Article Text

Abstract
Background Programmed death-1 (PD-1) and programmed death-ligand 1 (PD-L1) inhibitors can cause unique immune-related adverse effects due to non-specific immunological activation. However, less is known about adverse effects of these drugs in the eye.
Methods Two adverse event databases were retrospectively reviewed. The two databases consisted of a routine adverse event database and a serious adverse event database of expeditiously submitted reports. Patients with any malignancy who had ocular adverse events while on PD-1/PD-L1 inhibitor treatment were included. Patients received nivolumab, pembrolizumab, atezolizumab or durvalumab alone or in combination with other anticancer agents per each trial’s protocol. Databases were queried up to May 19, 2020.
Results In the routine adverse event database, 272 adverse events from 213 patients were reported and in the serious adverse event reporting database, 59 ocular adverse events from 47 patients were reported. A lower estimate of the prevalance from the routine adverse event database showed 259/7727 patients on study treatment arms reporting ocular adverse events (3.3% prevalence). Excluding trials that do not report lower grade adverse events to the routine adverse event database results in a higher end estimate of 242/3255 patients on study treatment arms reporting ocular adverse events (7.4% prevalence). Ocular events occurred early after drug initiation (routine database: median 6 weeks, IQR 0–16, serious adverse events database: median 11 weeks, IQR 6–21). The median Common Terminology Criteria for Adverse Events grade was grade 1 (mild) (IQR 1–2) and grade 2 (moderate) (IQR 2–3) for the routine database and the serious adverse events database, respectively. In-depth analysis of the serious adverse event reports revealed varying degrees of clinical workup, with 30/47 patients (64%) receiving ophthalmological evaluation and 16/47 (34%) of patients having to delay or discontinue treatment. However, 16/47 (34%) patients experienced resolution and 14/47 (30%) patients experienced at least some improvement.
Conclusions This is one of the largest analyses of ocular adverse events in patients treated with PD-1/PD-L1 inhibitors in the USA. We found ocular adverse events are rare complications of PD-1/PD-L1 inhibitor therapy, can be severe enough to cause treatment discontinuation/delay, and may not always be appropriately evaluated by eye specialists. Standardized plans for ophthalmology evaluation and management of ocular toxicities are needed in studies of patients treated with PD-1/PD-L1 inhibitors.
- immunotherapy
- inflammation
- drug therapy
- combination
Data availability statement
Data are available on reasonable request. Please contact ES, sharone@mail.nih.gov.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Background
The programmed death-1 (PD-1) and programmed death-ligand 1 (PD-L1) inhibitors are a class of checkpoint inhibitors approved to treat a variety of malignancies, including melanoma, Hodgkin lymphoma, non-small-cell lung carcinoma and other advanced-stage cancers.1 The first PD-1/PD-L1 drugs to be approved were pembrolizumab and nivolumab in 2014 for melanoma, followed by atezolizumab in 2015 for non-squamous small cell lung cancer, and durvalumab in 2017 for urothelial carcinoma.2 Since then, the number of clinical trials investigating these drugs have increased dramatically. An April 16 2020, search of clinicaltrials.gov reveals 3097 clinical trials testing the PD-1/PD-L1 inhibitors nivolumab, pembrolizumab, atezolizumab and durvalumab.
