variant to offset issues arising from tumor heterogeneity, a method we used previously as part of a study of vaccines in murine tumor models.18
HLA typing
HLA typing was performed by aligning sequencing reads to the ImMunoGeneTics/HLA db.19 HLA alleles were scored by a sum of coverage of sequencing over the entire HLA gene region and the amount of sequencing reads (depth) over the region as well as alignment scores. Highest scores were determined to be the correct HLA sequence.
Neoepitope–HLA binding affinity
NetMHC 3.4 (http://www.cbs.dtu.dk/services/NetMHC-3.4/)20–22 was used to predict whether a neoepitope would bind to a specific HLA allele. In general, neoepitopes with predicted binding affinities of <500 nM protein concentration underwent further analysis in the MDA-MB-231 and TILGen patient studies.
Neoepitope identification pipeline applied to the MDA-MB-231 BC cell line
The neoepitope identification method described previously for TCGA data was then applied to the MDA-MB-231 BC cell line,11 12 starting with sequencing and genomic analysis, with focus on HLA-A2. Thereby, 50 potential neoepitopes predicted to bind HLA-A2 were identified. Of these, 20 were synthesized for testing of HLA-A2 binding (online supplemental table S1), which was determined by incubation of neoantigen peptides with T2A2 cells23 and assessment of affinity by flow cytometry.
Supplemental material
Cytotoxic T-lymphocyte lysis assays
For use in cytotoxic T-lymphocyte assays, neoepitope-specific HLA-A2-expressing T cells, dendritic cells (DCs), and ultimately CD8+ cells from peripheral blood mononuclear cells (PBMCs) donated by healthy individuals were generated (method details in the online supplemental file).
Purified, stimulated CD8+ cells were used in a lysis assay using MDA-MB-231 (BC) or SW620 colon cancer cells24 25 as targets to assess specificity of the predicted neoantigens for a cancer tissue type. Targets were labeled with carboxyfluorescein succinimidyl ester and plated at 3000 cells/well (in 100 µL). Effector cells were added to targets at various effector:target ratios (in triplicate). Where indicated, target cells were incubated with either anti-HLA-A2 or control IgG antibodies (One Lambda) for 2 hours at 37°C prior to the addition of effector T cells. Assays were incubated overnight at 37°C. Following incubation, dead cells were stained with propidium iodide (1 µg/mL final concentration) and analyzed on a Celigo Imaging Cytometer (Nexcelom). Percent cell death is defined as (1−(livesample/livespontaneous))×100.
Preclinical validation of neoepitope prediction in patients with BC
Collection of peripheral blood and tumor tissue for analysis
Subsequently, we applied our neoepitope prediction method to samples from two subjects in the TILGen study.13 A brief description of the study and methods for peripheral blood and tumor tissue collection and analysis are presented in the online supplemental file. TILGen patient 1 had HER2+ BC and TILGen patient 2 had TNBC; both received neoadjuvant chemotherapy and had pathological complete remissions. The patients’ diagnoses were based on immunohistochemical (IHC) analysis rather than gene expression profiling to facilitate our workflow wherein the isolation of TILs requires rapid diagnosis.
Peripheral blood and tumor tissue were obtained after approval by the internal Institutional Review Board (IRB) and with informed patient consent according to the Declaration of Helsinki. Peripheral blood was used for isolation of germline DNA, antigen-presenting B cells to be immortalized with Epstein-Barr virus (EBV) to generate lymphoblastoid cell lines (LCLs),26 and monocyte-derived DCs (details in the online supplemental file or in Patient samples and methods). TILs were isolated from tumor tissue to generate the T-cell clones used for culture experiments as described further and in the online supplemental file.
Selection of TILGen peptides for synthesis
After sequencing the tumor and normal genome of the TILGen subjects, determination of possible neoepitopes, and filtering transcriptomic analyses, peptides were selected for synthesis based on predicted HLA affinity. Synthetic peptides for the predicted neoepitopes were obtained at purity above 70% from GeneCust (Luxembourg). For HLA class I, all predicted peptides were synthesized as 9-mers. For HLA class II, all neoepitopes comprising the same mutation were fused to one long peptide ranging from 15 to 29 amino acids (online supplemental tables S2 and S3).
Expansion and isolation of TILs
TILs were expanded from patient tumor tissue and cell culture experiments performed with TILGen subject samples to determine neoepitope recognition. Biopsies obtained from the two TILGen patients were cultured in Roswell Park Memorial Institute (RPMI) with 40 U/mL penicillin/streptomycin, 2 mM L-glutamine, 50 µM β-mercaptoethanol (all Gibco), and 5% fetal calf serum (FCS), 5% human serum, 0.4% vitamin solution, 1% minimal essential medium, and 1 mM sodium pyruvate (all PAN-Biotech, Germany) in the presence of 2.5×105 CD3/CD28 beads/mL (Gibco), 100 U/mL IL-2 (Proleukin) and 5 ng/mL IL-15 (Cellgenix, Germany) for 14–21 days.
Antigen presentation assays for BC-derived neoepitopes
EBV–LCLs and DCs were used as autologous antigen-presenting cells (APCs), HeLa cells after transduction with HLA molecules to determine HLA-restriction, and MCF-7 cells transduced with the respective HLA molecule and with neoepitopes or wild-type (wt) counterparts for processing and presentation assays.
