Article Text

Abstract
Background Homocitrullination is the post-translational modification of lysine that is recognized by T cells.
Methods This study identified homocitrullinated peptides from aldolase, enolase, cytokeratin and binding immunoglobulin protein and used human leukocyte antigen (HLA) transgenic mice to assess immunogenicity by enzyme-linked immunosorbent spot assay. Vaccine efficacy was assessed in tumor therapy studies using HLA-matched B16 melanoma expressing constitutive or interferon γ (IFNγ)-inducible major histocompatibility complex class II (MHC-II) as represented by most human tumors. To determine the mechanism behind the therapy, immune cell infiltrates were analyzed using flow cytometry and therapy studies in the presence of myeloperoxidase (MPO) inhibitor and T-cell depletion performed. We assessed the T-cell repertoire to homocitrullinated peptides in patients with cancer and healthy donors using flow cytometry.
Results Homocitrulline (Hcit) peptide vaccination stimulated strong CD4 T-cell responses and induced significant antitumor therapy in an established tumor model. The antitumor response was dependent on CD4 T cells and the effect was driven mainly via direct tumor recognition, as responses were only observed if the tumors were induced to express MHC-II. In vitro proliferation assays show that healthy donors and patients with cancer have an oligoclonal CD4 T-cell repertoire recognizing homocitrullinated peptides. Inhibition of cyanate generation, which mediates homocitrullination, by MPO inhibition reduced tumor therapy by the vaccine induced T cells (p=0.0018). Analysis of the tumor microenvironment (TME) suggested that myeloid-derived suppressor cells (MDSCs) were a potential source of MPO. The selected B16 melanoma model showed MDSC infiltration and was appropriate to see if the Hcit vaccine could overcome the immunosuppression associated with MDSCs. The vaccine was very effective (90% survival) as the induced CD4 T cells directly targeted the homocitrullinated tumor and likely reversed the immunosuppressive environment.
Conclusion We propose that MPO, potentially produced by MDSCs, catalyzes the buildup of cyanate in the TME which diffuses into tumor cells causing homocitrullination of cytoplasmic proteins which are degraded and, in the presence of IFNγ, presented by MHC-II for direct CD4 T-cell recognition. Homocitrullinated proteins are a new target for cancer vaccines and may be particularly effective against tumors containing high levels of MPO expressing MDSCs.
- CD4-positive T-lymphocytes
- immunity
- cellular
- immunization
- immunotherapy
- vaccination
Data availability statement
Data sharing not applicable as no datasets were generated and/or analyzed for this study. Data and materials are available upon reasonable request from the corresponding author.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Proteins can be subject to post-translational modifications (PTMs) which increase structural and functional diversity by modifying the functional groups on amino acids.1 These altered self-proteins can contain novel T-cell and B-cell epitopes which stimulate responses that do not cross-react with the wild-type (WT) peptides.2 We have previously shown that vaccines targeting the PTM citrullination can induce efficient CD4 T-cell-mediated antitumor therapy in vivo in a process mediated by autophagy.3–5 Another PTM associated with generating altered self-antigens is homocitrullination.6 Homocitrullination (or carbamylation) is the modification of lysine residues that occurs when cyanate, or its active form isocyanic acid, reacts with the amine (NH2) groups on the side chain of lysine residues yielding homocitrulline (Hcit). This leads to a change in the molecular charge of the amino acid altering the antigenic properties of the peptides thereby generating unique epitopes.1 7 During inflammatory conditions, homocitrullination is predominantly driven by the actions of the myeloperoxidase (MPO) enzyme which, in the presence of hydrogen peroxide (H2O2), converts thiocyanate to cyanate and isocyanic acid.8 9 MPO is produced by immune cells including neutrophils, monocytes, macrophages and myeloid-derived suppressor cells (MDSCs).10 Tumor cells can produce a high level of H2O2 as a result of oncogenic transformation associated with antioxidant imbalances.11 This results in DNA damage which in turn promotes H2O2 generation resulting in a vicious cycle of H2O2 production. We hypothesize that the combination of MPO from immune infiltrates and H2O2 from tumor cells produces cyanate which can induce protein homocitrullination within the tumor microenvironment (TME) thus providing potential targets for cancer therapies.
Our previous work on PTM antigens in tumor cells suggests that proteins which are abundant in cells such as cytoskeletal proteins or glycolytic enzymes constitute good targets for cancer immunotherapy.4 12 Here, we have studied aldolase A, binding immunoglobulin protein (Bip), α-enolase and cytokeratin 8 (Cyk8) as potential targets for cancer therapy. Aldolase A is a glycolytic enzyme and a crucial player in adenosine triphosphate (ATP) synthesis.13 It is highly expressed in a range of cancers including lung, renal and adrenocortical tumors.14 15 Bip is an endoplasmic reticulum chaperone protein that acts as a sensor of unfolded proteins.16 Upregulation of Bip is linked to survival of cancer cells in hypoxic conditions and is associated with tumor progression in mammary and colorectal tumors.17–19 α-Enolase is a glycolytic enzyme that is overexpressed in a wide range of tumors as a result of increased glycolysis in both normoxic and hypoxic conditions.20–22 Cytk8 is an intermediate filament protein important for the cytoskeleton of epithelial cells. Cyk8 has been identified as a potential self-antigen in some diseases.23 High Cyk8 expression is a predictor of poor prognosis for patients with lung adenocarcinoma and hepatocellular cancer.24 25
In this study, we examine Hcit peptides as targets for tumor therapy. We demonstrate vaccination with five Hcit peptides from aldolase, enolase, Cyk8 and Bip can stimulate Hcit-specific CD4 T-cell responses which efficiently mediate tumor therapy. We show that healthy donors and patients with lung cancer have a CD4 T-cell repertoire capable of responding to the Hcit aldolase peptides. We also demonstrate that homocitrullination in the TME is mediated by MPO produced by MDSCs. Despite the immunosuppression mediated by MDSCs vaccination with Hcit peptides generated a strong enough proinflammatory response to overcome the immunosuppression and provide antitumor immunity.
Materials and methods
Unless otherwise stated reagents were obtained from Sigma-Aldridge. Full antibody details are given in online supplemental table S3.
Supplemental material
Peptides
Peptides were selected based on immune epitope database (IEDB) (http://www.iedb.org/) binding predictions.26 Peptides (online supplemental table S1) were synthesized at >90% purity (GenScript), stored lyophilized at −80°C and then reconstituted in phosphate buffered saline (PBS) on day of use.
Animals and cell lines
Human leukocyte antigen (HLA)-A2.1+/+HLADP4+/+hCD4+/+ (HHDII/DP4; EM:02221, European Mouse Mutant Archive) transgenic mice aged 8–12 weeks were used.
The murine melanoma B16F1 cell line (ATCC) was engineered to knockout mouse major histocompatibility complex (MHC) and express HHDII (HLA-A2) alone or in combination with either constitutive HLA-DP4 or interferon γ (IFNγ)-inducible HLA-DP4 (iDP4) as previously described.3 5 Cells were cultured in RPMI medium 1640 with L-glutamine (2 mmol/L), 10% foetal calf serum (FCS) and appropriate antibiotics. Cell lines used were mycoplasma free, authenticated by suppliers (short tandem repeat (STR) profiling) and used within 10 passages.