The PD-1/PD-L1 inhibitors are known to cause unique immune-related adverse effects in almost every organ system due to their non-specific immune activation. Common immune-related adverse effects include pneumonitis, colitis and hepatitis.1 3 Endocrinopathies such as type 1 diabetes mellitus and neurotoxicity have also been reported.3 PD-1/PD-L1 inhibitor-associated adverse events (AEs) in the eye are also known to occur, but are rare and therefore less well-studied. Case reports, case series, safety analyses, and some observational studies have shown that these drugs can cause inflammatory eye disease, such as uveitis,3–9 scleritis,8 dry eye syndrome,4 5 9 10 optic neuropathy9 and orbital myopathy.11 Additionally, ipilimumab, a cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) checkpoint inhibitor in a similar class as PD-1/PD-L1 inhibitors, has been documented to cause potentially blinding conditions such as optic neuropathy,12 13 orbital inflammation,14 15 ophthalmopathy,16 and orbital apex syndrome.17 Though uveitis is a potentially blinding condition, only a few large-scale studies focusing on ocular AEs have been performed to date.7 11 18 19 So far, these studies have shown that inflammatory eye disease AEs tend to be rare, ranging from a prevalence of <1% of uveitis in drug manufacturers prescribing information20–24 to 3.6% in a large retrospective review of a large ophthalmology electronic health record registry.19
In lieu of large prospective observational studies, retrospective analyses of large ocular AE databases can be performed, such as the recent analysis of the Food and Drug Administration’s Adverse Events Reporting System Database (FAERS) conducted by Fang et al.18 or the analysis of the American Academy of Ophthalmology Intelligent Research in Sight Registry (IRIS).19 The FAERS database has a large sample size, but is limited by its lack of clinical and demographic information, while the IRIS registry has detailed ophthalmologic notes but less information on non-ocular conditions. In this study, we analyze two databases of AEs from the US National Cancer Institute’s Cancer Therapy Evaluation Program (CTEP), the largest public sponsor of oncology clinical trials globally. This study, therefore, aims to analyze ocular AEs associated with PD-1/PD-L1 inhibitors submitted to the two separate and overlapping CTEP databases.
Methods
CTEP databases
Study sites report AEs to CTEP through two pathways, resulting in two distinct and partially overlapping AE databases. The first database is composed of AEs submitted expeditiously to the National Cancer Institute as serious AEs (SAEs) and will be hereafter referred to as the SAE Database. The second database is composed of AEs reported as part of required clinical data submissions for routine general study monitoring and will hereafter be referred to as the routine AE database. Study sites use the Common Terminology Criteria for Adverse Events (CTCAE) as a guide when making AE submissions, and can submit reports of AEs such as watering eyes or blindness. These terms have strict definitions (eg, blindness is defined as 20/200 vision or worse in the affected eye).25
The SAE database is derived from the CTEP Adverse Event Reporting System (CTEP-AERS). CTEP-AERS is the formal mechanism for study sites to expeditiously report serious SAEs, for sponsor oversight of clinical trial safety and for determination of expedited reporting to the United States Food and Drug Administration. AEs that study sites consider to be serious are reported to CTEP-AERS within 24 hours to 10 days of the site learning of the AE. Reporting time frames vary based on severity and are defined in each trial’s protocol. Clinical trials of all phases submit AE reports through CTEP-AERS. For simplicity, events that were expeditiously reported to CTEP-AERS will be subsequently referred to as SAEs.
The SAE database is composed of reports that include clinical information such as patient demographics, brief oncological history, description of the SAE and select clinical notes, imaging, pathology and laboratory reports provided by the study site. Each report can include multiple AEs, for example, ‘uveitis’ and ‘eye pain’. In addition, investigators can include lower-severity ocular AEs in these reports if they occurred in conjunction with other serious events that required expeditious reporting.
In contrast to the SAE database, the routine AE database is derived from the CTEP Clinical Data Update System (CDUS), which obtains a full clinical trial dataset from most phase 1 and phase 2 trials conducted within the CTEP-supported networks on a quarterly basis. This dataset includes treatment, response, and AE data. The routine AE database includes patient demographics, the start date of the treatment course on which the ocular AE occurred, grading of its severity (see ‘AE Severity grading’), and the patient’s assigned treatment arm. If a patient experiences an AE event across multiple study visits and the study site reports them, multiple entries will appear in the CDUS database. Of note, there is partial overlap between the two databases. While not all studies submit routine AEs to CTEP, for those that do, the routine AE database includes both the AEs not requiring expeditious reporting as well as the SAEs submitted expeditiously through CTEP-AERS. Both databases were used for this analysis with duplicate AEs reconciled.
