Article Text

Abstract
M2 macrophages promote tumor progression and therapy resistance, whereas proimmunogenic M1 macrophages can contribute to the efficacy of cytostatic and immunotherapeutic strategies. The abundance of M2 macrophages in the immune infiltrate of many cancer types has prompted the search for strategies to target and eliminate this subset. From our prior experiments in syngeneic mouse tumor models, we learned that pharmacological inhibition of mitogen-activated protein kinase kinase (MEK) did not merely result in tumor cell death, but also in the modulation of the tumor immune infiltrate. This included a prominent decrease in the numbers of macrophages as well as an increase in the M1/M2 macrophage ratio. Investigation of the mechanism underlying this finding in primary murine macrophage cultures revealed that M2 macrophages are significantly more sensitive to MEK inhibition-induced cell death than their M1 counterparts. Further analyses showed that the p38 MAPK pathway, which is activated in M1 macrophages only, renders these cells resistant to death by MEK inhibition. In conclusion, the dependency of M2 macrophages on the MEK/extracellular-signal regulated kinase (ERK) pathway empowers MEK inhibitors to selectively eliminate this subset from the tumor microenvironment.
- macrophages
- drug evaluation
- preclinical
- immunomodulation
- tumor microenvironment
Data availability statement
Data are available upon reasonable request. Antibody lists used in DigiWest analyses can be provided upon request.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Immune checkpoint blockade (ICB) shows significant clinical efficacy against tumors harboring high numbers of somatic mutations, corresponding to elevated tumor immunogenicity resulting from the expression of mutanome-encoded neoantigens. The therapeutic impact of ICB on less immunogenic tumors is, however, still limited.1 Tumor immunogenicity can be enhanced through treatment with cytostatic drugs. This results in cell-death-related proinflammatory ‘danger’ signals and increases the availability of tumor antigen for dendritic cell-mediated cross-presentation to T cells.2 Therefore, regimens combining cytostatic drugs with ICB may be used to treat cancer indications that do not respond to either of the single-agent treatments. We and others recently reported that pharmacological inhibition of mitogen-activated protein kinase (MAPK) kinase (MEK) constitutes a highly promising cytostatic strategy in this respect, in that combination with immunostimulatory antibodies results in increased antitumor efficacy compared with single-agent treatment in syngeneic mouse tumor models. This was shown for ICB using PD-L1-blocking antibodies as well as for agonist antibodies addressing the immunostimulatory receptors OX40, CD137, and CD40.3–5
An important aspect of the mechanism of action of MEK inhibitors (MEKi) in this context is the induction of tumor cell death without hampering the function of dendritic cells and T cells. Furthermore, we found that the antitumor efficacy of MEK inhibition is not only limited to the induction of tumor cell death, but also involves modulation of the tumor immune cell infiltrate at several levels.5 This includes the elimination of the vast majority of macrophages, which are known to accumulate in many experimental and human cancer types, thereby contributing to the subversion of the antitumor T-cell response.6 7 As a consequence of MEKi treatment, the balance between macrophage subsets in the tumor microenvironment was found to shift from the tumor-nurturing M2 type toward the proimmune M1 type, suggesting that M2 macrophages are more sensitive to this drug than their M1 counterparts. In this study, we investigated the mechanism underlying this differential impact of MEKi on macrophage subsets. We found that activation of the p38 MAPK pathway in M1 macrophages makes these cells independent of the MEK/ERK pathway and thereby rescues them from death by MEK inhibition, while the survival of M2 macrophages critically depends on the MEK/ERK pathway.
Methods
Mice and in vivo experiments
C57BL/6-Ly5.1 mice were bred in animal facilities of the German Cancer Research Center. Detailed information on in vivo experiments, including the handling and dosing of small-molecule drugs and antibodies, can be found in online supplemental methods. All animal experiments were performed on the basis of prior approval by the ethical authorities. Mice were sacrificed if signs of distress were noticed, when termination criteria were reached, or analyses were performed at specific time points.
Supplemental material
Patient samples
Informed written consent was obtained from all participants before sample collection. Collection of human material and clinical data was approved by the local ethics committees and conducted in accordance with the regulations of the tissue biobanks and the Declaration of Helsinki.
