Article Text

Abstract
Background Pharmacological autophagy enhancement constitutes a preclinically validated strategy for preventing or treating most major age-associated diseases. Driven by this consideration, we performed a high-content/high-throughput screen on 65 000 distinct compounds on a robotized fluorescence microscopy platform to identify novel autophagy inducers.
Results Here, we report the discovery of picropodophyllin (PPP) as a potent inducer of autophagic flux that acts on-target, as an inhibitor of the tyrosine kinase activity of the insulin-like growth factor-1 receptor (IGF1R). Thus, PPP lost its autophagy-stimulatory activity in cells engineered to lack IGF1R or to express a constitutively active AKT serine/threonine kinase 1 (AKT1) mutant. When administered to cancer-bearing mice, PPP improved the therapeutic efficacy of chemoimmunotherapy with a combination of immunogenic cytotoxicants and programmed cell death 1 (PDCD1, better known as PD-1) blockade. These PPP effects were lost when tumors were rendered PPP-insensitive or autophagy-incompetent. In combination with chemotherapy, PPP enhanced the infiltration of tumors by cytotoxic T lymphocytes, while reducing regulatory T cells. In human triple-negative breast cancer patients, the activating phosphorylation of IGF1R correlated with inhibited autophagy, an unfavorable local immune profile, and poor prognosis.
Conclusion Altogether, these results suggest that IGF1R may constitute a novel and druggable therapeutic target for the treatment of cancer in conjunction with chemoimmunotherapies.
- immunotherapy
- breast neoplasms
- systems biology
Data availability statement
Data are available on reasonable request. All data relevant to the study are included in the article or uploaded as online supplemental information. All data supporting the findings of this study are available within the article and its online supplemental information files and from the corresponding author on reasonable request.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Macroautophagy (which we refer to as ‘autophagy’) is a complex intracellular phenomenon in which portions of the cytoplasm including entire organelles are engulfed in autophagosomes that subsequently fuse with lysosomes for the digestion of the luminal cargo. Genetic or acquired defects in this process are linked to a broad spectrum of human diseases ranging from neoplastic to cardiometabolic diseases, inflammatory syndromes and degenerative processes affecting virtually every organ system and cell type.1 At the physiological level, autophagy is part of a general stress response that facilitates cellular and organismal adaptation to changing external conditions.2 Genetic or pharmacological stimulation of autophagy can extend the healthspan and lifespan of model organisms, thus postponing the stigmata of disease and frailty.3 4 For this reason, there is an ever-increasing interest in identifying pharmacological autophagy enhancers.5 6
In the context of cancer, autophagy plays an ambiguous role.7 8 On one hand, autophagy is required for maintaining cellular homeostasis and genomic stability,9 as well as for facilitating anticancer immunosurveillance,5 meaning that the inhibition of autophagy spurs carcinogenesis10 11 and disabled autophagy actually constitutes a poor prognostic marker in some cancers.12 On the other hand, autophagy enhances the fitness of cancer cells and allows them to avoid cell death induction in response to cytotoxicants or targeted therapies.13 Thus, the effects of autophagy modulation on tumor progression are highly context dependent. For example, in pancreatic cancer, the induction of autophagy has been suggested as a therapeutic strategy, depending on the genetic makeup of the cancer cells and immune factors.14–16 Notwithstanding the ambiguous role of autophagy in carcinogenesis, attempts have been launched to stimulate autophagy for enhancing the therapeutic activity of immunogenic chemotherapies (for instance with anthracyclines and oxaliplatin (OXA)) and immunotherapies5 17–19 In this context, it appears that autophagy has two major effects. First, it can enhance the lysosomal secretion of adenosine triphosphate (ATP) from dying cancer cells, thus enhancing the extracellular ATP-mediated recruitment of dendritic cell precursors into the tumor.5 20 Second, autophagy can contribute to the maintenance of specific T lymphocyte stem cell properties.18
Driven by the aforementioned considerations, we set out to identify novel autophagy inducers. Here, we report the identification of picropodophyllin (PPP), a cyclolignan alkaloid derived from the mayapple plant, as a potent inducer of autophagy that acts through the inhibition of insulin-like growth factor-1 receptor (IGF1R). When administered to tumor-bearing mice, PPP enhanced the efficacy of immunogenic chemotherapy combined with immunotherapy and these effects relied on the induction of autophagy in malignant cells.
Material and methods
Cell culture and chemicals
Culture media and supplements for cell culture were obtained from Life Technologies (Carlsbad, California, USA) and plastic materials came) from Greiner BioOne (Kremsmünster, Austria) and Corning (Corning, New York, USA). Rat adrenal gland PC12 cells stably expressing doxycycline-inducible Q74-GFP were cultured in Roswell Park Memorial Institute (RPMI)−1640 containing 5% fetal bovine serum (FBS) and 10% horse serum.21 Human neuroglioma H4 cells, human osteosarcoma U2OS cells, MCA205 murine fibrosarcoma and all the other cells were maintained in Dulbecco’s modified Eagle’s medium, supplemented with 10% (v/v) FBS, 10 U/ mL penicillin sodium and 10 µg/mL streptomycin sulfate at 37°C in a humidified atmosphere with 5% CO2. Transcription factor EB (TFEB)-deficient (TFEB−/−), transcription factor 3 (TFE3)-deficient (TFE3−/−), TFEB and TFE3-double deficient (TFE DKO), autophagy related 5 (ATG5)-deficient (ATG5−/−), and eukaryotic translation initiation factor 2 alpha kinase 3 (EIF2AK3, better known as PERK)-deficient (PERK−/−) U2OS-green fluorescent protein (GFP)-microtubule associated protein 1 light chain 3 beta (MAP1LC3B, better known as LC3) cell lines and TFEB and TFE3-double deficient (TFE DKO) in H4-GFP-LC3 cells were generated by means of the CRISPR/Cas-mediated genome editing, as per manufacturer’s recommendations.17 U2OS cells stably expressing RFP-LC3 bearing a mutant non-phosphorylation of eukaryotic initiation factor 2α (EIF2A) (eIF2αS51A) were constructed using the CRISPR-Cas9 technology as previously detailed.22 In addition, U2OS cells stably expressing GFP-TFEB were generated by our group in the past.17 23 MCA205 cells stably expressing shRNAs interfering with the expression of TFE3/TFEB or ATG5, and overexpressing CD39 and a mutant phosphorylation AKTT308D/S473D were also constructed as the manufacturer.5 17 24 The polyphenol library and PPP were purchased from TargetMol (Boston, MA, USA); Torin1 (TOR), bafilomycin A1, mitoxantrone (MTX) and OXA were obtained from Sigma-Aldrich (St. Louis, MI, USA). Recombinant IGF1 were obtained from PROSPECBIO (CYT-216, Rehovot, Israel). Linsitinib (HY-10191) were obtained from MedChemExpress (Shanghai, China).
High-content microscopy
Human osteosarcoma U2OS and neuroglioma H4 cells stably expressing GFP-LC3, GFP-TFEB, GFP-AKT, GFP-AKT R25C, RFP-GFP-LC3-tandem or RFP-GFP-p62-tandem reporter and rat adrenal gland PC12 cells stably expressing doxycycline-inducible Q74-GFP were seeded in 384-well black imaging plates at a density of 2000 cells per well and allowed to adapt for overnight. Cells were treated with the indicated agents, then fixed with 3.7% paraformaldehyde (PFA, w/v in PBS) (F8775, Sigma-Aldrich) at 4℃ overnight and stained with 1 µg/mL Hoechst 33 342 in PBS. Subsequently, the fixative was exchanged to PBS and the plates were analyzed by automated microscopy. Image acquisition was performed using an ImageXpress Micro XL automated microscope (Molecular Devices, Sunnyvale, California, USA) equipped with a 20 X PlanApo objective (Nikon, Tokyo, Japan), followed by automated image processing with the custom module editor within the MetaXpress software (Molecular Devices). At least four view fields were acquired per well, and experiments involved at least triplicate assessment. Cellular regions of interest, cytoplasm and nucleus, were defined and segmented by using the MetaXpress software (Molecular Devices). After exclusion of cellular debris and dead cells from the dataset, parameters of interest were normalized, statistically evaluated, and graphically depicted with R software. Using R, images were extracted and pixel intensities scaled to be visible (in the same extent for all images of a given experiment). Cell viability was assessed as described before.25
Immunofluorescence
Human osteosarcoma U2OS cells were treated for 6 hour to detect TFE3, then were fixed with 3.7% PFA at 4°C overnight. For staining, fixed cells were permeabilized with 0.1% Triton X100 on ice, and blocked with 5% bovine serum albumin (BSA, w/v in PBS) for 1 hour. Next, cells were incubated with antibodies specific to TFE3 (#ab93808, 1:400, Abcam) at 4°C overnight. After washing with PBS twice, the cells were incubated with AlexaFluor conjugates (Thermo Fisher Scientific) against the primary antibody for 2 hour at ambient temperature. Then cells were washed and imaged by automated fluorescence microscopy as described above. The nuclear intensity of TFE3 was measured and normalized to Ctrl.