AE severity grading
For both databases, AEs are classified according to the organ system they affect (eg, ‘EYE DISORDERS’), and severity is graded per the CTCAE,25 a standard AE grading system in oncology drug development. The attribution of the study drug to the AE is assessed on a scale of ‘unrelated’, ‘unlikely’, ‘possible’, ‘probable’ or ‘definite’. In the SAE database, study investigators and CTEP physicians assess AE grading and attribution for trials conducted under CTEP sponsorship. Attribution is assessed on the level of the individual study agents. As per US Food and Drug Administration regulations, ultimate determination of attribution for an event requiring expeditious reporting is the responsibility of the sponsor, but site investigators are required to make an initial assessment. For the routine AE database, study investigators assess AE grading and attribution, and attribution is assigned to the study treatment in its entirety. However, these AE reports are regularly reviewed by the investigators and CTEP physicians overseeing the trial.
Eligibility criteria
We queried both CTEP AE databases for studies using at least 1 PD-1/PD-L1 inhibitor (search terms ‘nivolumab’, ‘pembrolizumab’, ‘atezolizumab’ and ‘durvalumab’) up to May 19 2020.
Results
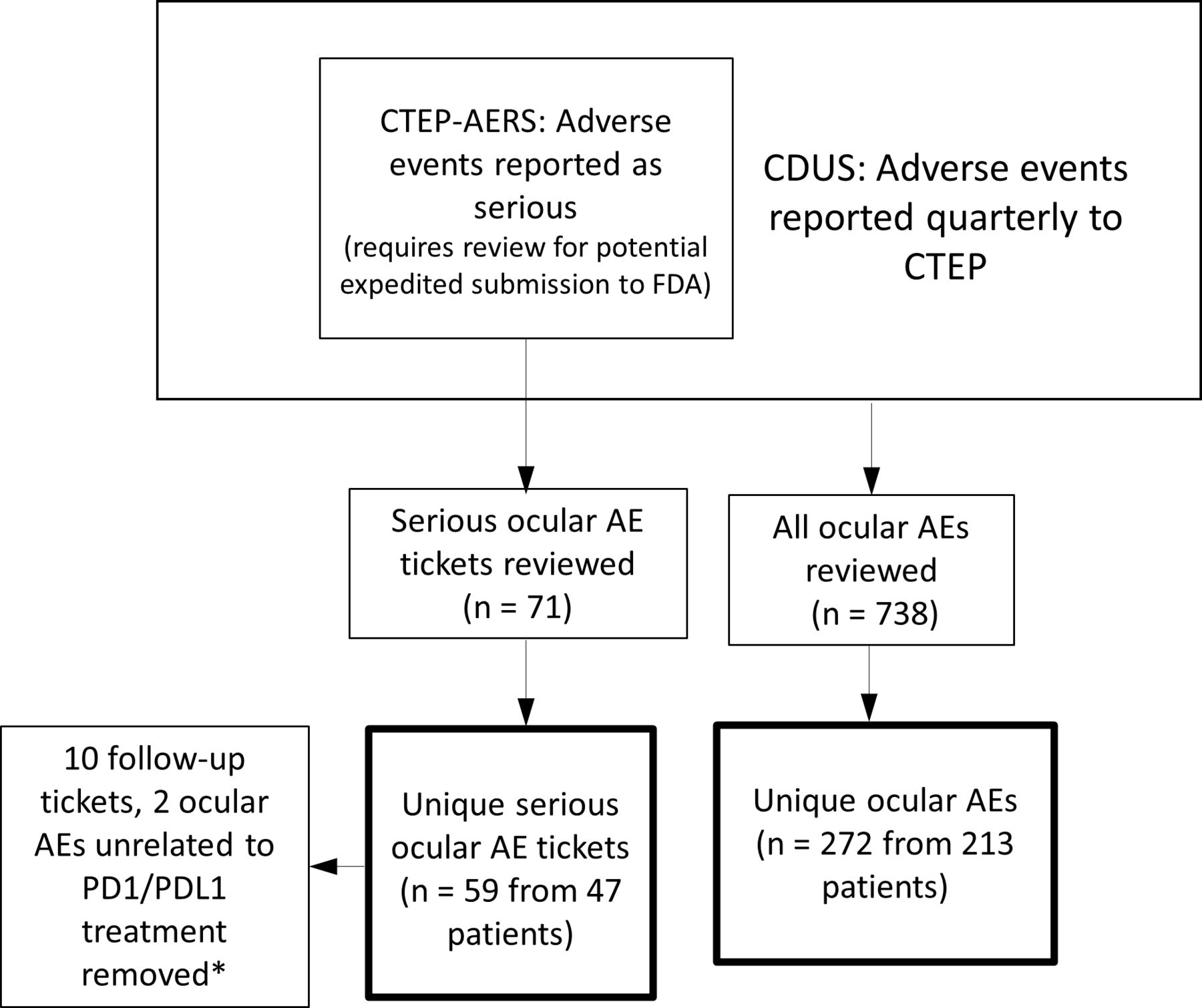
At the time of database queries, CTEP had opened 116 clinical trials using at least 1 PD-1/PD-L1 inhibitor as study drugs, either as monotherapy or in combination with other agents. From these 116 trials, 7727 patients were enrolled in interventional arms with at least one PD-1/PD-L1 inhibitor. Demographic information for the 7727 patients exposed to the PD-1/PD-L1 inhibitors is presented in table 1. A total of 272 unique ocular AEs from 213 patients were found in the routine AE database and a total of 59 unique SAEs from 47 patients were found in the SAE database (table 1, figure 1). The prevalence of all persons with reported ocular AEs is potentially as low as 259/7727 persons (3.3%).