Cell lines, culture, and cytotoxicity assays
Murine PDA30364 cells and bone marrow-derived macrophages were generated, cultured, and used in cytotoxicity assays as described in Baumann et al5 and online supplemental methods. Dual cytotoxicity assays were performed in the presence of 2 µM BIRB 796 and varying concentrations of MEKi GDC-0623.
Human monocytes were isolated from peripheral blood mononuclear cells using the pan monocyte isolation kit (Miltenyi Biotec) and differentiated ex vivo with cytokines into M1-like or M2-like macrophages as described in online supplemental methods. Cytokine concentrations are indicated in the figure legends. These cells were used in cytotoxicity assays as described before5 and in online supplemental methods.
Drugs and reagents
MEKi GDC-0623 (A-1181; Chemgood), p38 MAPKi BIRB 796 (S1574; DP, Selleckchem), murine multimeric CD40L (AG-40B-0020; Adipogen), poly I:C (tlrl-pic; InvivoGen), lipopolysaccharide (LPS) (L4391; Sigma), recombinant murine interferon-γ (IFNγ) (12343536; ImmunoTools), murine macrophage-colony stimulating factor (M-CSF) (12343115; ImmunoTools), murine interleukin (IL)-4 (12340042; ImmunoTools), murine IL-10 (12340105; ImmunoTools), anti-mouse CSF1R (BP0213; BioXCell), recombinant human M-CSF (11343115; ImmunoTools), human IFNγ (11343536; ImmunoTools), human IL-4 (11340045; ImmunoTools), human transforming growth factor-β1 (TGFβ1) (11343160; ImmunoTools), and human IL-10 (11340103; ImmunoTools). Chimeric agonist anti-mouse CD40 antibody (mIgG1, clone 3-23) was obtained through Martin Glennie (University of Southampton, UK) and produced as described in online supplemental methods.
Macrophage OT-I assay
Murine bone marrow-derived macrophages were generated, cultured, and MEKi-treated as described before.5 After 72 hours of MEKi treatment (100 nM or 1 µM), macrophages were pulsed with 1.25 ng/mL of SIINFEKL peptide for 3 hours. Remaining MEKi and SIINFEKL peptide were washed away, and peptide-loaded macrophages were co-cultured with carboxyfluorescein succinimidyl ester (CFSE)-labeled (5 µM) naïve OT-I cells freshly isolated from spleens and lymph nodes of OT-I mice and magnetic activated cell sorting (MACS) purified using the mouse CD8+ T-cell isolation kit (Miltenyi Biotec) according to the manufacturer’s instructions. After 72 hours of co-culture, OT-I cells were harvested and analyzed by flow cytometry. Staining and data analysis were performed as described in Baumann et al.5 CountBright absolute counting beads (Thermo Fisher Scientific) were added to determine absolute T-cell numbers.
Flow cytometry
Extraction of cells from tumor tissue and flow cytometry staining of cells from tumors or in vitro-generated macrophages were performed as described in online supplemental methods and in Baumann et al.5
Western blot analysis
Cells were incubated in the presence of 500 nM MEKi GDC-0623 or dimethyl sulfoxide for 1 hour before addition of stimuli. Stimulation periods and exact experimental setups are described in detail in the figure legends. Staining with primary antibodies was performed overnight at 4°C and with secondary antibodies for 2 hours at room temperature. The following antibodies and dilutions were used: anti-mouse pERK1/2 (clone 20G11, 4376, 1:1000; Cell Signaling), anti-mouse pp38 (D3F9, 4511, 1:1000; Cell Signaling), anti-mouse pSTAT1 (D4A7, 7649, 1:1000; Cell Signaling), anti-mouse GAPDH (GTX100118, 1:1000; GeneTex), and goat anti-rabbit IgG-HRP (7074, 1:3000; Cell Signaling). Clarity Western ECL substrate (170-5060; Bio-Rad) was used for chemiluminescence reaction according to the manufacturer’s instructions. Images were processed using the Image Lab Software (V.6.0.1; Bio-Rad).