Immunization protocol
Mice were injected subcutaneously (s.c.) with 25 μg of each peptide either as a pool or individually in combination with 6 μg of CpG ODN 1826 (CpG) and monophosphoryl lipid A (MPLA) (InvivoGen) on days 1, 8 and 15 and responses analyzed on day 21. Groups of n=3 selected through statistical power calculations were used on independent occasions.
For antitumor experiments, mice were implanted s.c. with tumor cells 3 days prior to immunization regime above unless otherwise stated. Implant doses were 5×105 cells/mouse for B16F1 HHDII/DP4 and 1×105 for B16 HHDII/iDP4 and B16 HHDII/MHC-II K/O. In relevant studies, 250 µg anti-Ly6G or anti-Ly6C antibodies were given intraperitoneally (i.p.) 8, 10, 12, 15, 17 and 19 days post tumor implant. Anti-CD4 or anti-CD8 antibodies were given i.p. at 500 µg on day 4 and 300 µg on days 8 and 11, 800 µg MPO inhibitor 4-aminobenzoic hydrazide was administered i.p. daily on days 8–12 and 15–19 post tumor implant. Tumor growth was monitored two times per week and mice were humanely euthanized once tumors reached ≥15 mm in diameter. Groups of n=10 selected through statistical power calculations were used.
Enzyme linked immunosorbent spot (ELISpot) assays
Murine ELISpot kits (Mabtech) were used with 5×105 splenocytes/well and 10 µg/mL synthetic peptide added to quadruplicate wells, 20 µg/mL anti-CD8 or anti-CD4 antibodies were added to appropriate wells for blocking studies. Plates were incubated at 37°C for 40 hours in an atmosphere of 5% CO2. Spots were analyzed using an automated plate reader (Cellular Technologies).
Isolation of animal tissues
Spleens were disaggregated and treated with red cell lysis buffer for 2 min. Tumors were harvested and mechanically disaggregated.
Bone marrow-derived MDSCs (BM-MDSCs)
BM-MDSCs were generated from mice as previously described.27 Cells were cultured in RPMI with L-glutamine, 10% FCS supplemented with 1 ng/mL granulocyte macrophage colony stimulating factor (Miltenyi) and 50 ng/mL interleukin 18 (IL-18) (BioLegend). On day 5 MDSCs were harvested from non-adherent cells.
For coculture experiments, B16F1 cells were seeded in a 24-well plate at 2×104 cells/well. MDSCs were then washed and added at 2×105/well with 100 µM potassium thiocyanate and 30 nM H2O2.
In vitro homocitrullination and detection
Recombinant human aldolase A protein or B16F1 cell lysates were in vitro homocitrullinated using the previously described method of incubation with 1M potassium cyanate (cyanate) at 37°C for 10 hours.28 Cell monolayers were treated with 1M cyanate overnight, cells were washed, removed using a cell scraper and lysed by repeated freeze/thaw cycles. All samples were dialyzed in dH2O. OXIselect carbamylation ELISA (Cell Biolabs) or anti-carbamylation antibody staining were used to detect Hcit.
Flow cytometric staining of animal tissue and cells
Cells were stained with extracellular antibodies for 30 min, washed, fixed and permeabilized using intracellular fixation/permeabilization buffers (Thermo Fisher). Intracellular antibody staining was performed in permeabilization buffer for 30 min before washing and fixing. Stained samples were analyzed on a MACSQuant10 flow cytometer.
Mass spectrometry
Samples were trypsin digested and identified by liquid chromatography with tandem mass spectrometry (LC-MS/MS) analysis using an Eksigent eKspert nanoLC 425 liquid chromatography system, running at 5 μL/min (Eksigent) using gradient elution (mobile phase A 0.1% formic acid; B acetonitrile with 0.1% formic acid) 5%–40% B over 30 min, coupled to a SCIEX TripleTOF 6600 mass spectrometer via a DuoSpray source operating in information dependent acquisition mode (top 30 ions).29 Total cycle time was 1.8 s, accumulation times of 250 ms for Time of Flight Mass Spectrometry (TOFMS) scan and 50 ms for product ions. Data were analyzed using PEAKS V.8.0 (Bioinformatic Solutions, Waterloo, Ontario, Canada) searching the SwissProt human database, mass error tolerance 25 ppm precursor, 0.1 Da fragment.30 Modification sites were identified as confident with a minimum ion intensity of 5%.
Peripheral blood mononuclear cells (PBMCs) isolation and processing
PBMCs were isolated from healthy donors and patients with cancer (online supplemental table S2) using histopaque-1077 then depleted of CD25 positive cells using anti-CD25 microbeads and MACS cell separation columns (Miltenyi). Isolated CD25-depleted PBMCs were loaded with 5 µM carboxyfluorescein succinimidyl ester (CFSE) (Thermo Fisher).4 Cells were resuspended at 1.5×106/ml, plated and stimulated with vehicle, phytohemagglutinin (PHA) (positive control, 10 µg/mL), soluble anti-CD3 (positive control, 30 ng/mL) or peptide (10 µg/mL). Peptide concentration was selected based on reports in the literature using similar post-translationally modified epitopes.31–33 On day 10, 500 µL of cells were stained and samples were analyzed immediately. For functional restimulation assays, cultures were established with peptide stimulation as above in OpTmizer serum free media and the addition of 2% pooled human serum, 5 ng/mL IL-15, 10 ng/mL IL-7 and 20 U/mL IL-2. On day 14, cells were harvested and assessed using human IFNγ ELISpot kits (Mabtech) where 1×105 cells/well were stimulated with 10 μg/mL peptide or PHA (10 µg/mL) in quadruplicate wells. ELISpot plates were incubated at 37°C for 24 hours in an atmosphere of 5% CO2 and then developed following the manufacturer’s instructions. Spots were analyzed using an automated plate reader (Cellular Technologies). All samples showed responses to PHA (data not shown).
T-cell receptor (TCR) α and β chains analysis of proliferating PBMCs
Stained CD4+/CFSEhigh and CD4+/CFSElow cell populations were bulk sorted into RNA protect (Qiagen) using a MoFlo XDP Sorter (Beckman Coulter). RNA was purified from sorted cells, reverse transcription polymerase chain reaction (RT-PCR) performed, the cDNA was subjected to amplicon rescued multiplex PCR using human TCR α and β 250 PER primers (iRepertoire, Huntsville, Alabama, USA).5 Ten assessed sample libraries were pooled and sequenced using the Illumina MiSeq platform (Illumina, USA). Tree maps and D50 diversity values were generated using iRweb software. Tree maps show each unique complementarity determining region 3 (CDR3) as a colored rectangle, the size of each rectangle corresponds to each CDR3s abundance within the repertoire and the positioning is determined by the V region usage. Diversity was measured using D50 immune repertoire diversity index. The D50 index is a quantitative measure of the degree of diversity of T cells within a sample. The D50 is the percentage of T-cell clones that account for the cumulative 50% of the total CDR3s counted in the sample. The more diverse a library, the closer the value will be to 50. Low diversity values are associated with decreased diversity. Data are presented as non-normalized (which takes into account the frequency of each unique CDR3).