Immunohistochemistry
A total of 49 formalin-fixed paraffin-embedded; tissue samples of triple-negative breast cancer were obtained from Renmin Hospital of Wuhan University, People’s Republic of China (online supplemental table S1). Patients did not receive financial compensation. Patients with at least 5-year follow-up were included in this retrospective study. All methods were performed in accordance with the relevant guidelines and local regulations. Immunohistochemical staining was performed by an automatic staining machine (Leica Bond3) or manually processed. Sections were dehydrated and antigenic epitopes were retrieved using a 10 mM citrate buffer and microwaving for 10 min. Specimen were then incubated with for LC3B (#3868, 1:2000, Cell Signaling Technology), phospho-IGF1R (#orb503127, 1:100, Biorbyt, UK), CD163 (#93498, 1:500, Cell Signaling Technology), Foxp3 ((#98377, 1:100, Cell Signaling Technology), CD8 (#70306, 1:200, Cell Signaling Technology). Primary antibody staining was detected by peroxidase-conjugated IgG (1:500 diluted P0448, Dako, Glostrup, Denmark). Positive cells were counted on nine randomly chosen tumor areas at 200 magnifications in a double blinded fashion. Quantitative analysis was performed using ImageJ software as described.26 The receiver operating characteristic analysis was used to determine the optimal cut-off values of all proteins expression levels for survival rate.
Supplemental material
Immunoblotting
Tissues (~30 mg) were dissociated in Precellys lysing tubes (#CK28_2 mL, Bertin Technologies SAS, France) containing 1 mL of radio immunoprecipitation assay buffer (RIPA) lysis buffer (#89901, Invitrogen, Carlsbad, California, USA) by using the Precellys 24 homogenizer (Bertin Technologies SAS) at 6500 rpm for 5 min, followed by spinning at 14 000 g for 15 min to collect the supernatant that contains soluble proteins. For cells, the protein extracts were dissolved in RIPA buffer and obtained by ultrasonicating for 3×10 s and centrifuging at 12 000 g for 15 min to collect the supernatant that contains soluble proteins. Protein concentration was measured by means of by the BCA Assay (Bio-Rad, Hercules, California, USA). The protein solution was mixed with 4X loading buffer (# NP0008, Invitrogen), and denatured at 100°C for 15 min before subjected to western blotting. The total protein (~30 µg) were resolved on 4%–12% NuPAGE Bis-Tris protein gels (#NP0322, Thermo Fisher Scientific) and electrotransferred to 0.2 µM polyvinylidene fluoride membranes (#1620177, Bio-Rad). The membranes were blocked with 0.05% Tween 20 (#P9416, Sigma Aldrich) v:v in Tris-buffered saline (TBS) (TBST) (#ET220, Euromedex) supplemented with 5% non-fat powdered milk (w:v in TBS), followed by an overnight incubation at 4°C with primary antibodies specific for LC3B (#2775, 1:1000, Cell Signaling Technology), HA (#ROAHAHA, 1:1000, Sigma-Aldrich), phospho-P70 (Thr389) (#9234, 1:1000, Cell Signaling Technology), P70 (#9202, 1:1000, Cell Signaling Technology), phospho-IGF1R (Tyr1135/1136) (#3024, 1:1000, Cell Signaling Technology), IGF1R (#9750, 1:1000, Cell Signaling Technology), pTFEB (Ser211) (#37681, 1:1000, Cell Signaling Technology),TFEB (#4240, 1:1000, Cell Signaling Technology), TFE3 (#ab93808, 1:1000, Abcam), phospho-AKT (Ser473) (#4060, Cell Signaling Technology), AKT (#4691, Cell Signaling Technology), phospho-mechanistic target of rapamycin (mTOR) (Ser2448) (#2971, Cell Signaling Technology), mTOR (#2983, Cell Signaling Technology), H3 (#9715, 1:1000, Cell Signaling Technology). Membranes were washed three times for 10 min with TBST before incubation with HRP-conjugated goat-anti-rabbit secondary antibody (CliniScience) for 2 hours at room temperature. Then the membranes were washed again and subjected to chemiluminescence detection with the Amersham ECL Prime detection reagent kit (GE Healthcare, Piscataway, New Jersey, USA) on an ImageQuant LAS 4000 software-assisted imager. The membranes were stripped and reprobed for loading control with anti-actin (# ab 20727, 1:10000, Abcam), anit-glyceraldeyde-3-phosphate dehydrogenase (GAPDH)-specific antibody (#2118, 1:5000, Cell Signaling Technology) or anti-vinculin antibody (#13901, 1:1000, Cell Signaling Technology). Quantification was performed by densitometry using the Image J software.
Plasmid transfection
Cells were seeded, let adhere for 24 hours, and following transfected with the CRISPR-Cas9 plasmid U6gRNA-Cas9-2A-RFP targeting IGF1R (50-ATGATGCGATT CTTCGACG-30) or a plasmid coding for AKT carrying the T308D/S473D mutation (#14751, Addgene, Watertown, Massachusetts, USA), according to the manufacturer’s advice.
Nuclear extraction experiment
U2OS-GFP-LC3 cells were collected and processed with the Nuclear Extraction Kit (#ab113474, Abcam) following the manufacturer’s methods. The GAPDH antibody (#2118, 1:1000, Cell Signaling Technology) was used as cytoplasmic control, and H3 (#9715, 1:1000, Cell Signaling Technology) was selected as nuclear loading and quality control.
Detection of protein deacetylation
U2OS-GFP-LC3 stable expressing cells (~2000 cells/well) were seeded in 384-well microplates overnight. After experimental treatments, cells were fixed with 3.7% PFA containing 1 µg/mL Hoechst 33 342 overnight at 4°C. Thereafter, cells were incubated with an antibody specific for acetyl-alpha-tubulin (#5335, 1:500, Cell Signaling Technology) in 5% bovine serum albumin (BSA, w/v in PBS) for 1 hour to block non-specific binding sites and acetylated tubulins, followed by overnight incubation at 4°C with specific antibody to detect acetylated proteins at lysines (#623402, 1:400, BioLegend, San Diego, California, USA). After washing three times with PBS, cells were incubated in AlexaFluor-568 conjugates (Life Technologies) against the primary antibody for 2 hours at room temperature. Fluorescent images were acquired and analyzed as described before.
ATP release assays
Intracellular ATP levels were detected by quinacrine staining (Calbiochem), subsequently the images of quinacrine were observed by means of high-content microscopy and the cytoplasmic intensity of quinacrine was quantitated as described above. Extracellular ATP levels were measured by the ENLITEN ATP Assay System Bioluminescence Detection Kit (Promega, Madison, Michigan, USA; #FF2000) following the manufacturer’s instructions. Luminescence was measured by means of a Paradigm I3 multimode plate reader (Molecular Devices).
Animal model
The animal experiments were approved by the Gustave Roussy ethics committee with the project number: 24 771–2020032413235413, and all procedures were performed under the governmental and institutional guidelines and regulations. All mice were maintained in a temperature-controlled and pathogen-free environment with 12 hours light/dark cycles, with food and water ad libitum.
For tumor growth experiments, 7 week-old female wild-type C57BL/6 mice or athymic female nude mice (nu/nu) were obtained from Envigo, France (Envigo, Huntingdon, UK). AT3 wild-type, TC1 wild-type, MCA205 wild-type (WT), overexpressing a CD39 transgene (CD39+) or a constitutive active variant of AKT T308D/S473D (4×105), MCA205 with ATG5 knockout (WT, 6×105) cells were subcutaneously injected into C57BL/6 hosts. When tumors became palpable, mice were treated with 20 mg/kg PPP dissolved in corn oil (Sigma-Aldrich), 25 mg/kg linsitinib (dissolved in 5% DMSO, 10% PEG300, 5% Tween 80, 80% PBS) or an equivalent volume of vehicle alone or in combination with 10 mg/kg (OXA, Sigma-Aldrich) or 200 µg anti-PD-1 antibody (Clone 29 F.1A12, BioXcell, West Lebanon, New Hampshire, USA), by intraperitoneal (i.p.) injection. On the following days, mice well-being and tumor growth were monitored and documented. Animals were sacrificed when tumor size reached endpoint or signs of obvious discomfort were observed following the EU Directive 63/2010 and our Ethical Committee advice.
Ex vivo–phenotyping of the tumor immune infiltrate
Tumors were harvested, weighed and transferred on ice into gentleMACS C tubes (Miltenyi Biotec, Bergisch Gladbach, Germany) containing 1 mL of RPMI medium. Tumors were dissociated first mechanically with scissors, then enzymatically using the mouse tumor dissociation kit (Miltenyi Biotec) and a GentleMACS Octo Dissociator according to the manufacturer’s instructions. The dissociated bulk tumor cell suspension was resuspended in RPMI1640, sequentially passed through 70 µm MACS Smart-Strainer (Miltenyi Biotec) and washed twice with PBS. Finally, bulk tumor cells were homogenized in PBS at a concentration corresponding to 250 mg of the initial tumor weight per mL. Prior to staining of tumor-infiltrating lymphocytes (TILs) for flow cytometry analysis, samples (~50 mg) were incubated with LIVE/DEAD Fixable Yellow Dead Cell dye (Thermo Fisher Scientific) to discriminate viable cells from damaged cells. Fc receptors were blocked with anti-mouse CD16/CD32 (clone 2.4G2, Mouse BD Fc Block, BD Pharmingen) before staining with fluorescent-labeled antibodies targeting T-cell surface markers. Surface staining of murine immune cell populations infiltrating the tumor was performed with the following fluorochrome-conjugated antibodies: anti-CD45-AF700, anti-CD3-BV421, anti-CD8-PE, anti-CD4-Percp.Cy5.5, anti-CD25-PE/Cy7, and anti-PD-1-APC/Cy7 (BioLegend). Then, cells were fixed and permeabilized in eBioscience Foxp3/Transcription Factor Staining Buffer (Thermo Fisher Scientific) and stained for intracellular Foxp3. Finally, stained samples were run through a BD LSR II flow cytometer. Data were acquired using BD FACSDiva software (BD Biosciences) and analyzed using FlowJo software (TreeStar). Absolute counts of leukocytes and tumor cells were normalized considering the following parameters: weight of the harvested tumor and total volume of the dissociated tumor cell suspension (cell concentration typically set to 250 mg/mL in PBS), proportion of the whole cell suspension and proportion of the cell suspension used for cytometry.