An adjustment to the denominator of patients within the CDUS database is necessary for assessment of the prevalance of ocular AEs of any grade. As the denominator of 7727 does not generally include AEs from phase III trials, (unless those AEs were expeditiously reported to CTEP-AERS) the 3.3% prevalence likely underreports the true prevalance. Excluding trials that do not report routine AEs to the CDUS database results in a total patient population of 3255 patients on treatment arms involving the study drugs of interest. Since the number of patients who had ocular AEs on this subset of trials is 242 patients, the estimate of the prevalence of ocular AEs could be as high as 242/3255 (7.4%).
Patient characteristics

Depiction of CTEP adverse event databases and workflow. *One ocular AE occurred before PD1/PDL1 exposure, one ocular AE occurred over 3 years after last PD1/PDL1 exposure.; CTEP-AERS, Cancer Therapy Evaluation-Program Adverse Events Reporting system; CDUS, Clinical Data Update System; FDA, Food and Drug Administration; PD-1 Programmed Death 1; PD-L1, Programmed-Death Ligand 1.
Within the routine AE database, the most common study drug associated with ocular AEs was nivolumab, reflecting the higher number of trials sponsored by CTEP using this agent. In the routine ocular AE group, 103 patients (48%) were exposed to both nivolumab and ipilimumab, a CTLA-4 inhibitor. Sixty-three patients (30%) were exposed to nivolumab alone. Twenty-seven patients (13%) were on pembrolizumab, 20 (9%) were on atezolizumab and 0 patients were on durvalumab. The median CTCAE grade was 1 (IQR 1–2).
In the serious ocular AE group, 10/47 patients (21%) were on nivolumab and 15/47 patients (32%) were on both nivolumab and ipilimumab. Fourteen patients (30%) were on pembrolizumab, six (13%) were on atezolizumab and two (4%) were on durvalumab. The median CTCAE toxicity grade for the events in the SAE database was 2 (moderate) (IQR 2–3).
For patients in the routine AE database, the median age at time of study enrollment was 58 (IQR 40–69). 107 (50%) patients were female and 175 (82%) were white (table 1). For the 47 patients in the SAE database, the median age at the time of ocular AE onset was 59 years (IQR 40–66). Of these 47 patients, 24 (51%) were female and 36 were white (77%) (table 1). Most patients in the SAE database had metastatic disease (32/47, 68%) and patients had a wide variety of malignancies (table 1). The most common malignancy was melanoma (eight patients), followed by invasive breast carcinoma (five patients). Ocular AEs occurred fairly early during PD-1/PD-L1 inhibitor treatment. In the routine AE database, the ocular AE occurred a median of 6 weeks (IQR 0–16) after starting the study treatment, and in the SAE database, the ocular AE occurred a median of 11 weeks (IQR 6–21) after study drug initiation (table 1).
Ocular AEs
Ocular AEs ranged from specific diagnoses, such as ‘uveitis (Vogt-Koyanagi-Harada syndrome, VKH)’, to descriptions of symptoms, such as ‘eye pain’. Ocular AEs that were attributed to the study treatment (as assessed by the study investigators and CTEP physicians, see the Methods section), are presented in more detail in online supplemental table 1.