Statistical analysis
Statistical analysis was performed using GraphPad Prism software V.7. Comparative analysis of the ELISpot results was performed by applying paired or unpaired ANOVA or Student’s t-test as appropriate with p values calculated accordingly. Comparison of tumor survival was assessed by log-rank test. The association between tumor growth and tumor-infiltrating lymphocyte (TIL) infiltration was assessed using linear regression. Human responses were analyzed by Wilcoxon matched-paired test. P values<0.05 were considered statistically significant.
Results
Modification-specific immune responses are generated to homocitrullinated peptides
Previous studies have shown that citrullinated peptides are presented on HLA-DP4, an HLA allele that is frequently expressed in the Caucasian population.5 We aimed to determine if homocitrullinated peptides could be presented on HLA-DP4 molecules on the cell surface. IEDB algorithm was used to predict potential peptides from aldolase, Bip, Cyk8 and enolase that have a strong or moderate predicted binding affinity to HLA-DP4 (DPA1*01/DPB1*04:01) and contain lysine residues in the predicted core binding regions.26 Homocitrullinated peptides were then synthesized with Hcit replacing lysine (online supplemental table S1). All peptides are homologous in mice and humans with the exception of Eno156-176Hcit, Aldo217-235Hcit and Aldo238-256Hcit which differ at a single amino acid.
To determine if peptides were immunogenic, HHDII/DP4 mice were immunized with individual homocitrullinated peptides and immune responses measured by IFNγ ELISpot (figure 1A). Three aldolase peptides, Aldo74-93Hcit, Aldo140-157Hcit and Aldo238-256Hcit, two Bip peptides, Bip328-346Hcit and Bip562-579Hcit, Eno156-176Hcit and Cyk371-388Hcit stimulated IFNγ responses. In contrast, three peptides, Aldo217-235Hcit, Aldo289-307Hcit and Eno400-420Hcit, failed to stimulate responses (online supplemental figure S1). There was no obvious correlation between predicted peptide binding and immunogenicity of the Hcit containing peptides. For peptides that were immunogenic responses to the homocitrullinated peptides were significantly higher than to their unmodified wild type (WT) equivalents (Aldo74-93Hcit (p=0.0033), Aldo140-157Hcit (p=0.0126), Bip328-346Hcit (p=0.0126) and Cyk371-388Hcit (p=0.0052)).

Immunization with homocitrullinated peptides can induce modification-specific immune responses. IFNγ ELISpot responses were assessed for HHDII/DP4 mice immunized with individual peptides containing Hcit residues (A) or lysine residues (WT) (B). Aldo238-256wt was not tested. Symbols represent mean response for individual mice, line represents median value between mice. Significant p values are shown. Data are representative of independent studies where n=3. ELISpot, enzyme-linked immunosorbent spot assay; Hcit, homocitrulline; IFNγ, interferon gamma; WT, wild type.
Immune responses in mice immunized with WT peptides were then assessed (figure 1B). Immunization with Aldo74-93wt, Bip328-346wt or Cyk371-388wt peptide induced an IFNγ response to the respective WT peptides but not the Hcit containing peptides (p=0.0105, p=0.0254 and p=0.0112, respectively). No significant responses were seen for Aldo140-157wt, Bip562-579wt or Eno156-176wt immunization. Therefore, WT peptides can stimulate responses, but these responses likely represent a different T-cell repertoire as little cross-reactivity to the homocitrullinated epitopes was observed.
Immune responses in mice are CD4 T-cell-mediated and show efficient tumur therapy
Since epitopes were selected on predicted HLA-DP4 binding, we sought to confirm whether the immune responses were mediated by CD4 T cells. Mice were immunized with the Hcit peptides and immune responses measured by IFNγ ELISpot in the presence of CD4 or CD8 blocking antibodies (figure 2A). Addition of a CD4 blocking antibody significantly blocked the immune responses generated by Aldo74-93Hcit (p<0.0001), Aldo140-157Hcit (p=0.0005), Cyk371-388Hcit (p=0.0137) and Eno156-176Hcit (p=0.0155) peptides, whereas the CD8 blocking antibody had no effect. Splenocytes from immunized mice showing lower frequency responses were first cultured in vitro to expand responses prior to blocking analysis. Responses were blocked by CD4 but not the CD8 blocking antibody for Bip328-346Hcit (p<0.0001) and Bip562-579Hcit (p=0.0001) (figure 2B). These responses are Th1 responses as splenocytes from mice immunized with Aldo74-93Hcit and Aldo140-157Hcit peptides did not generate any IL-17 response and only demonstrated low frequency IL-10 responses (online supplemental figure S2).

Hcit-specific immune responses are CD4 mediated and mediate efficient tumor therapy in vivo. IFNγ ELISpot responses were assessed for HHDII/DP4 mice immunized with homocitrullinated peptides. Splenocytes from Aldo74-93Hcit, Aldo140-157Hcit Cyk371-388Hcit, Bip328-346Hcit, Bip562-579Hcit or Eno156-176Hcit immunized mice were restimulated with peptides alone or in combination with anti-CD4 or anti-CD8 blocking antibodies ex vivo (A) or after 7-day culture (B). Data are collated from at least two independent studies where n=3. In vivo tumor survival studies were carried out by implanting mice with HLA-matched B16F1 cells with constitutive MHC-II (B16 HHDII/DP4) or IFNγ-inducible MHC-II (B16 HHDII/iDP4). Mice were then given three immunizations of homocitrullinated or WT peptides. Survival was assessed for aldolase peptides in mice implanted with the B16 HHDII/DP4 (C) and B16 HHDII/iDP4 (D) melanoma lines. In the B16 HHDI/iDP4 model, survival was also determined for Bip328-346Hcit, Bip562-579Hcit and Eno156-176Hcit (E) and Cyk371-388Hcit (F) peptides. Statistical analysis compared survival in immunized mice with unimmunized mice. A representative data set is shown in which for each group n=10 and significant p values are shown. ELISpot, enzyme-linked immunosorbent spot assay; Hcit, homocitrulline; HLA, human leukocyte antigen; IFNγ, interferon γ; MHC-II, major histocompatibility complex class II; WT, wild type.
We next investigated if homocitrullinated epitopes are presented on the tumor. Target epitopes require homocitrullination in the TME and presentation by MHC-II for successful tumor therapy. We implanted HHDII/DP4 mice with HLA-matched B16 cells and then immunized with homocitrullinated peptides. Mice immunized with Aldo74-93Hcit (90% p<0.0001), Aldo140-157Hcit (60% p=0.0170) peptides or the combination (90% p<0.0001) showed a significant survival benefit compared with unimmunized control mice (figure 2C). In contrast, immunization with Aldo238-256Hcit peptide showed no significant survival advantage despite inducing an immune response and also providing evidence that CpG/MPLA adjuvant has no antitumor effect in this model which is consistent with previous data.5 These results suggest that Aldo74-93 and Aldo140-157 peptides are naturally homocitrullinated and presented on MHC-II, whereas Aldo238-256 is either not naturally homocitrullinated or not presented in vivo.