EBV–LCL, HeLa and MCF-7 cells were cultured in RPMI with the same additives as described previously for TILs, but with 10% FCS. The T-cell clones were cultured in RPMI also with the same additives but 5% human serum, 5% FCS and 200 IU/mL IL-2, and were restimulated every 10–20 days with irradiated allogeneic PBMCs and 0.8 µg/mL phytohemagglutinin (Thermo Scientific).
Autologous DCs of the two patients with BC were generated from monocytes isolated from autologous PBMCs by magnetic separation as described in the online supplemental data.
Expanded T-cell lines were cocultured with autologous EBV-transformed LCLs (EBV–LCLs) or autologous DCs loaded with a peptide pool (1 µg/mL) of potential neoepitopes at an effector:target ratio of 4:1. Activated T cells were clonally isolated by flow cytometric cell sorting after 4.5 hours (interferon gamma (IFN-γ) secretion assay, Miltenyi Biotec) or 36 hours (CD137 expression determined by flow cytometry).
The stimulator cells (5×104 cells/well), including EBV–LCL, HeLa or MCF-7 cultured in RPMI with 10% FCS, were coincubated overnight in U-bottom 96-well plates with T-cell clones (5×103 cells/well), which were expanded after peptide stimulation from patient-derived TILs. Details of peptide pulsing can be found in the online supplemental data.
Retroviral transduction of neoantigens and HLA alleles, flow cytometry, T-cell receptor (TCR) sequencing, and clonotypical PCR
To confirm natural processing and presentation of neoantigens occurs after endogenous expression, sequences for the neoantigens were cloned into retroviral vectors for the transduction of EBV-LCL, HeLa or MCF-7 cells as described in the online supplemental file.
Methods for flow cytometry for either analysis or isolation of cells using labeling with CD3 (fluorescein isothiocyanate/FITC or Brilliant Violet 510/BV510), APC-Cy7 or APC CD8, BV421 CD4, PE CD137 and NGFR are detailed in the online supplemental file.
TCR sequencing of T-cell clones, and clonotypical PCR, including all plasmids and oligonucleotides used for cloning (online supplemental table S4), are described in the online supplemental file.
Results
Successive filtering by neoepitope burden, expression, and HLA binding leads to identification of high-probability actionable neoepitopes in TCGA TNBC cases
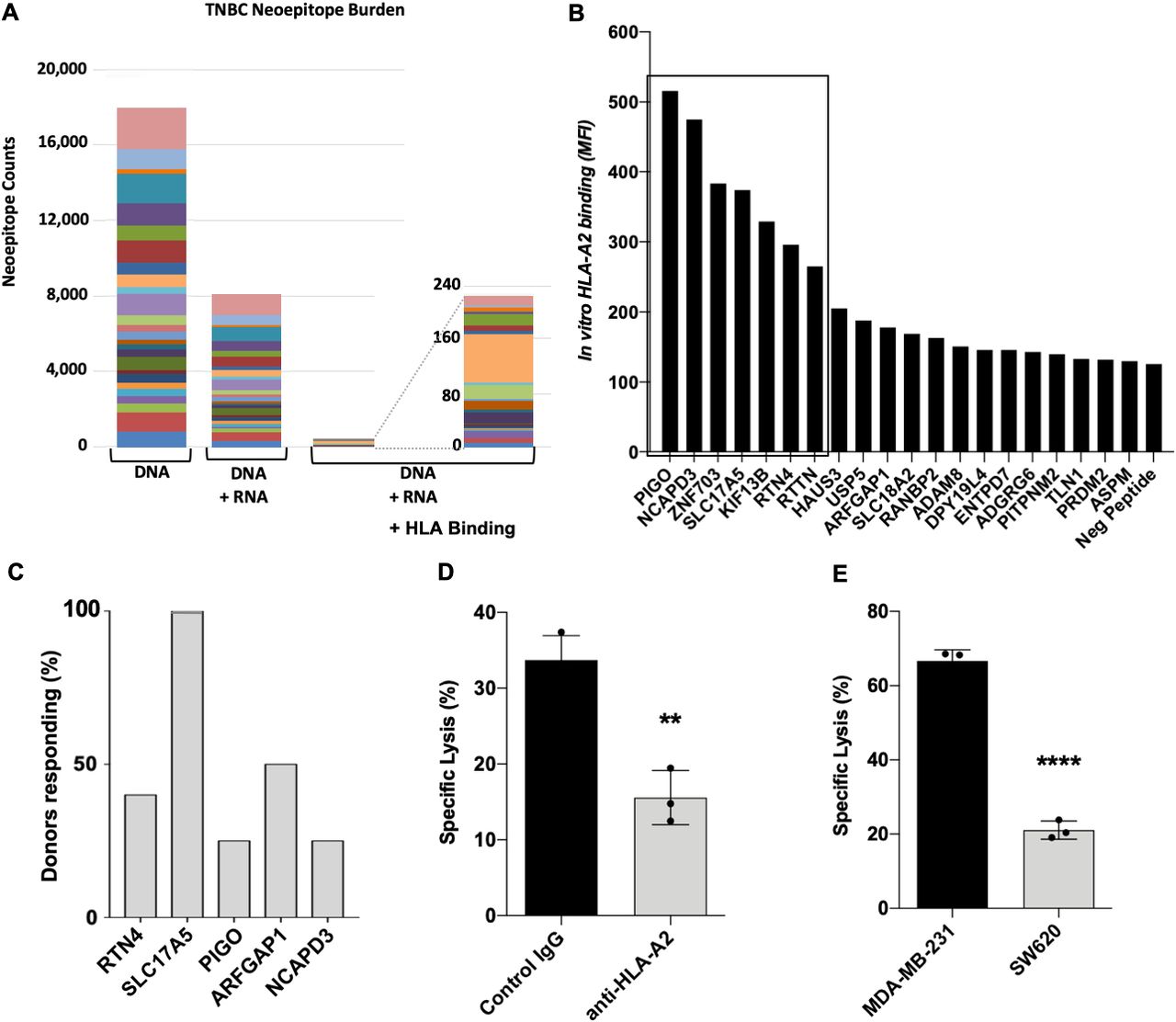
Analysis of TCGA data revealed that successive elimination of potential TNBC neoepitopes first identified by WGS sequencing for SNVs and INDELs, then filtered for RNA expression levels and finally potential HLA class I binding—as well as similarity to peptide sequences present elsewhere within the normal human proteome—eliminates the vast majority of potentially targetable mutations (figure 1A). This successive filtering allows prioritization of potential neoepitopes with the highest probability for being actionable for further testing.