Supplemental material
An analysis of the eight melanoma patients with serious ocular AEs found three patients on nivolumab and ipilimumab, one on nivolumab only and four on pembrolizumab. Of these eight melanoma patients, one patient had eye pain and blindness, one patient had eye pain and uveitis, one patient had anterior uveitis, two patients had rhegmatogenous retinal detachments requiring surgical repair, one patient had diplopia and two patients had blurred vision. No melanoma patients in this cohort were diagnosed with VKH disease.
A total of 52 patients from both databases had multiple ocular AEs, though only 10 patients had multiple ocular AEs that were definitive ophthalmological diagnoses (not merely visual symptoms). For the serious ocular AEs dataset, 7/47 patients had multiple serious ocular AEs, and only two patients had more than one definitive ophthalmologic diagnosis. Most patients with SAEs (28/47, 60%) had both ocular AEs and AEs from other organ systems. Their non-ocular AEs had a median CTCAE grade of 3 (IQR 2–3) and included a wide variety of conditions (online supplemental table 2). Three patients with serious ocular AEs died—one due to progressive disease, one due to a duodenal hemorrhage and one whose cause of death was not definitively determined but likely due to cancer progression.
Within the routine AE database, a total of 229/272 (84%) AEs were ‘possibly’, ‘probably’ or ‘definitely’ attributed to the study treatment. Meanwhile, 35/59 (59%) of the ocular events in the SAE database were ‘possibly’, ‘probably’ or ‘definitely’ attributed to the PD-1 or PD-L1 inhibitor.
In the routine AE database, the most common ocular AE attributed to study treatment was blurred vision (58 patients), followed by dry eye (44 patients), eye pain (15 patients), conjunctivitis (13 patients), periorbital edema and watering eyes (10 patients) and uveitis (9 patients) (online supplemental table 1). In the SAE database, the most common ocular AE attributed to a PD-1/PD-L1 inhibitor was uveitis (four patients), though patients also had other inflammatory ocular conditions such as episcleritis (three), scleritis (one), sclerouveitis (one), neuroretinitis (one) and filamentary keratitis (one) (online supplemental table 1).