DigiWest pathway activity analysis
DigiWest (NMI TT) was conducted as published in Treindl et al.8 In brief, proteins were blotted onto polyvinylidene fluoride membranes and biotinylated. Each sample lane was cut into 96 molecular weight fractions and proteins were eluted. Each molecular weight fraction was bound onto color-coded Luminex beads and incubated with primary, followed by secondary antibodies. Readout was performed on a Luminex FlexMAP 3D. Antibody lists can be provided upon request. For quantification of the antibody-specific signals, the analysis tool is described in detail in Treindl et al.8 Protein expression values were normalized on the total protein amount loaded onto one lane, and non-parametric testing was conducted using the MultiExperiment Viewer (MeV) V.4.9.0.9 Data were mean-centered and log2-transformed prior to utilization of the MeV software, and hierarchical clustering was performed on significant genes using Euclidean distance and complete linkage.
dsRNA immunofluorescence staining
PDA30364 tumor cells were treated for 48 hours with 1 µM GDC-0623. After treatment, cells were fixed in 4% paraformaldehyde and blocked for 1 hour in 5% bovine serum albumin (BSA)/phosphate-buffered saline. Staining for double-stranded RNA (dsRNA) was performed using the anti-dsRNA antibody K1 (Scicons) at a concentration of 1:200. As a secondary antibody, Alexa Flour 594 goat anti-mouse IgG was used at a concentration of 1:500. Nuclei were counterstained with Hoechst. Images were acquired with Leica TCS SP5 confocal microscope.
Immunohistochemistry
Murine tumors were fixed in 5% formaldehyde for 24 hours at 4°C, dehydrated in ethanol, and embedded in paraffin. Tissue blocks were sectioned into 4 µm slice using a rotatory microtome. Slides were deparaffinized in Roticlear (Carl Roth) and rehydrated in ethanol. Antigen retrieval was carried out by microwaving sections for 30 min in 10 mM citrate buffer (pH 6.0). Endogenous peroxidases were blocked by 3% H2O2. Blocking of sections was performed in rat serum (Vector Laboratories) and washed with Tris-buffered saline+0.1% BSA. Immunohistochemical (IHC) staining was performed with rat Vectastain ABC (PK-6104) according to the manufacturer’s instructions. F4/80-specific antibody (BM8; Abcam) was used at a dilution of 1:100. IHC stainings of human tumor specimens were performed and analyzed as described in Poschke et al10 using antibodies reactive against human CD68 (KP1; Abcam), CD163 (EDHu-1; AbD Serotec), and CD3 (PS1; Leica). Nuclei were counterstained with hematoxylin (Novocastra). After staining, slides were rehydrated using increasing ethanol concentrations, followed by Roticlear. Slides were mounted in Permount mounting medium (Thermo Fisher Scientific).