Potentially actionable neoepitopes are filtered by sequential DNA/RNA/HLA analyses. (A) Within the TNBC TCGA dataset filtered by WGS (DNA), then expression (RNA) and finally HLA-binding prediction (HLA); the final set of actionable neoepitopes showed no recurrent mutations. Different colors represent individual patient samples. (B) Application of DNA/RNA/HLA neoepitope filtering to MBA-MD-231 cell line resulted in identification of 50 potential HLA-A2-binding neoepitopes, synthesis of 20 9-mer neoepitopes, and determination of HLA-A2 binding using T2A2 cells revealed seven peptides with good binding (boxed, MFI>200). (C) Percentage of normal HLA-A2 donors responding by expansion of reactive T cells to five potential neoepitopes indicates SLC17A5 elicited a response from all donors (n=4–6 normal donors per peptide). (D) Lysis of MDA-MB-231 cells by SLC17A5-reactive T cells is blocked by an anti-HLA-A2 antibody. (E) Lysis of MDA-MB-231 and SW620 cells by SLC17A5-reactive T cells is greater for the BC cell line. Statistics performed using an unpaired Student’s t-test; **p<0.01, ****p<0.0001. MFI, Mean Fluorescence Intensity; TCGA, The Cancer Genome Atlas; TNBC, triple-negative breast cancer; WGS, whole-genome sequencing.
HLA-A2 binding and ability to activate T cells further narrow actionable neoepitopes
To test the reactivity of T cells to neoantigen candidates in vitro, we used our bioinformatics-based neoepitope identification method that includes sequencing to identify 50 potential neoepitopes in the MDA-MB-231 human BC cell line predicted to bind to HLA-A2. A library of 20 peptides was synthesized, comprising the top 14 based on predicted HLA-A2 binding affinity, the top 6 based on expression levels, and 6 peptides whose low expression levels or poor predicted HLA-A2 binding would presumably make them less than ideal candidates for immunological targeting (online supplemental table S1). We synthesized 20 of these potential neoepitopes and found that 7 of 20 were capable of binding HLA-A2 in vitro (figure 1B). Using peripheral blood from normal HLA-A2 human donors, we screened five peptides for their ability to expand reactive T cells. One identified neoepitope, SLC17A5 (GTIGIFWFV), was able to expand reactive T cells in all normal donors assayed (figure 1C). These SLC17A5-reactive T cells were capable of specifically lysing the MDA-MB-231 BC cell line in an HLA-A2-restricted manner (figure 1D,E).
Neoepitope prediction and validation for two patients with TILGen BC
An expanded neoepitope identification pipeline comprising HLA-binding prediction for HLA class I and II molecules of the respective patients was used for two TILGen subjects as shown in figure 2A. TILGen 1 was a patient with HER2+ BC and TILGen 2 was a patient with TNBC. Receptor status information was known previous to this study. WGS of these tumor samples revealed a higher TMB for subject 2, and comparison to corresponding reference DNA revealed 68 and 274 non-synonymous mutations, respectively (figure 2B). Based on genomic analysis only, 206 and 964 neoepitopes were predicted for subjects 1 and 2, respectively, but RNA-Seq expression analysis decreased this to 143 and 809; HLA class I binding prediction narrowed it further to 20 for subject 1 and 26 for subject 2; HLA class II binding prediction added another 8 for patient 1 and 39 for patient 2; see the list of potential neoepitopes for patients 1 and 2 in online supplemental tables S2 and S3, respectively.

Analysis workflow for neoepitope identification in TILGen subjects and subject genomic and expression information. (A) In the proposed workflow, WGS is performed on DNA from both peripheral blood and patient tumor biopsies for HLA typing, variant calling, and binding prediction; tumor RNA-Seq analysis is used to confirm expression. Autologous APCs are generated from peripheral blood, and TILs are expanded from tumor tissue. Peptides with confirmed expression and predicted binding to a patient HLA class I or II molecule are then synthesized and used for isolation of peptide-specific T cells. (B) When this workflow was applied to the two TILGen subjects, WGS-based TMB was found to be higher in subject 2, as were non-synonymous SNVs; both predicted neoepitopes and number of expressed potential neoepitopes were higher in subject 2. Hormone receptor status for these subjects was previously determined as part of the TILGen study, not the workflow shown here. APC, antigen-presenting cell; ER, estrogen receptor; HER2, human epidermal growth factor receptor 2; IMGT, ImMunoGeneTics; PR, progesterone receptor; RNA-Seq, RNA sequencing; SNP, single-nucleotide polymorphism; SNV, single-nucleotide variant; TIL, tumor-infiltrating lymphocyte; TILGen, Tumor-Infiltrating Lymphocytes and Genomics; TMB, tumor mutational burden; WGS, whole-genome sequencing.