Serious ocular AE clinical course
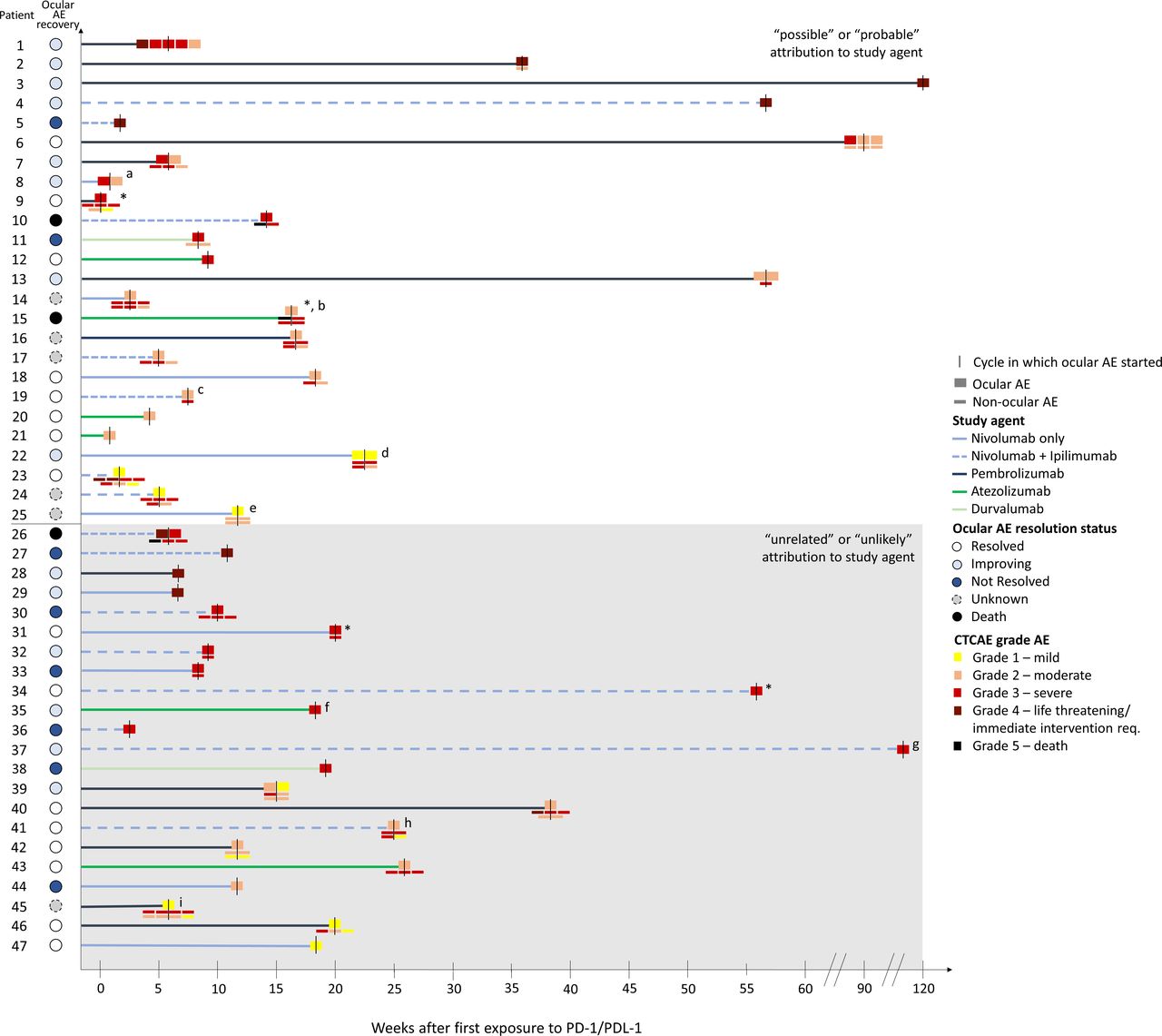
As patients had a wide variety of serious ocular AEs, their workup and treatment varied significantly (table 2, figure 2). A total of 31/47 patients (66%) required hospitalization or urgent intervention due to the ocular AE. However, though all patients with serious ocular AEs had eye-related concerns, only 64% were evaluated by an ophthalmologist. A total of 22/47 patients (45%) received some form of head, brain or eye imaging (MRI, CT, CT angiography, fluorescein angiography), 8 (17%) received a lumbar puncture and 2 (4%) received some form of eye biopsy. A total of 4/47 patients (9%) underwent special laboratory testing (table 2).
Clinical course, workup, treatment and recovery of patients with ocular AEs reported as serious
{kind=link}
{kind=link}
Graphical representation of all patients with serious ocular AEs. *Denotes patients with ocular AEs affecting the external eye (patient 9 periorbital swelling, patient 15 paralytic lagophthalmos of left eye, patient 31 right eye swelling, patient 34 preseptal cellulitis). aPatient 8 also on dacarbazine and doxorubicin, but ocular AE unlikely to be attributed to these agents. bPatient 15 was on Bevacizumab and Atezolizumab but their last dose of atezolizumab was 80 days before the ocular AE. Both agents were possibly attributed to the ocular AE. cPatient 19 was also on brentuximab vedotin. The ocular AE was probably attributed to Nivolumab or Ipilimumab, and possibly attributed to brentuximab vedotin. dPatient 22’s ocular AE also possibly attributed to Cabozantinib. ePatient 25’s ocular AE also possibly attributed to Etoposide, Carboplatin, and underlying small cell lung cancer. fPatient 35 also on Carboplatin and Etoposide, unrelated to ocular AE. gPatient 37 also on Cabozantinib, unrelated to ocular AE. hPatient 41 also on entinostat, entinostat unlikely to be attributed to ocular AE. iPatient 45 also on talimogene laherparepvec, unrelated to ocular AE. AE, adverse event; CTCAE, Common Terminology Criteria for Adverse Events.