The B16 HHDII/DP4 cell line and antigen presenting cells (APCs) constitutively expresses MHC-II. Expression of MHC-II in non-APCs including most cancer cells is noconstitutive but instead driven by the IFNγ-induced class II major histocompatability complex transactivator (CIITA) promoter pIV.34 To mimic natural MHC expression, we engineered B16 cells to express MHC-II under an IFNγ-inducible promoter (B16 HHDII/iDP4).3 5 HHDII/DP4 mice implanted with IFNγ-inducible B16 HHDII/iDP4 tumor and immunized with Aldo74-93Hcit and Aldo140-157Hcit peptides showed a significantly enhanced survival (80%) over control mice (p=0.0007) (figure 2D). Immunization with Aldo74-93wt and Aldo140-157wt peptides gave no survival advantage and was significantly worse than the Hcit peptide immunized group (p=0.0004) supporting evidence that the effects seen with the Hcit peptides are genuine and not an adjuvant-mediated effect.
Antitumor responses induced by immunization with the homocitrullinated enolase, Bip (figure 2E) and Cyk371-388Hcit (figure 2F) peptides were assessed in mice implanted with B16 HHDII/iDP4 cells. Immunization with Eno156-176Hcit, Bip562-579Hcit and Cyk371-388Hcit resulted in a significant survival advantage compared with unimmunized control (p=0.0005, p=0.0108, p=0.0273, respectively). Immunization with Bip328-346Hcit peptide did not generate any survival advantage, suggesting it is either not naturally homocitrullinated or not presented in vivo. Adjuvant only did not provide any significant survival advantage over unimmunized mice, which corresponds with published data.4 5 12
Tumor therapy requires direct action of CD4 cells on tumor cells presenting peptide via MHC-II
CD4 cells recognize homocitrullinated epitopes but the antitumor effect may be mediated by either modulation of the TME or stimulation of CD8 effector cells. To determine whether CD8 T cells play a role, the induced antitumor response following immunization was assessed following depletion of CD4 or CD8 T cells (figure 3A). Antitumor responses were significantly reduced in the presence of the anti-CD4 antibody (p=0.0124) but not with the anti-CD8 antibody (p=0.7051), indicating that CD4 T cells and not CD8 T cells were critical for the antitumor effect.

Tumor survival is dependent on MDSCs, CD4 cells and MHC-II presentation by tumor. Mice implanted with B16 HHDII/DP4 were treated with either anti-CD4 or anti-CD8 antibodies in combination with Aldo74-93Hcit and Aldo140-157Hcit immunizations. Survival was assessed in mice treated with antibodies (A). The statistical difference in survival between vaccinated mice and mice treated with each depletion antibody in combination with vaccination is shown. Mice implanted with B16 melanoma knocked out for mouse class II (B16 HHDII/MHC-II K/O) were also assessed for survival (B). For this study, statistical analysis compared control unimmunized mice with immunized mice, no statistical difference was observed. For all groups, n≥10. Hcit, homocitrulline; MDSCs, myeloid-derived suppressor cells; MHC-II, major histocompatibility complex class II.
CD4 T cells may act indirectly via MHC-II expressed on APCs. Therefore, we investigated whether the expression of MHC-II on tumor cells is needed for efficacy of the vaccine. HHDII/DP4 mice were implanted with B16 cells expressing matched MHC-I but not MHC-II (B16 HHDII/MHC-I/II KO). In this model, APCs express MHC-II and CD8 T cells can recognize the tumor via MHC-I. The antitumor effect of Aldo74-93Hcit and Aldo140-157Hcit peptides following immunization was completely lost (figure 3B) in this model, suggesting CD4 T cells are not indirectly targeting the APCs or other cells in the TME or acting through CD8 effector cells. These studies support presentation of the homocitrullinated peptides on MHC-II molecules on tumor cells and direct recognition by infiltrating CD4 T cells.
A repertoire of T cells that respond to homocitrullinated peptides is present in healthy human donors and patients with cancer
We then concentrated on further characterizing the Aldo74-93Hcit and Aldo140-157Hcit epitopes and next investigated whether humans have a repertoire of T cells specific to these peptides. Peptide-specific proliferative responses in PBMCs isolated from healthy donors and patients with cancer were measured (online supplemental table S2). Gating strategy is shown in figure 4A. Donors were considered responders if the percentage of proliferating cells was at least twice that observed in the media control. For all donors, proliferation was in the CD4 T-cell population with minimal proliferation observed in the CD8 T-cell population.

Humans have a repertoire of CD4 T cells that can recognize homocitrullinated peptides. PBMCs were isolated from 15 healthy donors and 11 patients with cancer and assessed for proliferative responses to homocitrullinated peptides. Example plots shown for healthy donor BD0051 (A) with gating strategy (i, iii), proliferation (CFSE) versus CD4 or CD8 gated on total population (ii) and proliferation gated on either CD4+ or CD8+ cells (iv). Values displayed in (iv) represent the per cent of proliferating cells among CD4+ or CD8+ populations. Graphs show percentage CD4 proliferation in healthy donors (B) and patients (E) and paired analysis comparing media control to each peptide stimulation. Restimulation assays were performed on healthy donor PMBCs that had been cultured with modified peptides for 14 days. IFNγ ELISpot showed restimulation responses to modified and wild-type peptides (C). Example spots and summary data with significant p values are shown. TCR α and β repertoire diversity was assessed in the CD4+ CFSEhigh/low cells in one healthy donor that showed responses to both peptides (D). Tree maps depict TCR α and β chain CDR3 clonotype usage in relation to repertoire size (i, ii). Each rectangle represents a unique CDR3 nucleotide sequence and rectangle size denotes the relative frequency of an individual sequence. Colors are randomly assigned and thus do not represent the same CDR3 sequence between plots. Diversity index for each population is shown in (iii). CFSE, carboxyfluorescein succinimidyl ester; ELISpot, enzyme-linked immunosorbent spot assay; IFNγ, interferon γ; PBMCs, peripheral blood mononuclear cells.
In PBMCs from healthy donors, 11/15 showed CD4 T-cell proliferation in response to at least one of the peptides (figure 4B) and 8/15 donors showed responses to Aldo74-93Hcit and 9/15 to Aldo140-157Hcit. Across all donors, Aldo74-93Hcit (p=0.0079) and Aldo140-157Hcit (p=0.0122) induced significant increases in CD4 proliferation compared with media only controls (figure 4Bi). Interestingly, responses were not restricted purely by the HLA-DP4 allele as HLA-DP4-negative donor BD0025 showed a proliferative response to Aldo74-93Hcit. In addition, four non-responding donors expressed the HLA-DP4 allele. Lack of response in these donors may be a reflection of the low frequency of specific T cells in a naive repertoire or perhaps a response restricted to a specific subtype of HLA-DP4 and further characterization would be necessary to determine this.
To determine whether the Hcit responses seen in healthy donors were modification-specific restimulation assays were performed. PBMCs from three donors were stimulated with the modified peptides and after 14 days cells were restimulated with either the modified or WT peptides (figure 4C). Two donors showed responses to the Aldo74-93Hcit peptide (BD0002 and BD150) and one donor to the Aldo140-157Hcit peptide (BD0016). All three responses were modification specific confirming that humans can generate Hcit-specific immune responses.