T-cell recognition of predicted neoantigens further refines actionable targets
The existence of TILs that recognize a neoantigen peptide further validates the neoantigen predicted by the workflow pipeline as likely actionable. To identify neoepitope-specific TILs, T cells were expanded from tumor biopsies of the two patients with BC. Expanded TILs were co-cultured with autologous EBV–LCLs or DCs loaded with a pool of peptides encoding the predicted neoepitopes for each patient. T cells were then sorted based on expression of either the activation marker CD137 or secretion of IFN-γ, and clonally expanded. Growing T-cell clones were retested for recognition of autologous EBV–LCL pulsed with the individual patient-specific peptide pool by IFN-γ ELISA as shown for TILGen patient 2 in figure 3A.

Identification of neoepitope-specific T-cell clones from BC biopsies, specificity and validation of natural processing. (A) Clonally expanded TILs from TILGen patient 2 were retested for recognition of the patient-specific HLA class I and HLA class II PP I/PP II by measuring IFN-γ secretion after stimulation with peptide-loaded autologous EBV–LCL in ELISA. Clones 23, 24, 25, 37, 38, 41, and 42 display specific recognition of the HLA class I peptide pool. For each identified neoepitope from TILGen subject 2 (B,C) or TILGen 1 (D), the Mut and wt variants of the peptides are tested for T-cell recognition after loading on autologous EBV–LCL as reflected by IFN-γ secretion. Representative T-cell clones for each antigen are shown: PNMAL1 clone 21 and CARS2 clone 55 (TILGen 2) and RBMX clone 3E1 (TILGen 1). Depicted are mean and SEM of triplicates. NC: unloaded autologous EBV–LCL. (E–G) T-cell recognition of Mut and wt peptides at decreasing concentrations as measured by IFN-γ ELISA is shown. Depicted are mean and SEM of duplicates. (H–J) T-cell recognition of retrovirally expressed full-length wt and Mut neoantigen proteins in HLA-matched EBV–LCL as measured in IFN-γ ELISA. Means and SEM of triplicates (PNMAL1) and of duplicates (RBMX and CARS2) are shown. BC, breast cancer; EBV, Epstein-Barr virus; IFN-γ, interferon gamma; LCL, lymphoblastoid cell line; PNMAL1, paraneoplastic Ma antigen family-like 1; CARS, cysteinyl tRNA synthetase 2; RBMX, RNA binding motif protein X-linked; Mut, mutated; NC, negative control; PP, peptide pool; TIL, tumor-infiltrating lymphocyte; TILGen, Tumor-Infiltrating Lymphocytes and Genomics; WT, wild type.
All clones that were not reactive against the unpulsed EBV-LCL but strongly recognized autologous EBV-LCL loaded with the peptide pool were further analyzed by first testing recognition of subpools of the peptides and finally individual peptides. Matrix subpools enable fast identification of the targeted epitope while consuming minimal amounts of material. Subpools were generated from each patient-specific peptide pool wherein each peptide was present in three subpools. Based on the combination of recognized subpools, the individual peptide could be identified.
Among TILs derived from TILGen patient 2, we were able to identify 31 CD8+ and one CD4+ T-cell clones recognizing the peptide pools. We note again for the TILGen subjects, neoepitope prediction was based on binding to both class I and II HLA molecules. All of the CD8+ T-cell clones and the CD4+ T-cell clone of this patient were derived from screening for secretion of IFN-γ. While three of the CD8+ T-cell clones could not be further expanded, the other 28 specifically recognized a mutated variant of PNMAL1 (P100R) as shown in figure 3B. The CD4+ T-cell clone specifically recognized a mutated variant of CARS2 (Q171H) (figure 3C and online supplemental figure S1A,B).
From TILGen patient 1, we characterized three CD4+ T-cell clones (3E1, E15 and G44), with all three recognizing the mutated variant of the X-chromosomally encoded RNA-binding motif protein RBMX (T55I) (figure 3D and online supplemental figure S1C,D).
Specificity for the mutated variants—PNMAL1 (P100R) and CARS2 (Q171H) for TILGen patient 2 and RBMX (T55I) for TILGen patient 1—was confirmed by a lack of recognition of the wt alleles (figure 3B–D, respectively; additional clones are shown in the online supplemental figure S2). At the peptide concentrations used (0.01, 0.1, and 1 µg/mL), the wt versions of PNMAL1 and RBMX neoepitopes did not stimulate the T-cell clones (figure 3E,G, respectively). The wt CARS2 neoepitope did stimulate the T-cell clones at 0.1 and 1 µg/mL (figure 3F), but the mutated variant CARS2 induced much stronger T-cell recognition.
To ensure the identified neoepitopes are naturally processed and presented, we transduced HLA-matched EBV–LCL cells to express either the full length wt or mutated protein. T cells were capable of specifically recognizing the endogenously expressed mutated but not wt proteins (figure 3H–J).
These studies validate the predicted neoantigens RBMX for TILGen patient 1 and PNMAL1 and CARS2 for TILGen patient 2 as potentially actionable.