Focusing on the 25 patients in the SAE database whose ocular AE was attributed to the PD-1/PDL-1 inhibitor, 13/25 (52%) required hospitalization or urgent intervention for their ocular AE, and 19/25 (74%) patients were seen by an ophthalmologist (table 2). Of the 35 ocular AEs experienced by these 25 patients, over half (18/35, 51%) were potentially vision threatening. Corticosteroids (whether in intravenous, oral or steroid eye-drop form) were the most common treatment. A total of 6/25 patients (24%) had to discontinue and 5 patients (20%) had to delay their PD-1/PD-L1 inhibitor treatment because of the ocular AE (table 2). Fortunately, most patients (16/25, 64%) with ocular AEs attributed to the study drug had resolution or improvement in their symptoms, with 8 patients having resolution and 8 patients having some improvement. More detailed summaries of the clinical courses for these 25 patients can be found in online supplemental table 3.
Discussion/Conclusion
This retrospective review of two AE databases is one of the largest analyses of serious ocular AEs due to PD-1 and PD-L1 inhibitors (nivolumab, pembrolizumab, atezolizumab and durvalumab) in the USA to date. Our study found a total of 27 AEs that were potentially vision-threatening inflammatory eye conditions (13 uveitis, 1 sclerouveitis, 3 episcleritis, 1 scleritis, 1 neuroretinitis, 1 iritis, 5 keratitis, 1 optic neuritis, 1 eye inflammation) from both databases.
The prevalence of uveitis in our cohort is 14/7727 patients (0.2%), which is higher than the estimated uveitis prevalence in general US populations (57–115 cases/100,000 persons, 0.05%–0.1%).26–28 However, we acknowledge that ocular AEs may have been underreported in our cohort, as Bitton et al11 report a 0.4% prevalence of uveitis in anti-PD-1/PD-L1 immunotherapy. Though not directly comparable to an analysis of reported AEs, Sun et al19 report that 3.6% of patients seen in ophthalmology offices on checkpoint inhibitors (both anti-CTLA-4 and anti-PD-1/PD-L1) experience immune-related eye AEs. However, our results are still consistent with drug manufacturers’ prescribing information for the PD-1/PD-L1 inhibitors, which report a less than 1% incidence of uveitis or <10% of iridocyclitis.20–24
One ocular AE that likely was underreported in our cohort was dry eye. We found 50 cases of dry eye (0.6% prevalence), though literature suggests the prevalence of dry eye in PD-1/PD-L1 inhibitors ranges from 1% to 24%.4 18 29 Dry eye is usually a CTCAE grade 1 event and participating CTEP study sites are not required to report grade 1 events if they are not felt to be related to study treatment and/or do not meet the definition of ‘serious’. Because of this, there may have been underreporting of dry eye, especially if patients experienced other AEs with higher CTCAE grades.
When looking at serious ocular AEs by each PD-1/PD-L1 inhibitor in the CTEP-AERS database, we found serious ocular AEs in 2 patients on durvalumab, compared with 10 patients on nivolumab, 15 patients on nivolumab and ipilimumab, 14 patients on pembrolizumab and 6 patients on atezolizumab. However, these proportions are roughly in line with the number of patients treated with these particular agents on CTEP-sponsored trials to date.
As noted, 103 patients in the routine AE database and 15 patients in the SAE database were on combination ipilimumab and nivolumab. Ipilimumab is a CTLA-4 inhibitor that has been previously described to cause ocular AEs,3 4 7 18 30 and the combination of nivolumab and ipilimumab is known to generally increase the rate of AEs.31 As the majority of ipilimumab use is now in combination with nivolumab, it would be difficult to tease out the relative contribution of PD-1/PD-L1 inhibitors versus CTLA-4 inhibitors and thus more research is needed.
We found that when ocular AEs were reported as serious, patients frequently had to discontinue or delay their PD-1/PD-L1 inhibitors. Of the 25 patients whose serious ocular AEs were attributed to their study agent, 5/25 (20%) discontinued their study agent due to the ocular AE. Though we did not find any studies looking at treatment discontinuation due to ocular AEs, previous studies report anywhere from 4% to 39% of patients on PD-1/PD-L1 inhibitors discontinue their drugs due to any treatment-related AEs.31 32
Due to our reliance on investigator-provided clinical information, follow-up data or even ophthalmology course notes were frequently unavailable. We did not have detailed clinical information in our routine AE dataset, and even in our SAE dataset with more detailed clinical information, information was sometimes incomplete. We acknowledge that this design limited our ability to make a detailed assessment of patients’ ocular clinical histories. We attempted to rectify this situation by requesting more information for 20 patients with ocular AEs reported as serious. We ultimately received satisfactory responses from 19 study sites.