Statistical analysis
Unless otherwise mentioned, data are reported as means±SD of triplicate determinations and experiments were repeated at least three yielding similar results. Statistical significance was assessed by Student’s t-test. TumGrowth and GraphPad were used to analyze in vivo data arising from murine models.27 The Kaplan-Meier method was used to calculate patient survival and the log-rank test was used to assess the heterogeneity for each prognostic factor. Univariate Cox proportional hazard regressions were used to obtain HRs and their respective 95% CIs to show the strength of the estimated relative risks. Pearson’s correlation was used to evaluate the correlation. P values of 0.05 or less were considered to denote significance (*p<0.05; **p<0.01; ***p<0.001; ns, not significant).
Results
Identification of PPP as a potent inducer of autophagic flux
Human osteosarcoma U2OS cells, which are often used as biosensor cell lines,28 were stably transduced with a GFP-LC3 fusion protein and subjected to rounds of selection (to ensure homogeneous GFP-LC3 expression in most cells) and quality control (to ensure adequate formation of GFP-LC3 puncta in response to autophagy induction by the positive control torin1). In a robotized high-content/high throughput screening platform,28 such cells were exposed to ~65 000 compounds (all tested at 10 µM) to identify agents that induce GFP-LC3 puncta at least as efficiently as the positive control (the MTOR inhibitor torin1 used at 300 nM) without cell loss (figure 1A,B). About 200 compounds fulfilled this criterion. We chose to follow-up (PPP, also known as AXL1717) for two reasons. First, in a validation experiment re-evaluating the top 400 compounds from the primary screen, in another smaller screen focusing on a library of polyphenols involving ~1000 compounds and in further low throughout experiments PPP was found to induce autophagic flux in cells expressing distinct flux biosensors, namely a RFP-GFP-LC3 (online supplemental figure S1A), a Q74-GFP construct (online supplemental figure S1B), a RFP-ATG4-GFP-LC3ΔG (online supplemental figure S2A,B), mCherry-GFP-p62 (online supplemental figure S2C,D) and HA-tagged p62 (online supplemental figure S2E-G), confirming that PPP indeed induces autophagic flux. Second, PPP has been described as an inhibitor of IGF1R29 30 with potent antitumor effects in preclinical models31–33 and acceptable toxicity in clinical phase I and II trials.34
Supplemental material
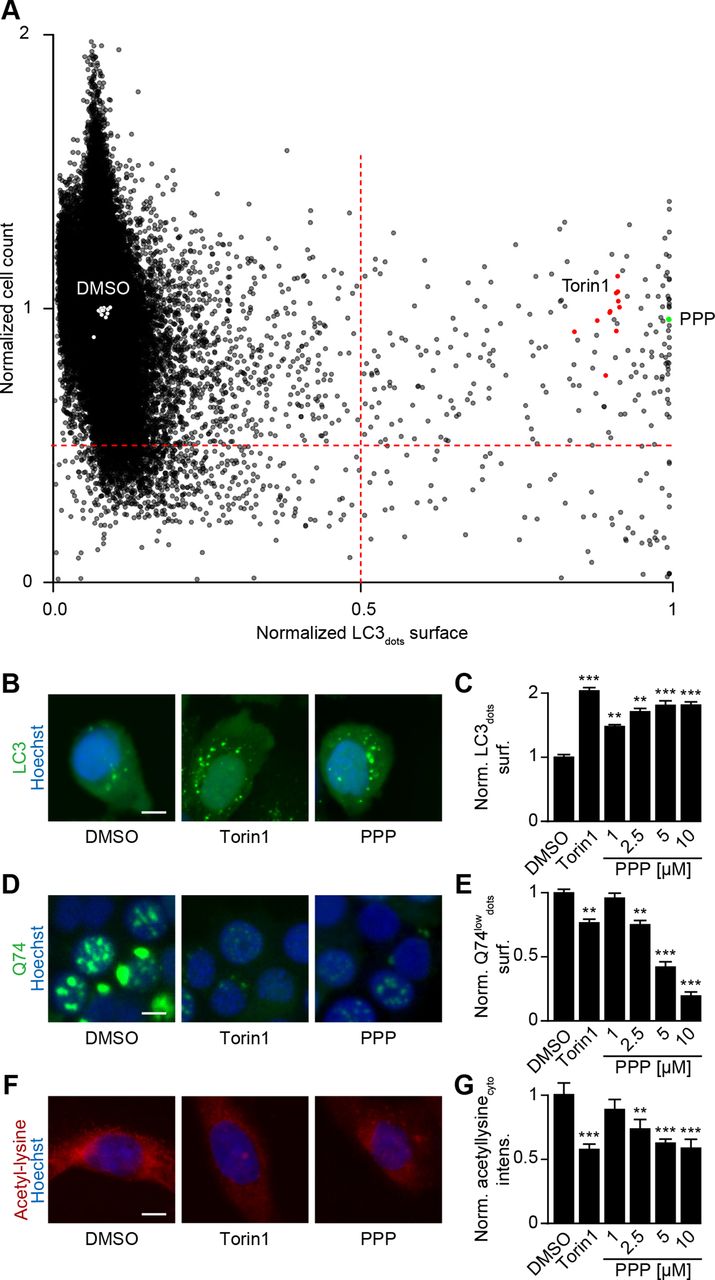
Identification of picropodophyllin (PPP) as autophagy inducer. (A) Human osteosarcoma U2OS stably expressing GFP-LC3 cells and were treated with a chemical library of 65 000 diverse compounds (10 μM) for 6 hours. Torin 1 at 300 nM was used as positive control for autophagy induction. The number of GFP-LC3 positive puncta was measured as a proxy for autophagy and the number of cells that confer a regular nuclear phenotype was assessed as an indicator for viability. We selected the agents that dramatically increase the expression of GFP-LC3 and whose viability is >0.5 times of its expression in control group, are potential autophagy activators. Data were normalized to controls, depicted as means of each campaign. (B, C) U2OS cells stably expressing GFP-LC3 were treated with PPP (1, 2.5, 5, 10 μM) and torin1 (300 nm) was used as control. the surface of GFP-LC3 positive puncta was measured as a proxy for autophagy (C) and representative images are depicted in B. scale bar equals 10 µm. (D, E) Rat adrenal gland PC12 cells expressing an inducible variant of Q74-GFP were treated with doxycycline (1 μg/mL) for 8 hours for the induction of Q74 expression. Then the medium was changed and PPP was added at 1, 2.5, 5 and 10 μM. Torin1 (300 nm) was used as positive control. data is depicted as barchart in E and representative images are shown in D. Scale bar equals 10 µm. (F, G) U2OS cells were treated with PPP and the positive control torin1 (300 nm) for 6 hours, followed by the incubation with specific antibodies to block acetylated tubulin. Thereafter, immunofluorescence was conducted with antibodies against acetylated lysine residues and appropriate AlexaFluor-conjugated secondary antibodies. representative images of acetylation are shown in F, and acetylation intensity in the cytoplasm was measured in G. Scale bar equals 10 µm. Data are means±SD of three replicates (**p<0.01, ***p<0.001 vs DMSO/Ctr, Student’s t-test).