To determine the clonality, TCR Vβ and Vα sequencing was performed on proliferating and non-proliferating CD4 T cells isolated from healthy donor BD0051 that had been stimulated ex vivo with Aldo74-93Hcit and Aldo140-157Hcit peptides. This donor showed minimal proliferation to the media control. The TCR α and β CDR3 tree maps (figure 4D) show decreased CDR3 diversity in the proliferating CD4+CFSElow cells combined with a dramatic increase in the relative frequency of subsets of CDR3 sequences, suggesting a more focused repertoire of cells responding to each peptide. The oligoclonal nature of the proliferating CD4 T cells is confirmed by the lower diversity index (D50) of the CDR3 sequences from the proliferating (CD4+CFSElow) compared with non-proliferating (CD4+CFSEhigh) populations (figure 4Diii). The D50 TCR α diversity was reduced in the proliferating population from 8.1 to 0.5 for Aldo 74-93Hcit and from 7.3 to 0.7 for Aldo140-157Hcit. The TCRβ diversity was reduced in the proliferating population from 5.8 to 0.2 for Aldo 74-93Hcit and from 5.8 to 0.6 for Aldo140-157Hcit. The reduction in diversity was not due to the culture period as clonal diversity in unstimulated non-proliferating populations was similar at start and end of culture (online supplemental figure S3). Clonality of the responses is greatly increased compared with polyclonal stimulus and more closely represent that seen from a response to a single peptide epitope from hepatitis B virus (online supplemental figure S3). These results suggest that peptide stimulation drives expansion of a specific CD4 T-cell repertoire that can proliferate in response to the homocitrullinated peptides.
The proliferative T-cell responses were also assessed in seven patients with lung, three patients with ovarian and one patient with breast cancer (figure 4Ei). The proliferative T-cell responses were lower in PBMCs from the patients when compared with the healthy donors. One out of eleven patients responded to Aldo140-157Hcit peptide and seven out of eleven patients responded to Aldo74-93Hcit peptide (p=0.0372) (figure 4Eii). These results demonstrate that both healthy donors and patients with cancer have a repertoire of CD4 T cells that can respond to homocitrullinated aldolase peptides.
Therapy is dependent on the action of MPO in the TME
To investigate the mechanism of tumor therapy, we examined the effects of immunization on the TME using an established tumor model. Tumor (B16 HHDII/iDP4)-bearing mice were immunized with Aldo74-93Hcit and Aldo140-157Hcit peptides combined as a single immunization on day 14 (figure 5A). Mice immunized with Aldo74-93Hcit and Aldo140-157Hcit peptides at this later timepoint showed a significantly enhanced survival (70% vs 20%, p=0.0037) compared with unimmunized controls. In light of this, we assessed the efficacy of immunization once the tumor reached a size of 5×5–8×8 mm. Mice were culled 7 days after immunization and TILs were analyzed by flow cytometry. Compared with the control unimmunized mice, the Hcit peptide immunized mice showed a delay in further tumor growth within 7 days following immunization (figure 5Bi). The change in tumor volume between the day of immunization and TIL analysis was significantly reduced after Hcit peptide immunization and was not seen in control mice (p=0.0275, figure 5Bi). Waterfall plots (figure 5Bii) show that immunized mice fall into two clusters, 11/13 mice showed minimal tumor growth or regression post vaccination, but 2/13 mice showed outlier tumor growth and were excluded from further TIL analysis. Despite CD4 depletion studies suggesting a vital role for CD4 T cells in tumor therapy there was not a significant correlation between increased CD4 infiltrate and regression (online supplemental figure S4). However, there was a correlation with CD45+ immune infiltrate (online supplemental figure S4) suggesting perhaps an altered phenotype or activation status of CD4 T cells is important for antitumor immunity rather than an increase in overall numbers of infiltrating CD4 cells and that this influences other immune infiltrates. Interestingly, tumor therapy also correlated with the increased expression of MHC-II on the CD45− cell population within the tumors providing further data for the important role of MHC-II epitope presentation in addition to the generation of a specific immune response (online supplemental figure S4).

MPO in the TME in necessary for the antitumor effect of homocitrullinated peptides. Survival was determined in mice where immunization was delayed till day 14 (A). Mice were implanted with B16 HHDII/iDP4, then given a single immunization once tumor size reached 5×5–8×8 mm. Tumors were removed 7 days later for analysis. Tumor growth prior to cull is shown for each group (Bi) and waterfall plot shows the change in tumor growth from vaccination to day mice were culled for analysis (Bii). Data are collated from two independent studies in which n>5. Post-translational homocitrullination occurs as a result of increased cyanate levels, either due to high urea concentrations or more commonly in tissue due to the breakdown of thiocyanate by the MPO enzyme. Build-up of cyanate and its active form isocyanic acid drives homocitrullination which converts lysine residues in to Hcit (C). Flow cytometry was used for analysis of tumor-infiltrating MPO+ cells (D). Correlation of change in tumor volume between immunization and tumor analysis to MPO expression was assessed by linear regression analysis and significant p values are shown. Mice implanted with B16 HHDII/iDP4 were treated daily with the MPO inhibitor daily for 2 weeks and/or Aldo74-93Hcit and Aldo140-157Hcit immunizations on days 4, 11 and 18 (E). Statistical analysis compared the combination with individual treatments, n≥10. Hcit, homocitrulline; MPO, myeloperoxidase; TME, tumor microenvironment.
Immune-mediated homocitrullination is largely driven by MPO (figure 5C). If this is the case, then vaccine-induced survival will be dependent on MPO activity. Analysis of tumors from immunized mice showed that tumor regression correlated with an increase in MPO expression (p=0.0414) (figure 5D) suggesting the importance of MPO for successful therapy. To confirm this, we administered an irreversible chemical MPO inhibitor35 in our therapy studies (figure 5E). Immunized mice showed a significant survival advantage compared with mice immunized in combination with the MPO inhibitor (p=0.0018). No significant difference was observed between mice given vaccine and MPO inhibitor compared with MPO inhibitor only (p=0.5536), thus confirming MPO has a role in the homocitrullinated peptide antitumor response.
Tumor-infiltrating cells including MDSCs are a source of MPO and are essential for tumor therapy
Given that MPO is necessary for an antitumor response, we investigated the source of MPO in the TME. Staining B16 cells that had been cultured in vitro confirmed that they do not express MPO (figure 6A), in contrast, MPO has been shown to be expressed by immune cells.36 We therefore analyzed immune infiltrates in tumors to see if they express MPO. Flow cytometry analysis showed MPO expression was absent from the CD45− fraction but was present in a population of CD45+ cells also expressing CD11b (figure 6B). Myeloid differentiation markers Ly6C and Ly6G revealed the MPO+ cells include two populations, Ly6G+Ly6Clow and Ly6G−Ly6Chigh. Tumor-associated Ly6G+Ly6Clow cells have been characterized as polymorphonuclear MDSCs (PMN-MDSCs)/neutrophils and Ly6G−Ly6Chigh cells as monocytic MDSCs (M-MDSCs)/monocytes.37 38 Examples of the MDSC gating strategy and MPO staining on ex vivo tumors are shown in online supplemental figure S5D,E.