Polyclonal T-cell responses to PNMAL1 and RBMX
To differentiate between a polyclonal T-cell response against the identified neoepitopes and repetitive detection of the same expanded T cells, we analyzed expression of the variable β-chain of the TCR (TILGen 2) or sequenced the CDR3 region of the TCR (TILGen 1) and observed that both PNMAL1 and RBMX elicited polyclonal T-cell responses (figure 4). This finding was supported by data obtained by testing truncated versions of the RBMX T55I. We observed that the minimal epitope of the recognized RBMX T55I peptide was shifted by one amino acid to the N-terminus or C-terminus among the different T-cell clones, but all required a core of seven to eight amino acids (online supplemental figure S3).

PNMAL1 P100R- and RBMX T55I-elicited polyclonal immune responses. (A) Flow cytometric analysis of vβ chain usage of three exemplary PNMAL1 P100R-specific T-cell clones is shown. (B) T-cell receptor sequencing of all three RBMX T55I-specific CD4+ T-cell clones demonstrated differential vβ chains and unique CDR3 regions. vβ, variable beta; FITC, fluorescein isothiocyanate; TRBV, T-cell receptor beta-chain variable; TRBJ, T-cell receptor beta joining; and TRBD, T-cell receptor beta-chain diversity genes.
These data confirm polyclonal T-cell responses to RBMX for TILGen patient 1 and to PNMAL1 for TILGen patient 2, indicating the immunogenicity of these neoepitopes.
Identified neoepitopes were patient-specific
To determine whether or not the neoepitopes we identified for TILGen patients 1 and 2 were frequently expressed in other patients with BC or were patient-specific, we analyzed 360 genomes from patients with BC, including 39 of the TILGen study, and were not able to find these mutations in any other patient, indicating that these neoepitopes are truly patient-specific.
Characterization of HLA class II-restricted neoepitopes
The RBMX T55I neoepitope identified in TILGen 1 was predicted to bind to three of the patient’s HLA class II alleles (HLA-DRB1*15:01, HLA-DPB1*02:01 and HLA-DPB1*04:01, respectively). To characterize the HLA restriction of these RBMX T55I-reactive T-cell clones, we first performed blocking experiments that confirmed their class II restriction (figure 5A) and subsequently expressed all three potential restriction elements in the HLA class II negative HeLa cells. We observed that clones 3E1 and G44 recognized RBMX T55I within the context of HLA-DPB1*04:01, whereas T-cell clone E15 recognized the neoepitope within the context of the HLA-DP*02:01 restriction element (figure 5B). None of the clones displayed recognition of the epitope in HLA-DRB1*15:01 (online supplemental figure S4B).

HLA class I or II restriction of identified neoepitopes. Blocking experiments with HLA class I and class II blocking antibodies on peptide-loaded autologous EBV–LCL confirmed restriction of the isolated (A) RBMX-specific and (D) CARS2- specific CD4+ T-cell clones to HLA class II. (B) T-cell recognition of HeLa cells retrovirally transduced with HLA-DPB1*02:01 and HLA-DPB1*04:01 was measured after loading with RBMX T55I peptide. Autologous peptide-loaded EBV–LCLs were used as controls. (C) HLA class I restricted neoepitope PNMAL1 P100R was predicted to bind in HLA-A1*11:01. Loading of PNMAL1 P100R peptide on HeLa cells transduced with HLA-A1*11:01 confirmed T-cell recognition in the IFN-γ ELISA. (E) T-cell recognition in IFN-γ ELISA of HLA-DQB1*02:02 transduced HeLa cells loaded with CARS2 Q171H. Shown are means and SEM of triplicates (C,D) and duplicates (A,B,E). RBMX, RNA binding motif protein X-linked; PNMAL1, paraneoplastic Ma antigen family-like 1; CARS, cysteinyl tRNA synthetase 2; EBV, Epstein-Barr virus; IFN-γ, interferon gamma; LCL, lymphoblastoid cell line; TILGen, Tumor-Infiltrating Lymphocytes and Genomics.
Epitope PNMAL1 (TILGen 2) was predicted to be presented in HLA-A1*11:01, which could be confirmed by retroviral transduction of HeLa cells with this restriction molecule (figure 5C). Similarly to RBMX, the CARS2 Q171H epitope was predicted to bind to two of the patient’s HLA molecules (DRB1*07:01 and DQB1*02:02). We again confirmed restriction to HLA class II by blocking studies (figure 5D) and determined HLA-DQB1*02:02 as the correct restriction molecule by retroviral transduction of HeLa cells (figure 5E and online supplemental figure S4).
Despite the technical limitations of HLA-binding prediction for HLA class II epitopes, all predicted HLA bindings could be experimentally confirmed for our identified neoepitopes.
Broadening of T-cell polyclonality is seen following treatment in TILGen 1
Chemotherapy or other treatment may affect neoantigen expression; thus, it is of interest to us to assess changes in predicted and validated neoantigens. This comparison was possible for TILGen patient 1, from whom tumor tissue was collected before and after treatment. On stimulation of TILs from the resected tumor of HER2+ TILGen 1 patient after chemotherapy with the peptide pool loaded autologous APCs, we again detected one RBMX T55I-specific T-cell clone (1A35). This T-cell clone also specifically recognized the mutated variant and was restricted to HLA-DPB1*04:01 (online supplemental figure S5A,B).
Sequencing of the TCR revealed a fourth independent clone that we had not detected at the time of diagnosis (online supplemental figure S5C). To unravel whether we missed this clone by screening of T-cell reactivities, we developed clonotypical quantitative PCRs for each of the identified clones. By this, we could show that the CDR3 regions of all four T-cell clones could be detected in the RNA of expanded T cells from the postchemotherapeutic tumor tissue. Furthermore, we could demonstrate that the CDR3 regions of the three T-cell clones isolated from core biopsy tumor tissue (prechemotherapy) could be detected in RNA of expanded T cells from peripheral blood at the time point of diagnosis, but the CDR3 of clone 1A35 could not be reliably detected at that time point (online supplemental figure S5D).