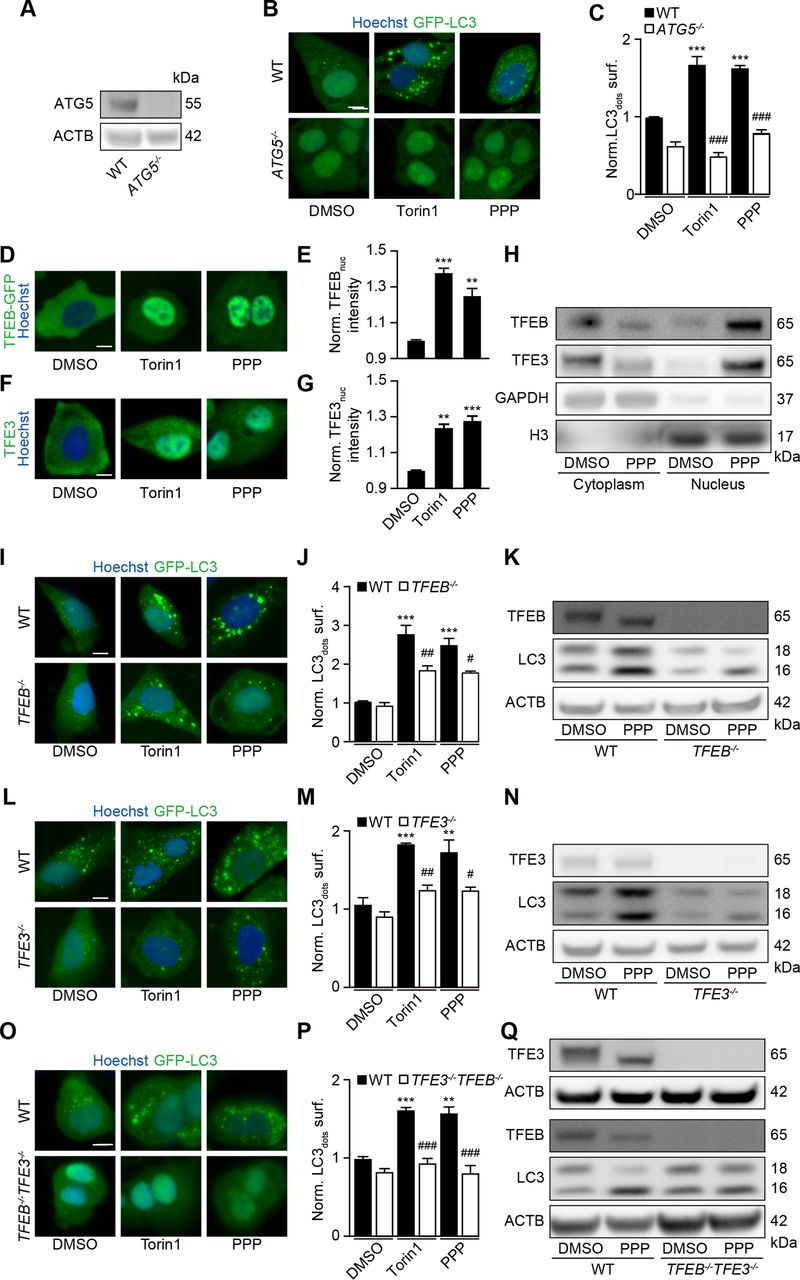
Further experiments confirmed that PPP induced GFP-LC3 puncta (figure 1B,C) and reduced the abundance of the autophagic substrate Q74-GFP (figure 1D,E) in a dose-dependent fashion. In addition, PPP reduced cytoplasmic protein acetylation, as determined by quantitative immunofluorescence staining (figure 1F,G). These effects were detectable at PPP concentrations of 2.5 to 10 µM, which did not affect cellular viability (online supplemental figure S2H). Of note, induction of GFP-LC3 puncta or LC3-II by PPP was lost in ATG5-/- U2OS cells (figure 2A–C), was accompanied by the translocation of TFEB and TFE3 into nuclei (figure 2D–H, (online supplemental figure S3A,B), was partially reduced in TFEB-/- or TFE3-/- cells, and strongly inhibited in TFEB-/- TFE3-/- double-knockout cells (figure 2I–Q, (online supplemental figure S3C–E). Hence, PPP stimulates autophagic flux through a canonical, ATG5 and TFEB/TFE3-dependent pathway.
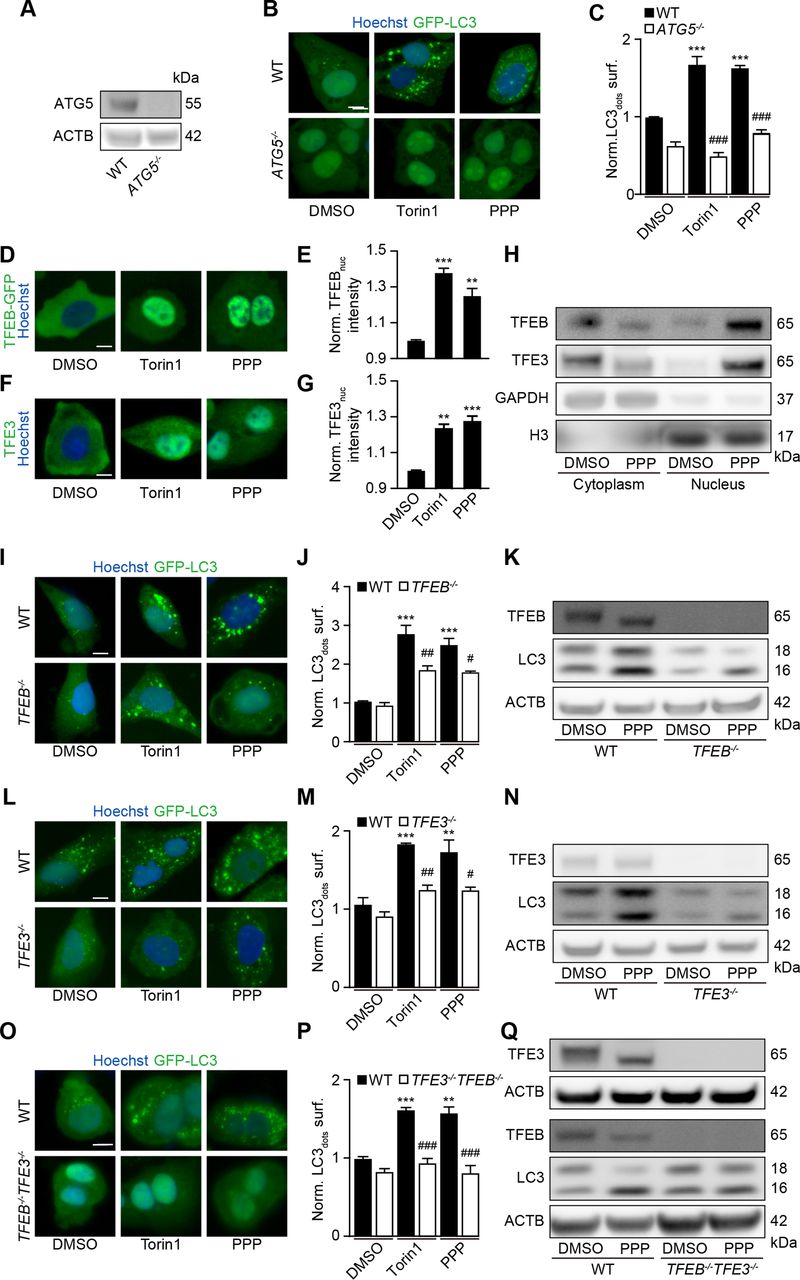
TFEB and TFE3 mediate PPP-induced autophagy. (A–C) U2OS-GFP-LC3 wild-type (WT) or ATG5 knockout cells were treated with PPP (10 μM) or torin1 (300 nM) for 6 hours. Then the cells were fixed and GFP-LC3 dots were quantified. Scale bar equals 10 μm. Data are means ± SD of four replicates ***p<0.001 vs untreated control; ###P<0.001 vs WT; Tukey’s multiple comparisons test). (D, E) U2OS cells stably expressing GFP-TFEB fusion protein were treated with PPP (10 μM) for 16 hours and torin1 was used as positive control. nuclear GFP intensities were measured (E) and representative images are shown in D. Scale bar equals 10 µm. Data are means±SD of three replicates (**p<0.01, ***p<0.001 vs DMSO/Ctr, Student’s t-test). (F, G) U2OS cells were treated with PPP (10 μM) for 16 hours and torin1 was used as positive control and then endogenous TFE3 translocation was assessed by immunostaining (F) and TFE3 nuclear intensities are depicted (G). Scale bar equals 10 µm. Data are means±SD of three replicates (**p<0.01, ***p<0.001 vs DMSO/Ctr, Student’s t-test). (H) U2OS cells were treated with PPP (10 μM) for 16 hours or were left untreated. Cytoplasmic and nuclear fractions were separated and assessed for nuclear translocation of TFEB and TFE3 by SDS-PAGE. band intensities of TFEB, TFE3, GAPDH and H3 were assessed and their ratio (TFEB or TFE3/GAPDH, and TFEB or TFE3/H3) was calculated (online supplemental figure S3). (I–Q) U2OS-GFP-LC3 cells WT or single as well as double knockout for TFEB and TFE3 were treated with PPP (10 μM) or torin1 for 16 H. LC3 II expression and TFEB/TFE3 deficiency by knockout were checked by SDS-PAGE and parallel immunoblot (K, N, Q). Band intensities of LC3-II and β-actin (ACTB) were assessed, and their ratio (LC3-II/actin) was calculated (online supplemental figure S3). representative images are shown in (I, L, O), and GFP-LC3 dots were quantified as indicator of autophagy (J, M, P). Scale bar equals 10 μm. Data are means±SD of four replicates (***p<0.001 vs untreated control; #P<0.05, ##P<0.01, ###P<0.001 vs WT; Tukey’s multiple comparisons test). PPP, picropodophyllin SDS-PAGE, sodium dodecyl sulfate polyacrylamide gel electrophoresis.
PPP induces autophagy through IGF1R inhibition
PPP is known to block the tyrosine kinase activity of IGF1R.29 30 Accordingly, PPP-induced GFP-LC3 puncta were not prevented by addition of IGF1 (figure 3A,B), the agonist ligand of IGF1R. PPP efficiently blocked IGF1-induced IGF1R autophosphorylation, the activating phosphorylation of protein kinase B (best known as AKT), the phosphorylation of MTOR, and the activity of MTOR complex 1 (MTORC1), evaluated by assessing the phosphorylation of the MTORC1 substrates p70S6K and TFEB (figure 3C). Knock-out of IGF1R rendered U2OS cells resistant to the autophagy-inducing effects of PPP (figure 3D–F, online supplemental figure S3F). Conversely, transgenic expression of a constitutively active AKT mutant (T308D/S473D)35 abolished the proautophagic activity of PPP (figure 3G–I, online supplemental figure S4A,B). IGF1 stimulated the membrane translocation of a GFP-AKT fusion protein (but not that of a GFP-AKTR25C mutant that fails to translocate to phosphatidyl inositol-rich membranes),36 and this effect was blocked by PPP (figure 3J,K). Of note, on i.p. injection into mice, PPP inhibited phosphorylation of IGF1, AKT and mTOR, P70S6K and enhanced the abundancy of LC3-II in the liver and in the heart (online supplemental figure S5A-K). LC3-II was also enhanced in the brain (online supplemental figure S5L,M). Altogether, these results indicate that PPP induces autophagy through the inhibition of IGF1R and its downstream signals AKT and MTORC1.