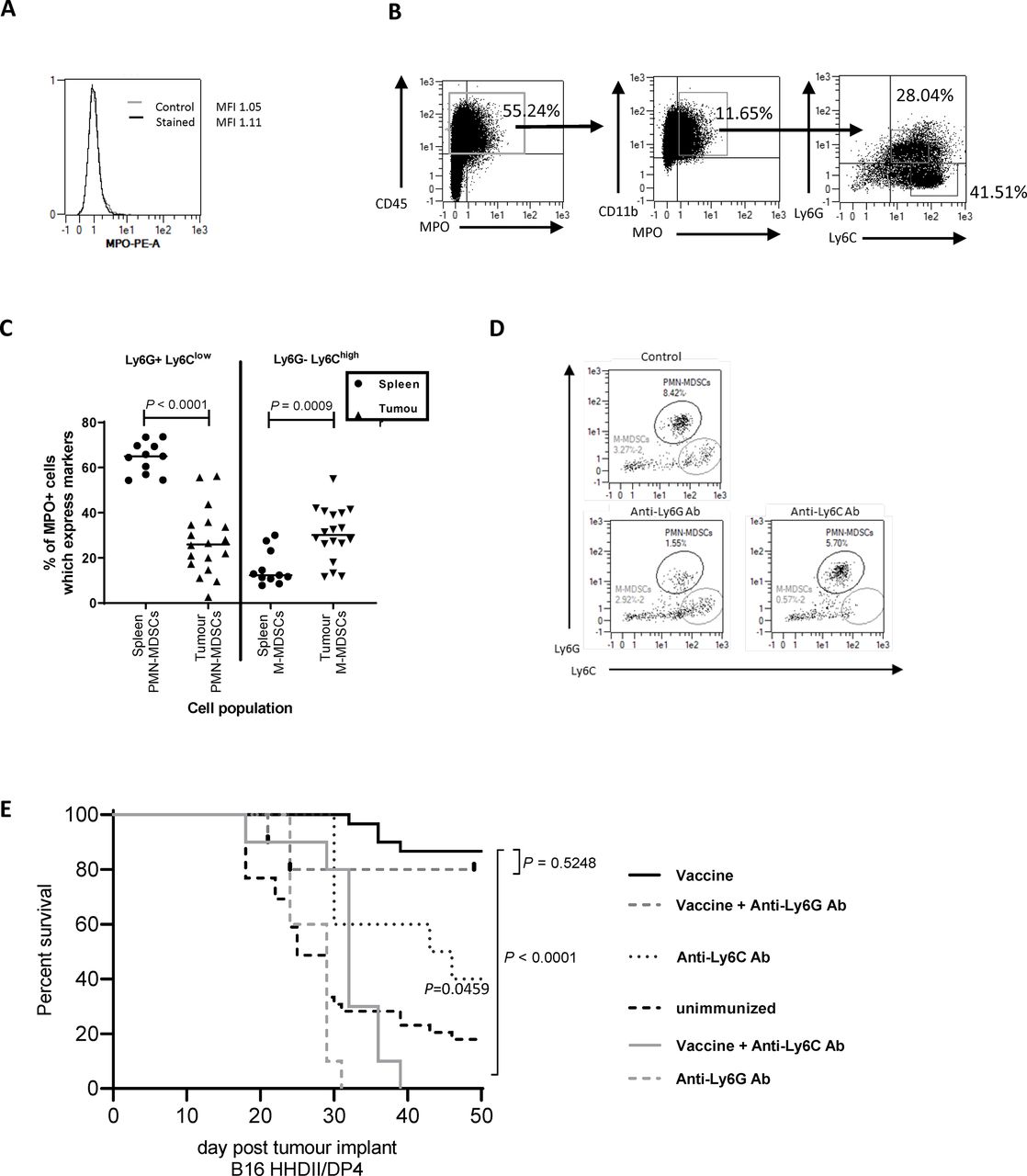
Tumor-infiltrating MDSCs are a source of MPO and essential for vaccine-mediated tumor therapy. Flow cytometry was used to assess MPO expression on in vitro grown B16F1 HHDII/DP4 (A) or CD45+ tumor-infiltrating (B) cells from in vivo grown B16F1 HHDII/DP4 cells. The proportion of MPO+ cells which express PMN-MDSC (Ly6G+Ly6Clow) or M-MDSC (Ly6G−Ly6Chigh) markers was assessed in the spleen and tumor (C). Mice implanted with B16 HHDII/DP4 were treated with either anti-Ly6C or anti-Ly6G antibodies in combination with Aldo74-93Hcit and Aldo140-157Hcit immunizations. Staining shows the populations of splenic Ly6G+Ly6Clow (PMN-MDSCs) or CD11b+Ly6G−Ly6Chigh (M-MDSC) after treatment with depletion antibodies (D). Survival was assessed in mice treated with antibodies (E). MDSCs, myeloid-derived suppressor cells; M-MDSC, monocytic MDSC; MPO, myeloperoxidase; PMN-MDSCs, polymorphonuclear MDSCs.
Neutrophils are the main source of MPO in the periphery, this might be different in the TME. We stained for MPO expression in the tumor and splenic myeloid populations. In the spleen, MPO is predominantly produced by Ly6G+Ly6Clow cells (median 65% of MPO+ cells) with a minimal contribution of Ly6G−Ly6Chigh cells (median 12% of MPO+ cells) (figure 6C). In contrast, in tumors, the expression of MPO is seen on both Ly6G−Ly6Chigh cells (median 36%) and Ly6G−Ly6Clow cells (median 22%). This suggests both PMN-MDSC and M-MDSC populations contribute to MPO production in tumors.
Since MDSC populations appeared to be the MPO producing cells within the TME, we next looked at whether MDSCs or a subset of MDSCs were critical for antitumor therapy. The B16 HHDII/iDP4 melanoma model selected for these experiments shows evidence of MDSC infiltration which can be depleted by antibodies (figure 6D). Removal of MDSCs provided an antitumor effect (figure 6E, p=0.0459). This model is thus appropriate to see if the Hcit vaccine could overcome the immunosuppression associated with MDSCs. Vaccine-mediated tumor therapy was assessed in the presence of an anti-Ly6G depletion antibody that removes neutrophils/PMN-MDSCs cells or an anti-Ly6C antibody that removes monocytes/M-MDSCs cells (figure 6E). Hcit peptide immunized mice showed an 86% survival advantage over control unimmunized mice. In immunized mice given anti-Ly6G antibody, there was no significant decrease in survival (80% survival, p=0.5248) indicating PMN-MDSCs/neutrophils are not critical for the antitumor response. However, immunized mice given anti-Ly6C antibody showed a significantly worse survival of 20% (p<0.0001) indicating that monocytes/M-MDSC play a critical role in the Hcit vaccine-mediated tumor therapy. Overall, these results indicate an important role for monocytes/M-MDSCs in tumors as the source of MPO which initiates the homocitrullination of proteins.