This finding suggests further induction of T-cell responses against neoepitopes in response to chemotherapy.
Neoepitopes are recognized when expressed endogenously in transduced MCF-7 BC cells
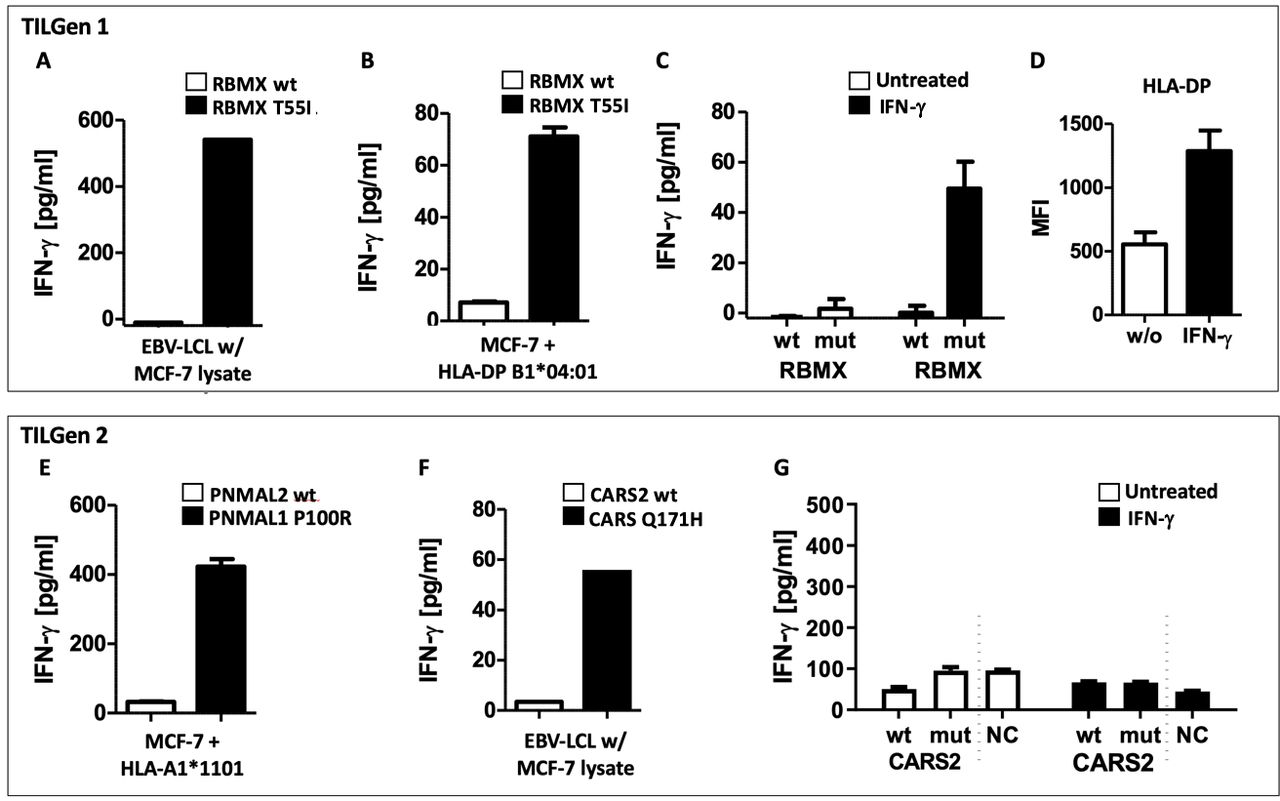
Finally, to analyze whether our newly identified epitopes could also be recognized by the T-cell clones when expressed in BC cells, we retrovirally transduced the MCF-7 human BC cell line with our full-length neoepitopes and the respective HLA molecules. We found RBMX T55I was presented both directly on MCF-7 cells and, as BC cells normally do not/rarely express HLA class II, after exposure of EBV-LCL to antigen-expressing tumor cell lysates (figure 6A,B). In addition, because MCF-7 cells endogenously harbor the HLA-DPB1*04:01 restriction molecule of RBMX T55I, we see activation of the T-cell clones after IFN-γ induced upregulation of HLA-DP even without transduction of the HLA molecule (figure 6C,D).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
T-cell recognition of neoepitopes expressed in MCF-7 BC cells. (A) IFN-γ ELISA of RBMX T55I-specific T cells cocultured with autologous EBV–LCL loaded with lysates of retrovirally transduced MCF-7 cells. (B) IFN-γ ELISA of T-cell recognition of MCF-7 cells retrovirally transduced with RBMX T55I or RBMX wt and the HLA-DP restriction molecule. (C) T-cell recognition as measured by IFN-γ ELISA of RBMX specific T-cell clone and RBMX T55I/wt transduced MCF-7 cells with and without prior IFN-γ treatment. (D) Surface expression of HLA-DP on MCF-7 cells with and without IFN-γ treatment as measured by flow cytometry. (E) T-cell recognition of HLA class I restricted antigen PNMAL1 P100R as well as wt variant retrovirally transduced in MCF-7 cells as measured by an IFN-γ ELISA. (F) IFN-γ ELISA of CARS2 Q171H-specific T cells cocultured with autologous EBV–LCL loaded with lysates of retrovirally transduced MCF-7 cells. (G) IFN-γ ELISA of T-cell recognition of IFN-γ treated and untreated MCF-7 cells transduced with CARS2 Q171H or CARS2 wt and the HLA-DQ restriction molecule NC: untransduced MCF-7 cells. Means with SEM of duplicates (B–E,G) are shown. BC, breast cancer; EBV, Epstein-Barr virus; IFN-γ, interferon gamma; LCL, lymphoblastoid cell line; RBMX, RNA binding motif protein X-linked; PNMAL1, paraneoplastic Ma antigen family-like 1; CARS, cysteinyl tRNA synthetase 2; Mut, mutated; NC, negative control; TILGen, Tumor-Infiltrating Lymphocytes and Genomics; wt, wild type.