Results
M2 macrophages are highly sensitive to MEK inhibition
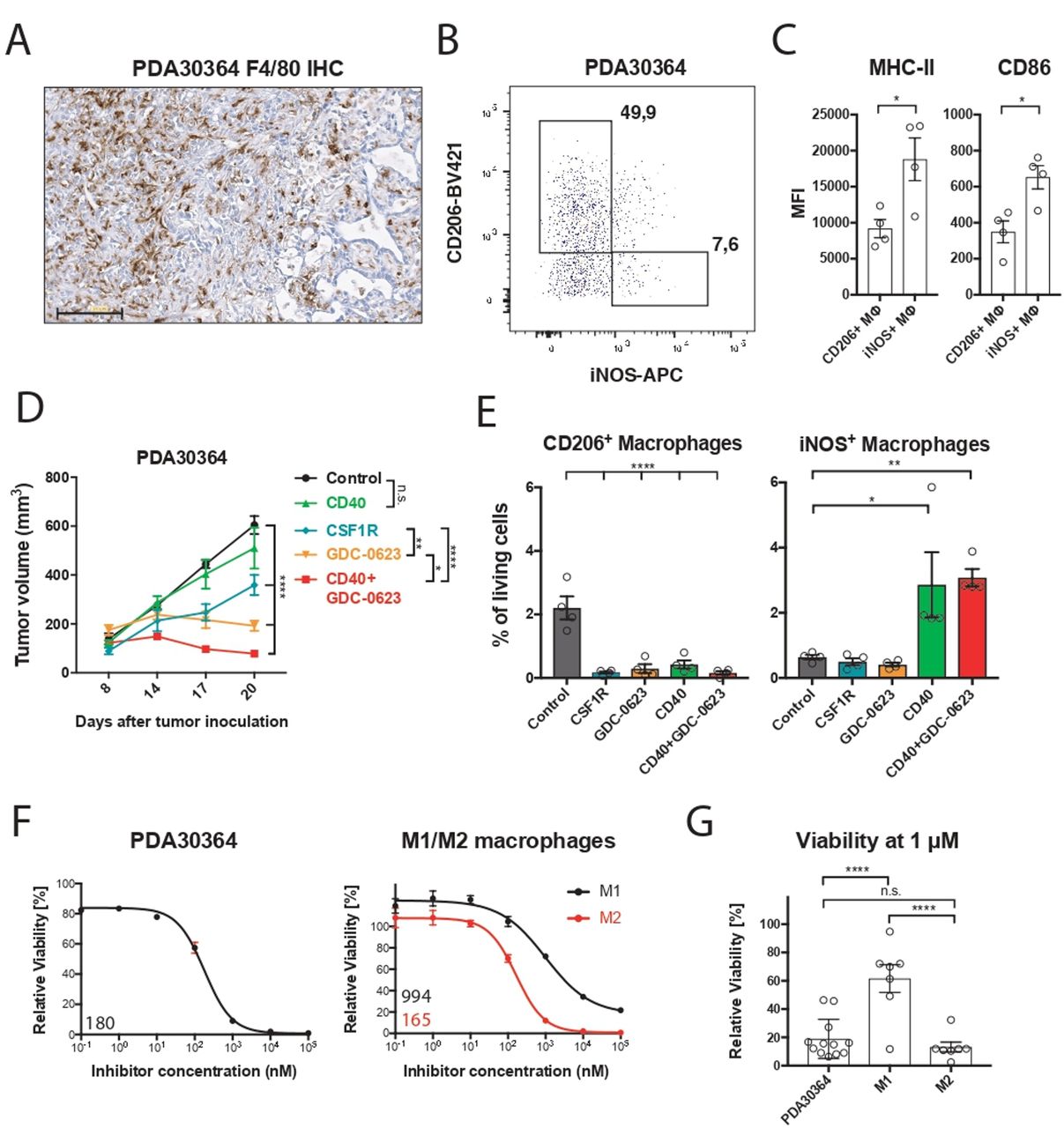
We examined the mechanism of action of MEKi toward the macrophage infiltrate in our transplantable pancreatic cancer model PDA30364, one of the three models used in our prior study,5 because these tumors—like human pancreatic ductal adenocarcinoma6 7 (see also below)— are highly infiltrated with M2-like macrophages (figure 1A,B), which are characterized by the marker CD206+ and lower surface levels of major histocompatibility complex (MHC)-II and CD86 compared with inducible nitric oxide synthase (iNOS)+ M1-like macrophages (figure 1C; online supplemental figure S1A). The tumor cell line PDA30364 was derived from a pancreatic tumor that arose in a genetically engineered mouse model featuring pancreas-specific expression of the KRAS-G12D oncogene as well as mutant P53-R172H5 (online supplemental methods). As a result, this tumor line also provides a benchmark for the cytotoxic impact of MEK inhibition. In line with our previously published data,5 single-agent MEKi treatment results in tumor stasis in this model (figure 1D). Ablation of macrophages with an anti-CSFR1 antibody also significantly suppresses tumor growth in this experiment, in line with findings reported by others,7 11 pointing at the protumorigenic role of tumor-associated macrophages in these tumors. Analysis of the immune infiltrate confirmed that both MEKi and anti-CSF1R antibody profoundly reduced the numbers of CD206+ intratumoral macrophages while having only a limited effect on the iNOS+ counterparts (figure 1E). As shown in our previous report, complementation of MEKi with agonist anti-CD40 antibody resulted in superior antitumor efficacy compared with either single-agent treatment (figure 1D). Notably, CD40 antibody treatment equally increased the frequency of iNOS+ macrophages in the absence or presence of MEKi (figure 1E), further indicating that the viability of this macrophage subset is not significantly affected by MEK inhibition.