Homocitrullination can be induced in tumor cells via cyanate or MDSCs
MPO is responsible for driving homocitrullination through the generation of cyanate, our results show MDSCs produce MPO in the TME. Anti-Hcit ELISAs confirm that cyanate is sufficient to drive homocitrullination of recombinant aldolase protein and B16 tumor lysates, showing a significant increase in Hcit residues (p<0.0001, p=0.0050, respectively) similar to that seen with the FCS control (figure 7A).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
In vitro recombinant aldolase and B16F1 cell lines can be homocitrullinated FCS, recombinant aldolase protein, B16F1 cell lysate or whole B16F1 cells were incubated for 10 hours with media only or cyanate. Anti-Hcit ELISA was performed to determine the level of homocitrullination on control of cyanate treated samples (A). Mean, SD and p values are shown, n≥3. Homocitrullinated lysine residues were identified by mass spectrometry analysis (B). Three independent experiments were performed each with multiple injections. Homocitrulline residues (gray) are shown. Some lysine residues were not covered by the stochastic analysis (hashed). In vitro bone marrow-derived MDSCs (BM-MDSCs) were generated from transgenic mice. Cell phenotypes were defined as either CD11b+Ly6G+Ly6Clow (PMN-MDSCs) or CD11b+Ly6G−Ly6Chigh (M-MDSC). Example plots are shown (C). B16F1 cells were incubated with untreated BM-MDSCs, H202 and thiocyanate overnight. The proportion of CD45− cells that stained positive for anti-Hcit antibody was then assessed by flow cytometry (D). Mean and individual experiments are shown after normalization to the untreated sample. Statistical analysis was performed. FCS, foetal calf serum; Hcit, homocitrulline; LPS, lipopolysaccharide; MDSCs, myeloid-derived suppressor cells.
Since MPO is not produced by tumor cells, we next investigated whether extracellular cyanate can induce homocitrullination in B16 cells. Analysis of Hcit in cells lysed after culture demonstrated homocitrullination was significantly increased in cells after incubation with cyanate (p<0.0001) (figure 7A). This demonstrated that proteins from viable tumor cells can undergo homocitrullination implying that cyanate/isocyanic acid can cross the cell membrane to induce intracellular homocitrullination. In addition, mass spectrometry analysis of recombinant aldolase showed the presence of Hcit residues at lysine sites after treatment with cyanate (figure 7B). Aldo74-93Hcit epitope contains one Hcit at residue 87 and Aldo140-157Hcit epitope contains three Hcits at residues 140, 147 and 153 after treatment with cyanate.
We next investigated whether PMN-MDSCs and M-MDSCs were capable of inducing homocitrullination in tumor cells in vitro. Analysis of in vitro grown BM-MDSCs showed a mixed population containing both PMN-MDSCs and M-MDSCs (figure 7C). Staining for MPO showed that all MPO+ cells fell within both the PMN-MDSCs and M-MDSCs populations and MPO expression could be increased following treatment with lipopolysaccharide (LPS)-positive control (figure 7C). Example staining for in vitro grown MDSCs and MPO expression is shown in online supplemental figure 5A,B. The increase in MPO expression on LPS treatment could be attributed to the differentiation of immature MDSC subsets into neutrophils or macrophages, therefore LPS treatment of MDSCs was not used prior to further coculture experiments. Neutrophils drive homocitrullination through production of MPO and H2O2 in the presence of potassium thiocyanate.39 We next set up coculture experiments to determine whether proteins from B16 cells can be homocitrullinated in the presence of in vitro grown MDSCs, thiocyanate and H2O2. Homocitrullination of B16 cells was determined by flow cytometry and normalized to staining on unstimulated cells (figure 7D). No homocitrullination was observed on B16 cells in the absence of the secondary antibody (first bar) but B16 cells showed a low level of homocitrullination even in the absence of additional thiocyanate and H2O2 which significantly increases after incubation with thiocyanate and H2O2 (p=0.0253) but not thiocyanate only. These results suggest that MDSCs are a viable source of MPO but unlike activated neutrophils, MDSC-mediated homocitrullination requires H2O2 in the microenvironment. This confirms that MDSCs can produce MPO which can subsequently induce homocitrullination of tumor antigens.
Discussion
Cancer vaccination aims to induce immune responses that target cancer-specific antigens and destroy tumor cells. We have previously demonstrated that targeting PTM epitopes can be beneficial in eradicating advanced aggressive tumors in animal models.3 4 Here, we demonstrated the potential of targeting homocitrullinated proteins in tumor therapy. We identified homocitrullinated epitopes from four abundant cytoplasmic proteins, aldolase, Bip, Cyk8 and enolase, that are ubiquitously expressed in many cancer types. Potential peptides were identified through prediction of MHC-II binding using IEBD prediction algorithm and strong and moderate binding peptides selected. We have demonstrated that peptide vaccination stimulates homocitrullinated peptide-specific CD4 T-cell responses in HLA-DP4 transgenic mice. Immunogenicity did not obviously correlate with predicted binding scores. It is likely this is due to the predictions being based on the native sequence as well as the T-cell repertoire available. The prediction algorithms are not able to account for the Hcit PTM and we anticipate these modifications may have an influence over binding affinity. We have also shown that healthy donors and patients with cancer have a repertoire of CD4 T cells that can respond to homocitrullinated aldolase peptides. Patients with lung cancer represent ideal candidates to receive therapies targeting homocitrullinated proteins since smoking has been shown as a major contributor to elevated homocitrullination levels in vivo.8 We have yet to confirm the HLA restriction of these responses in humans, the majority of responding healthy donors studied expressed the HLA-DP4 allele and it is possible that this will prove to be the main restricting allele. It is likely that homocitrullinated peptides can stimulate responses restricted through other HLA alleles. We have seen (unpublished data) responses to homocitrullinated peptides restricted by the HLA-DR4 and HLA-DR1 alleles suggesting a broader application to the targeting of homocitrullinated proteins. Data shown here in the murine studies suggest responses stimulated by Hcit peptide vaccination are Hcit specific but the WT peptides are also capable of stimulating T-cell populations. We have shown evidence in humans that CD4 T cells stimulated by the Hcit peptides are Hcit specific but this should be further investigated through TCR isolation and characterization or T-cell cloning from immunized patients.
Our data have demonstrated that vaccination with Hcit peptides stimulated specific CD4 responses that mediated efficient antitumor therapy in an aggressive mouse B16 melanoma model where MDSCs play an immunosuppressive role. We have shown that not all homocitrullinated peptides that were tested were capable of inducing antitumor responses. The Aldo238-256Hcit and Bip328-346Hcit peptides stimulated Hcit-specific T-cell responses but failed in tumor therapy in the B16 tumor model. This implies that not every lysine is converted to Hcit in vivo or not every homocitrullinated peptide is presented by tumors in association with MHC-II. Mass spectrometry analysis also demonstrated that not every lysine within aldolase-A is homocitrullinated.