To allow presentation of HLA class I‒restricted antigen PNMAL1, we additionally transduced the restriction molecule HLA-A1*11:01 and tested recognition by the PNMAL1 specific T-cell clone. As depicted in figure 6E, PNMAL1 P100R and HLA-A1*11:01 positive MCF-7 cells strongly activated the respective T-cell clone.
Interestingly, we found that autologous EBV-LCLs were capable of indirectly presenting the CARS2 after pulsing with tumor cell lysates (figure 6F), but we did not observe activation of the T-cell clone by the wt antigen. However, we were not able to demonstrate direct recognition of HLA-DQB1*02:02 transduced MCF-7 cells retrovirally expressing the wt or mutated CARS2 either with or without IFN-γ pretreatment (figure 6G). Lack of T-cell recognition was not due to insufficient expression of transduced CARS2 as western blot analysis revealed strong expression (online supplemental figure S6A) and CARS2 peptide loading resulted in effective T-cell recognition, indicating sufficient expression of the HLA-DQ restriction molecule (online supplemental figure S6B).
The recognition of endogenously expressed RBMX and PNMAL1, but not CARS2, suggests the former two should be prioritized for further analysis of actionable neoantigens in patients 1 and 2, respectively. Lack of processing of endogenously expressed CARS2 might explain T-cell recognition of the exogenously loaded wt version.
Discussion
The data presented herein show that by using our prediction algorithm, we can detect neoepitopes that are specifically recognized by TILs in TNBC and HER2+ patients with BC. Furthermore, these neoepitopes prove to be patient-specific both in our analysis of TCGA data as well as in our analyzed patients.
The specific targeting of tumor neoepitopes recognized by TILs and the development of individualized immune-based therapies would likely enhance checkpoint therapy, which has been disappointing as anti-PD-1 or PD-L1 monotherapy,5 27 and only somewhat improved by combination with atezolizumab/nab-paclitaxel. Given the rather modest mutational burden of BC,28 boosting tumors to an immunologically ‘hotter’ state either by adoptively transferring neoepitope-specific T cells or vaccination with immunogenic neoepitopes either as adjuvant treatment after conventional chemotherapy or in combination with checkpoint inhibition has the potential to vastly improve response rates.29 30
To identify actionable neoepitopes, we present here a prediction algorithm based on WGS, RNA expression data and HLA-binding prediction that allowed for successful identification of neoepitope-specific TILs for the first time in treatment-naive patients with BC. In contrast to other studies,9 31 we purposefully included HLA-binding prediction in our algorithm to reduce the number of potential epitopes, and while this may exclude some actionable neoepitopes, specifically for high TMB tumors, it increases feasibility. In combination with the use of long overlapping peptides for potential HLA class II epitopes, HLA-binding prediction led to a reduction of potential epitopes from 143 to 28 in the first patient (18%) and from 809 to 65 in the second (8%). Although we cannot guarantee that we did not miss any neoepitope-specific response, we clearly demonstrate that this method is feasible and leads to identification of polyclonal immune responses. For patients with low mutational burden, HLA prediction can easily be excluded from the workflow. The comparison of our neoantigen prediction method to other reported methods such as MHC analysis with recurrent integrated architecture (MARIA),32 will be part of future studies.
Knowledge of both tumor and normal tissue gene sequencing is vital for accurate variant detection and the basis for accurate neoepitope predictions; our pipeline here is distinguished from others in the literature by performing such tumor:normal comparisons. The bioinformatics method used here is also highly sensitive and specific for identification of the most likely variants and filters out low-probability variants. The ranking and prevalence of coding mutations within the dataset presented here fell within previously reported ranges that are representative of primary cancers.33 34
During cancer evolution, many mutations occur and may not persist, and such mutations as well as those not in known oncogenes are likely passenger mutations. Therefore, to circumvent targeting of only certain tumor subclones, we determined the allelic frequency of any candidate target neoepitope mutation of the tumor by deep sequencing.
To further lower the risk of escape variants and to generate a sustained antitumor effect, broad targeting of both CD4+ and CD8+ T-cell neoepitopes should be used. It is therefore of note that despite technical limitations of predicting HLA class II-restricted epitopes due to the varying peptide length within the open HLA II binding groove, we were able to identify neoepitope-specific CD4+ T-cell clones in both of our analyzed patients. The deep learning-based algorithm created by Chen et al32 may be a more robust approach to predicting HLA class II, and we will run a comparison in future studies. Nonetheless, our identification of these CD4+ T-cell clones allowed for combined CD4+ and CD8+-based immunotherapy. This is of particular interest as evidence has arisen that CD4+ T cells alone can actually drive therapeutic immune responses to cancer,35 36 including neoantigen recognition by CD4+ T cells as reported by Linnemann et al.37 Although typically most BC cells do not express HLA class II, the combined application of tumor-specific CD4+ and CD8+ T cells might be crucial for a sustained antitumor response. This also highlights one of the advantages of our method as compared with direct identification of neoepitopes presented on tumor cells by mass spectrometry (MS).38 39 While MS approaches ensure the presentation of the identified epitopes on the cell surface, their sensitivity is generally too low to identify HLA class II-restricted antigens in solid tumors as they are usually only presented on bystander APCs. MS could be used in conjunction with our method as a means to confirm presentation but not necessarily to exclude predicted neoantigens.