PPP induces autophagy via IGF1R/AKT signaling. (A, B) U2OS-GFP-LC3 cells were incubated in the absence of FBS overnight and were further treated with PPP (10 μM) or torin1 (300 nm) in the presence or absence of IGF1 (10 nm) for 6 hours. After fixation, GFP-LC3 dots were quantified in B. Scale bar equals 10 μm. Data are means±SD of three replicates (**P<0.01, vs DMSO/Ctr, Student’s t-test). (C) Immunoblot exploration of the IGF1R signal pathway. After U2OS cells were incubated in the absence of FBS overnight, then the cells were treated with PPP (10 μM) together with or without IGF1 (10 nm) for 6 hours. Proteins were separated by SDS-PAGE and parallel immunoblots of pIGF1R (Tyr1135/1136), IGF1R, pAkt (Ser473), Akt, pmTOR (Ser2448), mTOR, pp70 (THR389), p70, pTFEB (Ser211), TFEB, LC3-II and were performed in parallel instances and β-actin (ACTB) was used as loading control (C). (D, E) U2OS wild-type (WT) or IGF1R knockout cells were treated with PPP (10 μM) for 6 H. SDS-PAGE and immunoblots of IGF1R, LC3 and ACTB were performed (D), band intensities of LC3-II and ACTB were assessed, and their ratio (LC3-II/ACTB) was calculated (online supplemental figure S4). In parallel U2OS-GFP-LC3 WT or IGF1R knockout cells were treated with PPP (10 μM) for 6 hours then the cells were fixed and GFP-LC3 dots were quantified by microscopy (F). Representative images are shown in E and scale bar equals 10 μm. Data are means±SD of three replicates (***P<0.001 vs untreated control; ##P<0.01 vs WT; Tukey’s multiple comparisons test). (G–I) U2OS cells were transfected with constitutive active AKTT308D/S473D and were treated with PPP (10 μM) for 6 hours. SDS-PAGE and immunoblots of pAKT, AKT, LC3 and ACTB were performed (G), band intensities of LC3-II and ACTB were assessed, and their ratio (LC3-II/ACTB) was calculated (online supplemental figure S4). In parallel U2OS-GFP-LC3 WT or AKTT308D/S473D-expressing cells were treated with PPP (10 μM) for 6 H then the cells were fixed and GFP-LC3 dots were quantified by microscopy (I). Representative images are shown in H and scale bar equals 10 μm. Data are means±SD of three replicates (**P<0.01 vs untreated control; ##P<0.01 vs WT; Tukey’s multiple comparisons test). (J–K) U2OS cells stably expressing GFP-AKT or GFP-AKTR25C were incubated in absence of FBS overnight, then the cells were treated with IGF1 (10 nm) or PPP (10 μM) combined with IGF1 (10 nm). After 5 min, the membrane translocation of GFP-AKT was detected by microscopy (J) and the membrane intensity of AKT was measured (K). Scale bar equals 10 μm. Data are means±SD of three replicates (***P<0.001 vs DMSO/Ctr, Student’s t-test). IGF1, FBS, fetal bovine serum; IGF1, insulin-like growth factor-1; PPP, picropodophyllin; SDS_PAGE, sodium dodecyl sulfate polyacrylamide gel electrophoresis.
IGF1R activation as a negative prognostic factor in breast cancer
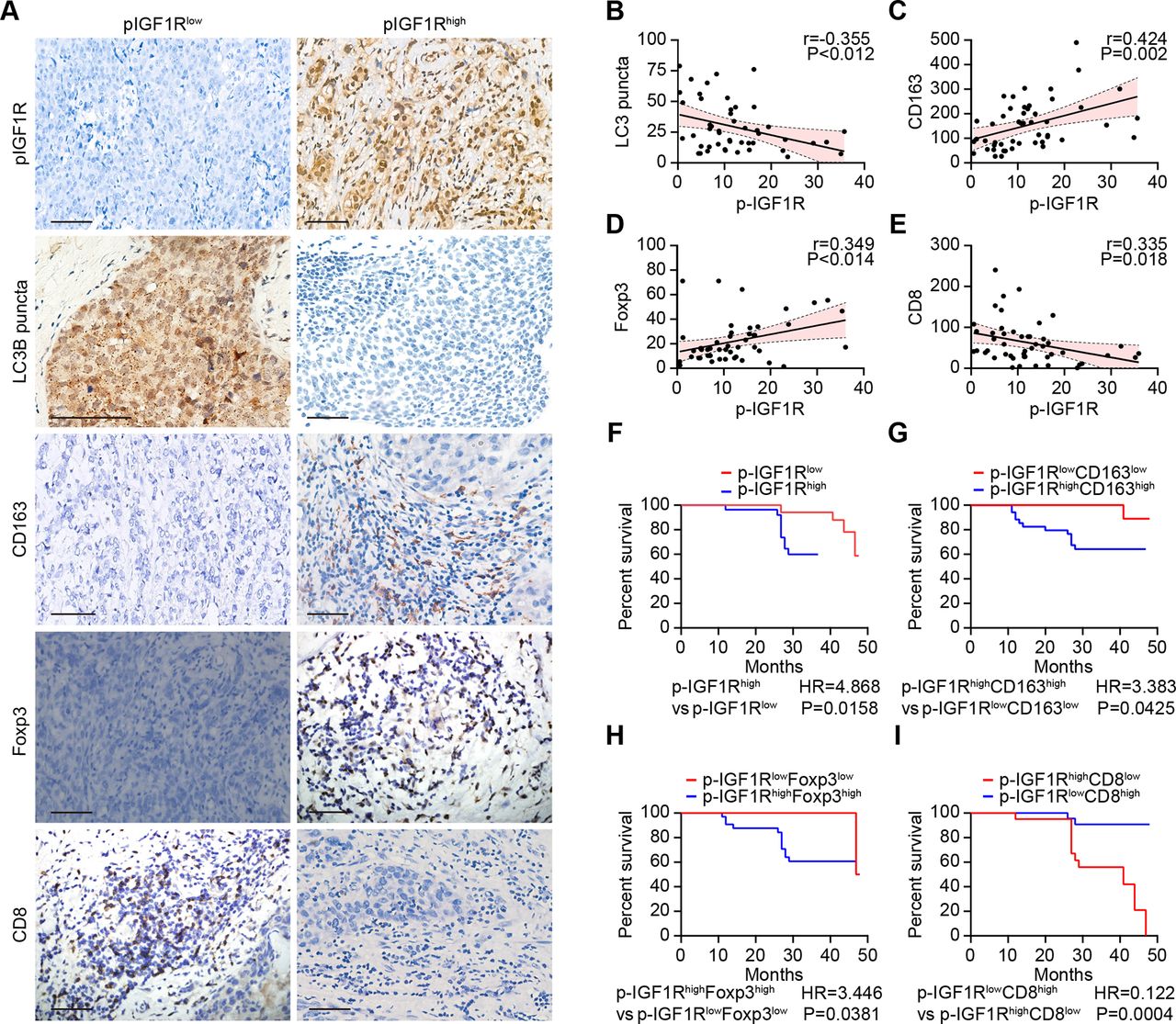
A paucity in LC3B puncta, reflecting disabled autophagy in malignant cells, is associated with dismal prognosis of breast cancer, as well as an unfavorable ratio of tumor infiltrating CD8+ cytotoxic T lymphocytes (CTLs) over FOXP3+ regulatory T cells (Tregs) or CD163+ tumor-associated macrophages (TAMs).12 37 In a series of 49 stage negative breast cancer patients treated by surgical resection (online supplemental table S1), the activating phosphorylation of IGF1R detectable by immunohistochemistry correlated negatively with the density of LC3 puncta and CD8+ CTLs but positively with FOXP3+ Tregs and CD163+ TAMs (figure 4A–E). Phosphorylation of IGF1R above the median level was associated with poor overall survival compared with low IGF1R phosphorylation (figure 4F). The risk stratification of breast cancer patients could be further improved by including the characteristics of the immune infiltrate. Thus, patients with phospho-IGF1Rhigh CD8low, phospho-IGF1Rhigh FOXP3high and phospho-IGF1Rhigh CD163high breast cancer exhibited the worst overall survival (figure 4G–I). Altogether, these results indicate that activation of IGF1R signaling might affect autophagy as well as breast cancer immunosurveillance in a clinically relevant fashion. We, therefore, decided to evaluate the effects of IGF1 inhibitors on cancer immunosurveillance.