The fact that NCI uses two overlapping databases makes it somewhat challenging to calculate the prevalence of ocular AEs. As noted, we identified a total of 259 ocular AEs out of 7727 patients who were exposed to PD-1/PD-L1 inhibitors in trials of all phases. However, the CDUS database likely underreports the prevalence, as this routine AE database does not generally include adverse events from phase III trials unless those AEs were expeditiously reported to CTEP-AERS. Thus, we estimate the 3.3% prevalence likely represents the lower estimate of the true prevalence of ocular AEs. An adjustment to the CDUS database denominator was necessary to obtain the higher estimate of the true prevalence of ocular AEs. By excluding trials that do not report routine AEs to the CDUS database, we were able to find a total patient population of 3255 on treatment arms involving the study drugs of interest. Since the number of patients who had ocular adverse events on this subset of trials is 242, an estimate of the prevalence of ocular AEs could be as high as 242/3255 (7.4%). This range of 3.3% to 7.4% is the best estimate of the true prevalence available from the CDUS database, but even this range could be affected by underreporting, as the database relies on investigator-provided data.
We also acknowledge that reporting of ocular AEs varies significantly. Because the CTCAE includes several descriptions of ocular symptoms and study investigators used the CTCAE as the basis for their submission, several ocular AEs were descriptions of ocular symptoms and not formal ophthalmologic diagnoses (eg, ‘decreased vision’, ‘blurred vision’ and ‘eye pain’). Because not every patient received a formal ophthalmological examination, study sites may have only been able to report eye symptoms instead of specific diagnoses. Furthermore, variability in ocular symptom reporting could have led to delays in detection and appropriate treatment. For example, one patient had blurry vision for a week but only mentioned it during a routine study visit. Her nivolumab was held and she underwent an emergency ophthalmology consultation where it was found that she had a retinal detachment and glaucoma. Though the nivolumab was ultimately determined to be unrelated to the patient’s ocular AEs, this example illustrates how a treatment-related ocular AE could have easily been missed. In addition, we found the surprising statistic that only 64% of patients with a serious ocular AE were evaluated by ophthalmology, indicating that 36% of patients were not evaluated by ophthalmology. These points reinforce the importance of educating patients receiving immune checkpoint inhibitors about the necessity of monitoring their eyes and vision, and having study sites help patients with eye symptoms seek prompt ophthalmological evaluation.
Overall, this analysis of two large AE databases found that ocular AEs in PD-1/PD-L1 therapy are rare, can cause inflammatory ocular disease and can cause treatment delays or treatment discontinuation. Patients’ symptoms tended to improve or resolve with corticosteroid therapy. However, due to significant variability in ophthalmologic consultation, workup, treatment and reporting of these AEs, standardized clinical evaluation guidelines for ocular events and enhanced protocol guidelines on reporting of ocular AEs are needed to guide future PD-1/PD-L1 clinical trials or clinical use.
Data availability statement
Data are available on reasonable request. Please contact ES, sharone@mail.nih.gov.
Ethics statements
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Twitter @Helen Chen
Contributors LY, HS, HC, HNS and ES contributed to the conception and design of the study. LY, JM, SF, ES contributed to the acquisition of data. LY, SF, HS, HC, HNS, ES contributed to the analysis and interpretation of the data. LY, HNS, SF, ES wrote the original draft of the manuscript. All authors gave final approval of the version of the manuscript to be published.
Funding This research is supported by the NIH intramural research program. This research was also made possible through the NIH Medical Research Scholars Program, a public–private partnership supported jointly by the NIH and contributions to the Foundation for the NIH from the Doris Duke Charitable Foundation, Genentech, the American Association for Dental Research, the Colgate-Palmolive Company and other private donors.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.