An additional feature of our identification protocol based on clonally expanded T cells is that it allows for direct characterization of the reactive TCR and also for clones with low initial frequencies, enabling the generation of TCR transgenic T cells for adoptive transfer.
While targeting of neoepitopes allows for high-avidity T-cell responses with a low risk of autoimmunity, induction of autoreactive antibodies40 41 or cross-reactivity with the wt version is possible. It is therefore advisable to test T-cell recognition of the wt version to at least avoid cross-reactivity on use of a neoepitope-based vaccine or T-cell transfer. In regard to CARS2, which shows a limited amount of cross-reactivity against the wt epitope, we suggest that for this particular antigen, discrimination between the mutated and wt antigen be determined by differential processing,42–44 because retroviral expression of full-length CARS2 in MCF-7 cells did not lead to T-cell recognition and exposure of APCs to tumor cell lysates resulted in specific presentation of mutant CARS2.
The neoepitopes identified here appeared to be patient-specific, and while we could find no role for CARS2 or PNMAL1 in cell proliferation or survival, RBMX plays a role in response to DNA damage. It has been reported that siRNA downregulation of RBMX decreases homologous repair to 7% of control—levels comparable to those of depletion of BRCA2.45 Whether RBMX T55I actually influences functionality of RBMX merits further evaluation.
It has been described recently that neoadjuvant chemotherapy increases the number of stromal TILs.46 We report here for the first time, however, the persistence and even expansion in polyclonality of a T-cell response against a defined HLA class II-restricted neoepitope during neoadjuvant chemotherapy. It would be of interest whether the general expansion of TILs consists mainly of CD4+ T cells induced by HLA class II-restricted antigens, as on tissue damage (1) tumor antigens are released and can be presented on surrounding APCs as we demonstrated for RBMX, and (2) local inflammation upregulates HLA class II expression on the tumor cells themselves, allowing direct targeting by the neoepitope-specific T cells. Besides trastuzumab and pertuzumab, the patient analyzed here was treated with docetaxel and carboplatin, a chemotherapy known to cause immunogenic cell death,47 which is in line with the polyclonal expansion of the immune response.
Immunotherapy clinical trials in BC have not shown clinical responses as robust as those in equivalent trials in both lung and skin cancer.27 Several factors may come into play in the poor responses observed. Checkpoint inhibitor therapies are currently typically used for TMB high cancers such as melanoma or non-small cell lung carcinomas.48 49 In BC, there is a lower average TMB and, in general, the tumor environment is less immunogenic.27 Our work has shown that there are TILs that recognize HLA-restricted neoepitopes and that they are immunologically active both in TNBC and HER+ BC. However, progressive tumor growth demonstrates the inability of these TILs to eradicate the malignant cells. Recognition of these specific personalized neoepitope sequences occurs ‘upstream’ of any potential checkpoint regulation, and any clinical use of neoepitopes may need to be combined with checkpoint inhibitors. However, patients may still benefit from immune therapy by checkpoint inhibitors in combination with cellular therapy or vaccination in a personalized fashion. This further emphasizes the need for -omics analysis in combination with the algorithm for identification of target neoepitopes described here to guide future therapies.
Data availability statement
Data are available upon reasonable request. Data have largely been presented in the text; other data are available upon request.
Ethics statements
Ethics approval
Peripheral blood and tumor tissue were obtained after approval by the internal institutional review board and with informed patient consent according to the Declaration of Helsinki.
Acknowledgments
The authors thank all of the patients who participated in the Tumor-Infiltrating Lymphocytes and Genomics study, as well as Bettina Knörr, Silke Landrith and Sonja Öser for excellent technical assistance. We also thank Uwe Appelt, Stefanie Gross, Markus Mroz, and Dagmar Schönhofer (Core Unit Cell Sorting and Immuno-monitoring, Erlangen, Germany) for fluorescence activated cell sorting (FACS).
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Twitter @gulleyj1
Contributors HR, EDvdM, JB, and SK performed all the in vitro experiments for identification of neoepitopes from patients with breast cancer; AN and JZS analyzed whole-genome sequencing and RNA-sequencing data and generated binding predictions; CJV reviewed the data; SCB supervised the analysis; KN, AM, PAF, PS-S, and ANK (with SR, MWB, and JS) conceived the study; MR coordinated the biopsy sampling and DNA isolation; AH coordinated and obtained clinical data and specimen; MG assisted with in vitro studies; JS directed the analysis at NCI; JLG advised on and reviewed the analysis; KLL assisted with in vitro studies; DHH performed in vitro studies and analyzed the data; PS-S created the figures and edited the manuscript; HR, AM, MWB, MG, PAF, and ANK wrote the manuscript.
Funding We thank the Interdisciplinary Center for Clinical Research (IZKF) (project D26 to AM and PAF) and the Wilhelm-Sander Foundation (project 2019.044.1 to ANK) for funding.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.