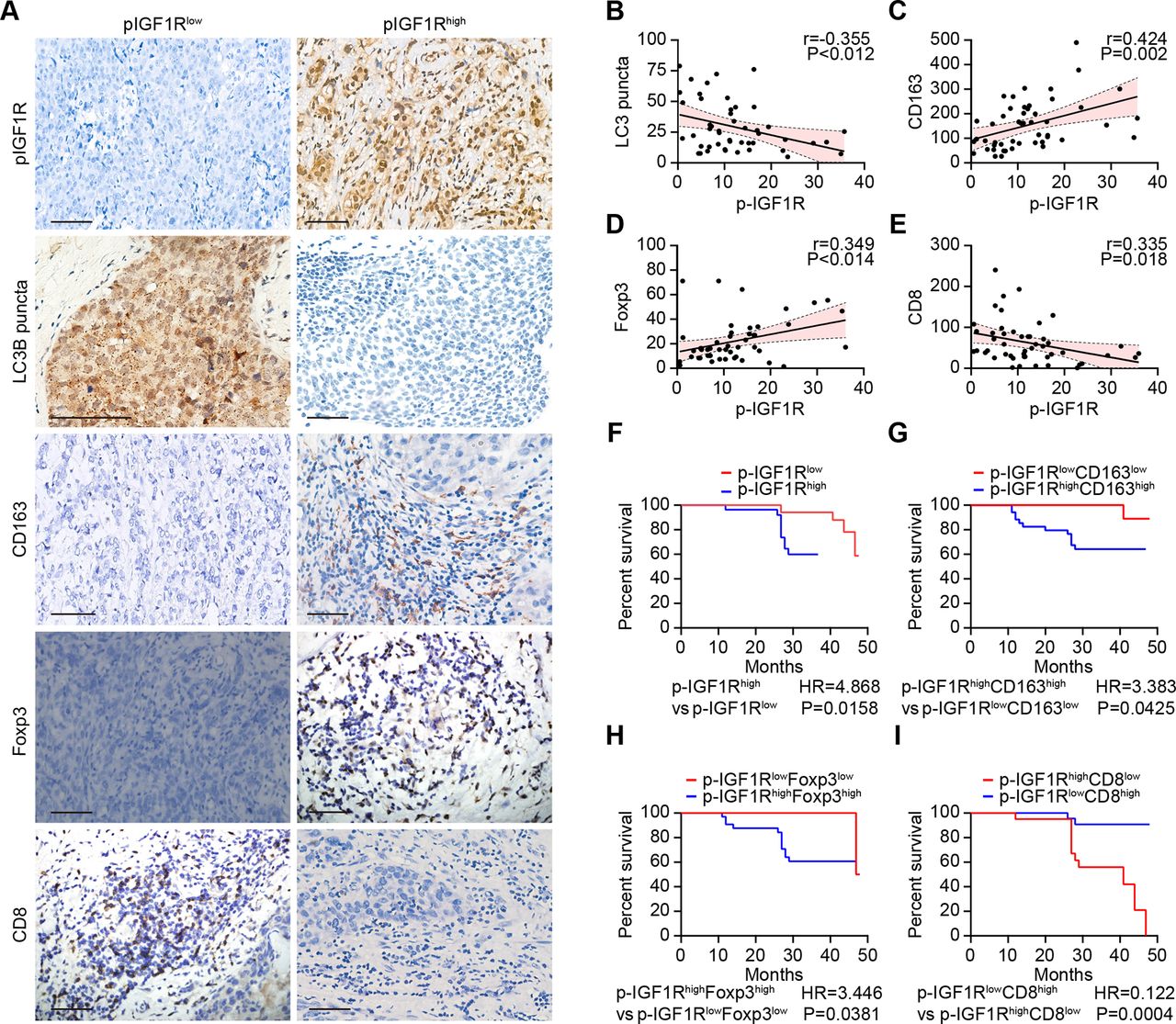
IGF1R signaling correlates with immunosuppressive markers and decreased survival in breast cancer. (A–I) The expression of CD163, FOXP3 and CD8 as well as the phosphorylation of IGF1R and the dot formation of LC3 were quantified in paraffin-embedded biopsies obtained from 49 triple-negative breast cancer patients by ImageJ after staining with specific antibodies. Representative images of phosphorylated-IGF1R, dotted LC3B, and CD163, FOXP3 and CD8 expression are shown in A. The scale bar indicates 100 μm. Correlation analyses (determined by the Spearman method) of the analyzed parameters for each patient are depicted in B–E. (F–I) Kaplan-Meier survival analysis of patients with biomarker-positive and biomarker-negative immunohistochemistry staining. IGF1R, insulin-like growth factor-1 receptor.
Immune response-amplifying effects of IGF1R inhibition
Autophagy induction can increase the immunogenicity of anthracycline or OXA-induced cell death by favoring the release of ATP.5 38 Accordingly, addition of PPP to U2OS cells enhanced the release of ATP from cells, causing a diminution of ATP-sensitive quinacrine fluorescence (figure 5A,B) and an increase in ATP concentrations in culture media in response to low-dose MTX. PPP similarly stimulated the release of ATP in response to low-dose OXA (online supplemental figure S6A,B). The low-dose chemotherapy-induced ATP release was inhibited by knockout of ATG5, knockout of eIF2α kinase 3 EIF2AK3 (best known as PERK, which is required for autophagy induction by MTX),22 the S51A knockin mutation in eIF2α that renders it unphosphorylable (and blocks autophagy induction),22 39 as well as the expression of a constitutively active AKT mutant, AKTT308D/S473D (figure 5C–F).
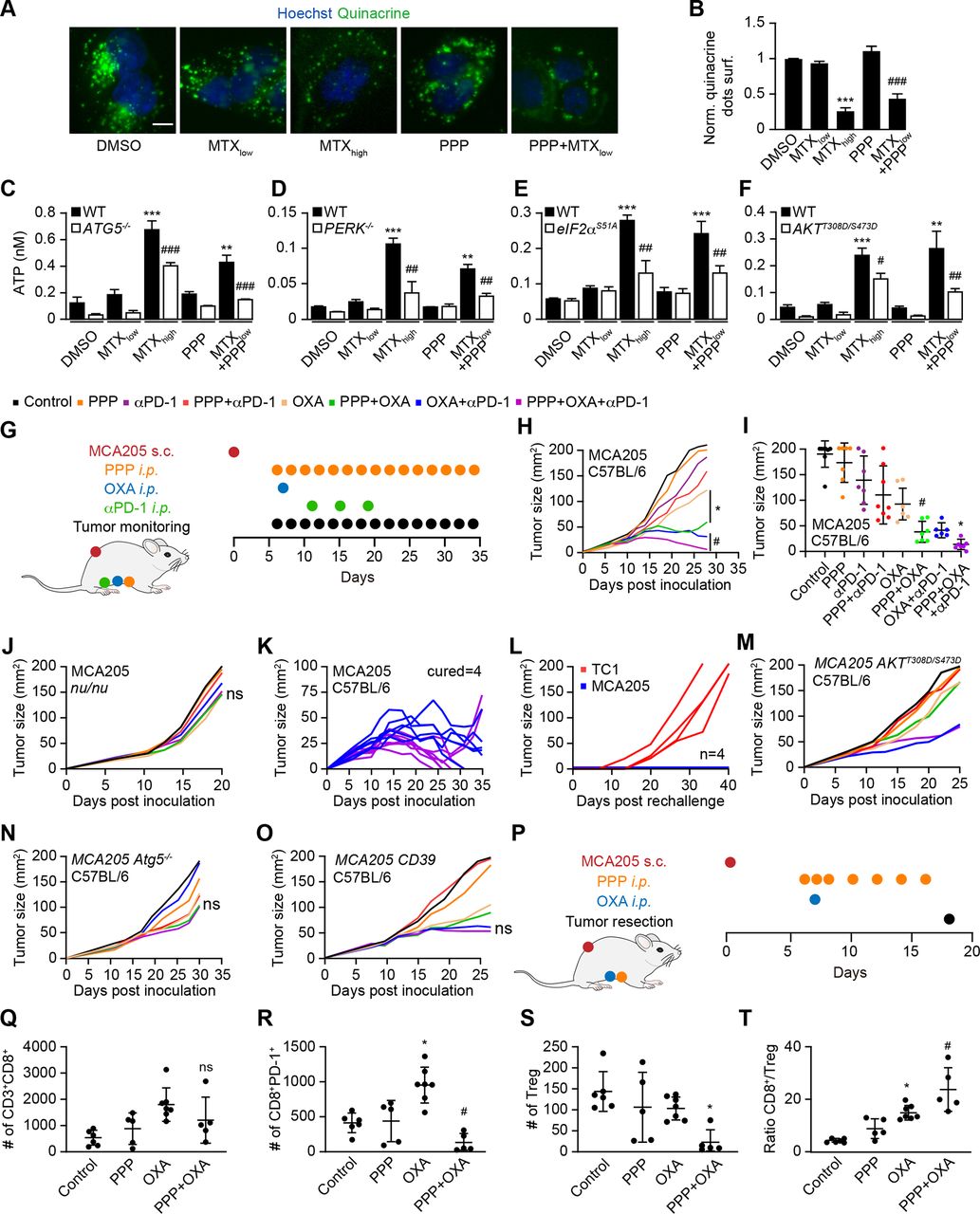
PPP improves autophagy-dependent anticancer chemotherapy in a T lymphocyte-dependent manner. (A, B) Human osteosarcoma cells were treated with PPP (10 μM) in the presence of a low dose of the ICD inducers mitoxantrone (MTX, 1 μM) for 6 hours. High-dose MTX (5 μM) was used as positive controls. Quinacrine staining was used to monitor intracellular ATP content. Representative images are shown in A and quinacrine dots were quantified in B. Scale bar equals 10 μm. Data are means±SD of three replicates (***p<0.001 vs untreated control; ###P<0.001 vs PPP; Tukey’s multiple comparisons test). (C–F) Human neuroglioma H4 cells wild-type or Atg5 knockout, human osteosarcoma U2OS wild type, PERK knockout or PEIF2α S51A knockin cells, murine fibrosarcoma MCA205 wild-type or AKTT308D/S473D knockin cells were treated with PPP (10 μM) alone or in combination with low doses of the ICD inducers MTX (1 μM) for 6 H as described above. High-dose MTX (5 μM) was used as positive controls. the extracellular ATP levels were measured in C–F. Data are means ± SD of three replicates (**P<0.01, ***P<0.001 vs untreated control; #P<0.05, ##P<0.01, ###P<0.001 vs WT; Tukey’s multiple comparisons test). (G–O) In vivo treatment of implanted murine MCA205 fibrosarcoma with oxaliplatin (OXA), anti-PD-1 antibody and PPP, alone or in combination (schematic view in G). (H, I) Growth kinetic of murine fibrosarcoma MCA205 cells evolving in immunocompetent C57BL/6 mice or athymic nu/nu mice (J) or MCA205 fibrosarcoma expressing constitutive active AKTT308D/S473D (M), Atg5 deficient MCA205 ATG5-/- (N) or MCA205 expressing the ectoATPase CD39 (O) evolving in immunocompetent C57BL/6 mice were treated as indicated in (G). When tumors became palpable, mice received systemic intraperitoneal (i.p.) injections of ppp alone or together with OXA or PD-1 blocker. Data are depicted as growth curves (mean±SD) (H, L–O) and tumor size distributions at day 24 (I). Individual tumor growth curves of mice treated with OXA and PPP, combined or not with PD-1 blockade are shown (K). The generation of immunological memory was assessed in cured animals by rechallenge with MCA205 and TC-1. Individual tumor growth curves are depicted (L). Data were analyzed with TumGrowth. n≥6 for mice in each group. (*P< 0.05 or ns, not statistically significant vs OXA; #P<0.05 or ns, not statistically significant vs OXA+PD-1 blockade, Student’s t-test, survival plots in online supplemental figure S7). (P) Schematic overview of the treatment of implanted murine MCA205 fibrosarcoma with OXA and PPP, alone or in combination. (P–T) Cytofluorometric analysis of tumor-infiltrating lymphocytes (TIL): CD3+CD8+ cytotoxic T lymphocytes (Q), CD8+PD-1+ T lymphocytes (R), CD4+FOXP3+CD25+ regulatory T cells (Treg) (S), and the ratio of CD3+CD8+ T cells over Treg (T). Data are means±SD (n≥5) (*p<0.05 or ns, not statistically significant vs control; #P<0.05 or ns, not statistically significant vs OXA, Student’s t-test). PPP, picropodophyllin.