Supplemental material
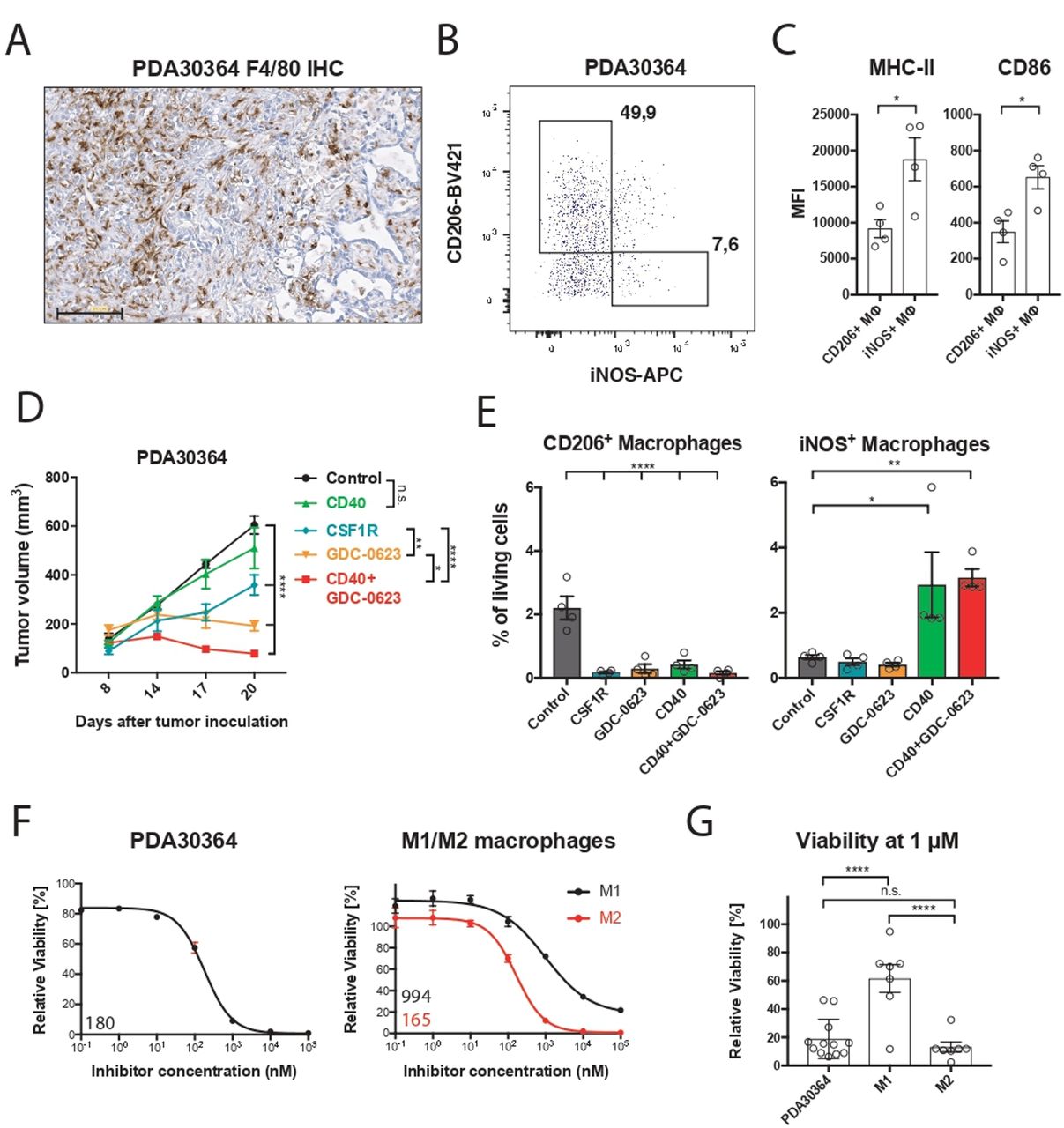
M2 macrophages are highly sensitive to MEK inhibition in vivo and ex vivo. (A) Immunohistochemistry of a PDA30364 tumor using an F4/80-specific antibody. Scale bar: 100 µm. (B) Flow cytometric identification of iNOS+ M1 and CD206+ M2 macrophages in a freshly dissociated tumor sample upon gating on the live CD45+, CD11b+, F4/80+ cell subset. (C) MHC-II (IA/IE) and CD86 surface levels of macrophages gated in (B). Mean±SEM, n=4. (D) Mice bearing PDA30364 tumors were treated with MEKi 30 mg kg−1 GDC-0623 (daily), anti-CSF1R antibody (1 mg once, followed by 0.5 mg every other day), or anti-CD40 antibody (200 µg on days 10, 12, 14, and 17 after tumor inoculation). Mean±SEM, n=5. Two-way ANOVA with post hoc Tukey test. (E) Flow cytometry-based quantification of M2 (CD206+) and M1 (iNOS+) macrophages in PDA30364 tumors from mice treated as described in (C). Mean±SEM, n=4. One-way ANOVA with post hoc Dunnett test. (F) Dose–response curves of PDA30364 and M1 (20 ng mL−1 M-CSF+1 ng mL−1 IFNγ/LPS) and M2 (20 ng mL−1 M-CSF+2.5 ng mL−1 IL-4) polarized murine macrophages treated with GDC-0623. Mean±SEM, n=3. Numbers indicate best-fit inhibitory concentration (IC)50 values. (G) Viability of PDA30364 and murine M1 and M2 macrophages treated with GDC-0623 at 1 µM. Mean±SEM, n>4. One-way ANOVA with post hoc Tukey test. Significance levels are indicated by asterisks (*p≤0.05; **p≤0.01; ***p≤0.001: ****p≤0.0001). ANOVA, analysis of variance; IFNγ, interferon-γ; IL, interleukin; LPS, lipopolysaccharide; M-CSF, macrophage-colony stimulating factor; MEK, mitogen-activated protein kinase kinase.
We zoomed in on the differential MEKi sensitivity of macrophage subsets by performing reductionist in vitro experiments with iNOS+ M1-like and CD206+ M2-like macrophage cultures. For this, primary mouse macrophage cultures were generated from bone marrow precursors in the presence of, respectively, IFNγ/LPS and IL-4 (online supplemental methods; online supplemental figure S1B). Monitoring of the viability of these cultures in the presence of MEKi demonstrated that