In the mouse models, we assessed the potential of this tumor therapy using B16 melanoma cell line that constitutively expresses MHC-II, we showed a 50%–90% survival benefit in immunized mice. In the TME responding CD4 T cells can be activated by APCs presenting tumor antigen. Activated CD4 T cells subsequently release cytokines that can have a cytostatic or cytotoxic effect on the tumor cells and/or recruit other immune effector cells such as CD8 T cells.40 41 In this environment, the Hcit peptide-specific response should be effective against MHC-II-negative tumors but in the presence of APCs expressing MHC-II. However, this was not the case as no antitumor responses were observed in mice bearing MHC-II-negative tumors that were immunized with the homocitrullinated aldolase peptides . Tumor therapy was largely dependent on direct recognition of tumor cells suggesting the Hcit peptides are presented by MHC-II on the tumor cells. As most tumors do not constitutively express MHC-II molecules, it was important to examine the therapy in a more physiologically relevant model. The expression of MHC-II on non-APCs can be induced by IFNγ.34 We next engineered the B16 cell line to express MHC-II under the control of an IFNγ-inducible promoter. In this model, tumor therapy was still effective suggesting the potent Th1 CD4 T cells secrete sufficient IFNγ to upregulate MHC-II. Indeed, this was shown to be correct as there was a direct correlation between MHC-II expression and tumor regression.
The cyanate necessary to induce homocitrullination can be produced from spontaneous degradation of urea or through the action of the MPO enzyme.8 9 MPO knockout mice have significantly decreased levels of homocitrullination following the induction of acute inflammation compared with WT mice; thus, suggesting that MPO has a major role in protein homocitrullination in vivo.42 Our data suggest MPO plays a role in intratumoral homocitrullination since MPO expression correlates with tumor regression in vaccinated mice and the inhibition of MPO abrogates the effect of the vaccine. MPO is produced by subsets of CD11b+ cells including neutrophils, macrophages and monocytes that release MPO either into phagosomes or the extracellular space.8 43 Flow cytometry staining confirmed that B16 cells do not express MPO, but expression of MPO is detected on CD11b+ cells within the tumor. Further analysis of CD11b+MPO+ cells in the TME shows the presence of two subsets, Ly6G+Ly6Clow and Ly6G−Ly6Chigh. These subsets have previously been defined as PMN-MDSCs and M-MDSCs, respectively.37 44 Ly6G+Ly6Clow is a phenotype shared with neutrophils, Ly6G+Ly6Chigh is found on macrophages, with many additional markers proposed to differentiate these populations.44 45 Our staining correlates with reports suggesting the expression of MPO is upregulated in tumor-associated MDSCs.37 Depletion of MDSCs via antibodies that target Ly6G and Ly6C attenuates the growth of lung carcinoma cell lines in vivo.46 Depletion studies in our tumor model revealed that removal of the Ly6G+ (PMN-MDSCs) fraction did not affect tumor growth and had no significant effect on vaccine-mediated therapy. This suggests Ly6G+ cells play a minimal role in MPO production in the TME. Depletion of the Ly6C+ (M-MDSCs) cells had a more profound effect on tumor growth, providing a significant survival advantage as a standalone treatment. The Ly6C+ M-MDSC has enhanced suppressive capacity in tumors47 perhaps explaining the effect seen with the depletion of this population. MDSCs can reduce priming of Th1 responses in tumors and there are reports of reduced priming after systemic vaccination, however, this effect is dependent on MHC-II expression levels.48 49 We have shown that Th1 responses can be stimulated by vaccination in the presence of tumor suggesting that circulating MDSCs do not influence the stimulation of Hcit vaccine-specific Th1 responses. However, immune suppressive activity of MDSCs could impact CD4 T cells in the TME. We would like to highlight that despite this, in the aggressive B16 model, the Hcit vaccine is able to overcome the immune suppressive activity of MDSCs and mediate strong tumor therapy, suggesting that the CD4 T-cell responses are not affected by MDSC suppression. We therefore propose that a strong Th1 response can potentially help switch the suppressive tumor environment inflammatory which mediates tumor regression.
The depletion of Ly6C (M-MDSCs) cells significantly reduced the efficacy of the Hcit vaccine to the level seen in the control group that had antibody depletion alone. This suggests a role for the Ly6C+ cell fraction, in particular the Ly6Chigh cells, as the source of MPO and homocitrullination in tumors. Interestingly, MPO production in tumor was associated with Ly6C+ cells, whereas in spleen we have shown that this population does not produce MPO, implying some tissue specificity exists. Infiltration of MPO expressing cells in colorectal and ovarian cancer is a positive prognostic indicator,50 but the oxidants produced by MPO can cause DNA damage and promote mutations.51 Whether MPO substrate, H2O2, is produced by tumor or MDSCs remains to be shown, but we have demonstrated that cyanate can diffuse into tumor cells and promote homocitrullination of proteins. Like citrullinated proteins, homocitrullinated proteins could be digested during autophagy and presented on MHC-II.3
Autoimmune associated homocitrullination is thought to be driven by neutrophils not MDSCs,9 39 but both mechanisms result in the same modification. Therefore, there is the potential for on target/off tumor effects specifically on inflamed tissues. In spite of this possibility no toxicity was observed in our murine studies up to 50 days post immunization. Future studies with Hcit peptides will need to carefully monitor on-target off-tumor autoimmune effects, although in rheumatoid arthritis, debate surrounds the relative importance of homocitrullination as a driver of disease.6 28 Smokers have increased serum thiocyanate levels which drives homocitrullination and increases the risk of autoimmune disease, but this potentially also provides a target for vaccination.52 The possibility that autoimmunity may be exacerbated in patients that already have the disease should not be underestimated and signs of autoimmunity should be monitored and patients with known underlying autoimmunity excluded from any future clinical trial.
This study provides the first evidence that homocitrullinated peptide vaccination generates Hcit-specific CD4 T-cell responses, leading to a potent antitumor effect. Tumor therapy is largely mediated via direct recognition mechanisms. We further demonstrate that MPO-producing MDSC-like cells are likely to be involved in homocitrullination within the TME. We therefore conclude that homocitrullinated proteins are excellent targets for cancer therapy especially those cancers which recruit MDSCs.
Data availability statement
Data sharing not applicable as no datasets were generated and/or analyzed for this study. Data and materials are available upon reasonable request from the corresponding author.
Ethics statements
Ethics approval
All studies using human blood were approved by the local ethics committee and informed consent was obtained from all subjects involved. Samples were obtained from patients (ethics review: Nottingham Biobank APC170 no. 13-EM-0218 or IRAS project ID 238448) at Nottingham City Hospital and healthy donors (ethics review: 161-1711) from the University of Nottingham following University of Nottingham review and ethical approval. All animal work was carried out under a UK Home Office approved project license and in accordance with EU Directive 2010/63/EU.
Acknowledgments
The authors would like to thank Dr Tina Parsons, Dr Samantha Paston and Dr Mireille Vankemmelbeke for help in proofreading the manuscript.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors LGD and VAB directed the study. KWC, WX, PS, ID, MG, DB, CC, AKM, SS, RHC, PV, DW and RLM performed experiments. LGD, VAB, KWC and WX designed experiments, analyzed the data and wrote the paper.
Funding This work was funded by Scancell Ltd.
Competing interests KWC, WX, VAB and LGD have ownership interest in the patent. LGD is a director and shareholder in Scancell Ltd. All authors are employees of Scancell Ltd. except DB, CC and AKM.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.