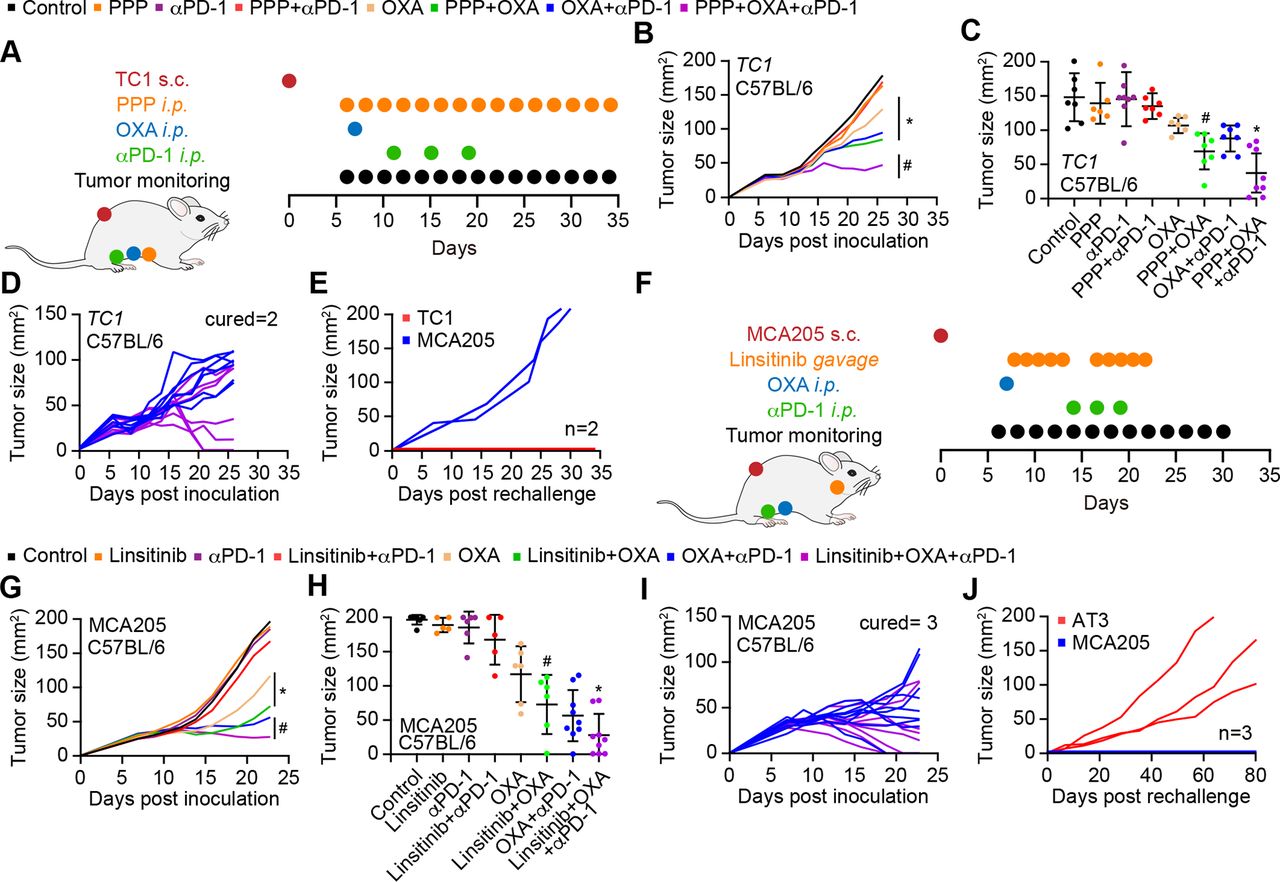
Considering the fact that PPP induced autophagy in mouse cancer cell lines (online supplemental figure S6C,D), we evaluated the capacity of PPP to improve the efficacy of chemoimmunotherapy against cutaneous MCA205 fibrosarcomas that were orthotopically implanted in histocompatible C57BL/6 mice. PPP alone did not reduce tumor growth. PPP failed to improve the efficacy of immunotherapy with PD-1 blockade, but improved that of chemotherapy with OXA. Moreover, the triple combination of OXA, PD-1 blockade and PPP was more efficient than chemoimmunotherapy with OXA and PD-1 blockade alone (figure 5H–I, online supplemental figure S7A). These effects depend on the immune system because no therapeutic efficacy could be measured in athymic nu/nu mice (that lack mature T lymphocytes) (figure 5J, online supplemental figure S7B). Immunocompetent mice that had been cured from MCA205 fibrosarcoma by the triple combination (OXA, PD-1 blockade and PPP) were resistant against rechallenge with MCA205 cells but readily developed antigenically unrelated TC1 lung cancer, meaning that they developed immune memory (figure 5K,L).
Inhibition of PPP-induced autophagy by AKTT308D/S473D or knockout of Atg5, as well as the expression of the ecto-ATPase CD39 abolished the anticancer effects of PPP (figure 5M–O, online supplemental figure S7C-E), supporting the involvement of autophagy-dependent ATP release in PPP effects. PPP did not affect the MCA205 tumor immune infiltrate on its own, but did reduce PD-1 induction by OXA on CD8+ T cells (figure 5P–R). Moreover, the combination of PP OXA depleted Tregs from the tumor environment (online supplemental figure 5S) and improved the CD8/Treg ratio (figure 5T).
These immune response-associated effects of IGF1R inhibition were also observed for TC1 non-small cell lung cancer, in which PPP similarly improved the effects of OXA or those of an OXA+ anti-PD-1 combination (figure 6A–C, online supplemental figure S7F). Of note, survival of mice with TC1 cancers was similarly extended by PPP +OXA and PPP +OXA+anti-PD-1 (online supplemental figure S7F). Mice that had been ridden from their TC1 tumors became resistant against rechallenge with the same tumor but not MCA205 fibrosarcomas (figure 6D,E). In mice-bearing AT3 breast cancers, PPP also improve the effects of OXA +PD-1 blockade (online supplemental figure S6E-G, S7G). Moreover, PPP could be replaced by another IGF1R antagonist, linsitinib, to ameliorate the outcome of chemotherapy with OXA or a combined OXA +anti-PD-1 regimen (figure 6F–H, online supplemental figure S7H). The triple combination (OXA +anti-PD-1+linsitinib) induced several complete remissions as well as immune memory against the tumors that had been eliminated (figure 6I,J). These results underscore the capacity of IGF1R antagonist to enhance the efficacy of chemoimmunotherapy in preclinical models.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
IGF1R inhibition improves the anticancer efficacy of immunotherapy. (A) Schematic overview of the in vivo treatment of murine lung cancer Tc1 cells with oxaliplatin (OXA), anti-PD-1 antibody and PPP, alone or in combination. (B–E) Growth kinetic of Tc1 cells evolving in immunocompetent C57BL/6 mice, treated as indicated in (A). When tumors became palpable, mice received systemic intraperitoneal injection of PPP alone or together with OXA or PD-1 blocker. Data ae depicted as (B) growth curves (mean±SD); (C) tumor size distributions at day 24; (D) individual tumor growth curves of mice treated with OXA and PPP, combined or not with PD-1 blockade. The generation of immunological memory was assessed in cured animals by rechallenge with MCA205 and Tc1. Individual tumor growth curves are depicted (E). Data were analyzed with TumGrowth. n≥ 6 for mice in each group. (*P<0.05 or ns, not statistically significant vs OXA; #P<0.05 or ns, not statistically significant vs OXA+PD-1 blocker, Student’s t-test, survival plots in online supplemental figure S7). (F) Schematic overview of the in vivo treatment of murine fibrosarcoma MCA205 cells with OXA, anti-PD-1 antibody and the selective of IGF1R inhibitor linsitinib (Lins), alone or in combination. IGF1R, insulin-like growth factor-1 receptor; PPP, picropodophyllin.
Discussion
Together with the insulin receptor, IGF1R is one of the most important trophic receptor tyrosine kinases, stimulating the uptake of nutrients into cells as well as a variety of anabolic reactions.40 Inhibition of IGF1R itself or that of the signal transduction cascade acting downstream of IGF1R (the PI3K/AKT/MTOR pathway) potently stimulates autophagy as well as other stress-adaptive mechanisms.41–43 Indeed, a vast literature suggests that chronic inhibition of this pathway by caloric restriction, pharmacological inhibitors or loss-of-function mutations has the capacity to extend the healthspan and lifespan in model organisms3 44–46 and perhaps in humans as well.47
Here, we identified a pharmacological IGF1R inhibitor, PPP, as a potent inducer of autophagic flux that acts on-target, as suggested by several observations. First, PPP inhibited all elements of the signaling cascading, hence reducing the activating phosphorylation of IGF1R, AKT and the MTOR substrate S6K, both in vitro, in cultured human cell lines and in vivo, in the liver and heart from mice, commensurate with the induction of autophagy. Second, PPP lost its capacity to induce autophagy after knockout of IGF1R, which obliges the cells to rewire their metabolism to support by other trophic receptors.48 49 Third, most convincingly, artificial activation of the inhibited cascade by expressing a transgene-encoded constitutively active AKT mutant, abolished autophagy induction by PPP. These results unequivocally demonstrate that PPP is activating autophagy through a specific mode of action, without major off-target effects. Of note, as for other autophagy inducers,22 50–52 this pathway involved the obligatory phosphorylation of eIF2α as part of the integrated stress response. Thus, cells bearing a non-phosphorylable eIF2α mutant or lacking the eIF2αkinase EIF2AK3/PERK were unable to activate the autophagic pathway in response to PPP.
Stimulation of autophagy by fasting or by the administration of CRMs enhances the efficacy of immunogenic chemotherapies (for instance with anthracyclines and OXA) as well as combination regimens of chemotherapies with immune checkpoint inhibitors targeting PD-15 19 53 54 Accordingly, we found that PPP enhanced the efficacy of anticancer chemotherapies with MTX and OXA, alone or in combination with PD-1 blocking antibodies. PPP on its own had little or no tumor growth inhibitory effects against MCA205 fibrosarcomas, TC1 non-small cell lung cancers and AT3 triple-negative breast cancer. The anticancer effects of PPP were only detectable in combination with chemoimmunotherapy and were lost in tumors that lacked essential genes/proteins involved in the autophagic process (due to knockout of ATG5 or knock-in mutation of eIF2α) or were rendered resistant to PPP (by transfection with constitutively active AKT). Moreover, these anticancer effects were accompanied by an increased infiltration of tumors by CTLs together with improvement in the local CTL/Treg ratio, and were lost in immunodeficient mice lacking mature T lymphocytes. Of note, it appears that the autophagy-dependent increase in extracellular ATP from tumor cells38 plays a rate-limiting role in the therapeutic efficacy of PPP, which indeed was lost when cancer cells were genetically manipulated to express the ATP-degrading enzyme CD39 on their surface. Importantly, in one tumor model (TC1), PPP improved the outcome of OXA-based chemotherapy, and this effect was not further improved by PD-1 blockade. This might prove therapeutically relevant in situations where anti-PD-1 cannot be administered.
PPP could be replaced by another IGF1R inhibitor, linsitinib, which has undergone evaluation in clinical trials.55–57 Linsitinib increased the efficacy of immunochemotherapy in mice, suggesting that this type of clinical grade IGF1R inhibitor should be evaluated in patients for similar combination effects. Indeed, linsitinib has been administered to patients with cancer either alone55–57 or in combination with other anticancer agents thought to mediate direct effects on cancer cells such as the MTORC1 inhibitor everolimus,58 the EGFR inhibitor erlotinib59 or the chemotherapeutics paclitaxel60 and irinothecan,61 with variable results. However, linsitinib has not been investigated for its potential immune effects and has not been combined with any kind of immunotherapy including PD-1 blockade.
At the clinical level, we observed that the activating phosphorylation of IGF1R detectable by immunohistochemistry correlated with a reduction of LC3B-positive puncta in triple-negative breast cancer patients. This IGF1R phosphorylation also correlated with poor local immunosurveillance as indicated by scarce infiltration by CD8+ CTLs but high abundance of FOXP3+ regulatory T cells and immunosuppressive CD163+ macrophages infiltrating the tumors, as well as dismal prognosis. These results confirm the negative effects of IGF1R signaling on immunosurveillance and disease control in breast cancer patients. A recent report on patients with colorectal cancer treated with chemotherapy together with EGFR or VEGF inhibitor revealed that overactivation of the IGF1R also constitutes a poor prognostic factor, particularly in patients bearing RAS wild-type tumors.62 These findings echo prior observations that high expression of IGF1R (though without proof of its activation) is a poor prognostic biomarker in gastric63 and breast cancer.1
In conclusion, excessive antiautophagic signaling via IGF1R has a major negative effect on anticancer immunosurveillance, thus reducing patient prognosis. However, IGF1R and its downstream signals are amenable to pharmacological inhibition and subsequent improvement of cancer control by the immune system. These considerations should be incorporated into the future design of clinical trials in which inhibition of the IGF1R/PI3K/AKT/MTOR pathway will be combined with adequate immunostimulation with ICD inducers and/or immune checkpoint inhibitors.
Data availability statement
Data are available on reasonable request. All data relevant to the study are included in the article or uploaded as online supplemental information. All data supporting the findings of this study are available within the article and its online supplemental information files and from the corresponding author on reasonable request.
Ethics statements
Ethics approval
All patients included in the study wrote the informed consent, approved by the Institutional Ethics Committee of the Renmin Hospital of Wuhan University (approval no. 2018K-C09). Animal experiments were conducted in compliance with the European Union (EU) Directive 63/2010 and protocols 2019_030_20590 and were approved by the Ethical Committee of the Gustave Roussy Campus Cancer (CEEA IRCIV/IGR no. 26, registered at the French Ministry of Research).
Acknowledgments
We are grateful to the support of the Gustave Roussy, Université Paris-Saclay, Plateforme Imagerie et Cytométrie (PFIC), UMS AMMICa INSERM US23-CNRS 3655.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors QW, A-LT and BL performed most of the experiments; QW, A-LT and WX performed in vivo experiments, ML, SF, PH and WG conceived and performed the large chemical drug screen, FM, YT, OK and GK conceived the study; PL, LZ, SZ and HP designed (parts of) the study; OK and GK wrote the paper.
Funding QW, A-LT, WX and HP were supported by the Chinese scholarship council. OK receives funding by the Île de France DIM ELICIT initiative. This work was partially supported by a Natural Science Foundation of Hubei grant (Grant No: 2020CFA026) to YT. GK is supported by the Ligue contre le Cancer (équipe labellisée); Agence National de la Recherche (ANR)—Projets blancs; AMMICa US23/CNRS UMS3655; Association pour la recherche sur le cancer (ARC); Association 'Ruban Rose'; Cancéropôle Ile-de-France; Fondation pour la Recherche Médicale (FRM); a donation by Elior; Equipex Onco-Pheno-Screen; European Joint ProgrammeProgram on Rare Diseases (EJPRD); Gustave Roussy Odyssea, the European Union Horizon 2020 Projects Oncobiome and Crimson; Fondation Carrefour; High-end Foreign Expert Program in China (GDW20171100085), Institut National du Cancer (INCa); Inserm (HTE); Institut Universitaire de France; LabEx Immuno-Oncology (ANR-18-IDEX-0001); the RHU Torino Lumière; Seerave Foundation; SIRIC Stratified Oncology Cell DNA Repair and Tumor Immune Elimination (SOCRATE); and SIRIC Cancer Research and Personalized Medicine (CARPEM). This study contributes to the IdEx Université de Paris ANR-18-IDEX-0001. FM is grateful to the Austrian Science Fund FWF (SFB LIPOTOX F3007 and F3012, W1226, P29203, P29262, P27893, P31727) and the Austrian Federal Ministry of Education, Science and Research as well as the University of Graz for grants 'Unkonventionelle Forschung-InterFast and Fast4Health' as well as 'flysleep' (BMWFW-80.109/0001-WF/V/3b/2015). We acknowledge the support of the field of excellence BioHealth, of NAWI Graz and the BioTechMed-Graz flagship project “EPIAge”.
Competing interests PH and WG are full-time employees of Samsara Therapeutics. GK, FM and OK are cofounders of Samsara Therapeutics.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.