the M1 macrophages are relatively resistant; even very high concentrations of MEKi induce only partial death. In contrast, M2 macrophages are highly sensitive, comparable to the mutant Kras-transformed PDA30364 tumor cells (figure 1F,G). As alternative M2 activation in the tumor microenvironment can occur in the presence of TGFβ and IL-10,7 we included this M2-like subtype in our experiments, showing that also these macrophages were highly sensitive to MEK inhibition (figure 2A,B). The survival of M1 macrophages under MEK inhibition raised the question of whether these cells had maintained their proinflammatory state. As shown in figure 2C, MEKi-treated M1 macrophages maintained high levels of CD86 and CD40 and even showed somewhat increased levels of iNOS. Interestingly, the remaining M2 macrophages after MEKi treatment showed reduced expression of the M2 marker CD206 and increased CD86 expression, suggesting partial reprogramming under the influence of MEK inhibition (figure 2D). When loaded with the peptide SIINFEKL derived from chicken ovalbumin, MEKi-treated M1 macrophages maintained their capacity to present antigen to SIINFEKL-specific OT-I T cells, as well as the expression of proinflammatory markers (figure 2C–F). Under the same experimental conditions, the numbers of M2 macrophages were decimated, resulting in the loss of antigen presentation in the OT-1 T-cell assay (figure 2E,F).

M1 macrophages are more resistant to MEK/ERK inhibition and maintain their proinflammatory state. (A) Dose–response curves of M1 (20 ng mL–1 M-CSF+1 ng mL–1 IFNγ/LPS) and M2 (20 ng mL–1 M-CSF+2.5 ng mL–1 IL-4 or 20 ng mL–1 TGFβ+10 ng mL–1 IL-10) polarized murine macrophages treated with GDC-0623. Mean±SEM, n=3. (B) Viability of murine M1 and M2 macrophages treated with 1 µM GDC-0623. Mean±SEM, n=6 (two independent experiments). One-way ANOVA with post hoc Tukey test. (C, D) Flow cytometric analysis of surface markers and costimulatory molecules on macrophage cultures as described in (A). Mean±SEM, n=3. Two-way ANOVA with post hoc Sidak test. (E) Co-culture OT-I T cells with SIINFEKL peptide (1.25 ng/mL) pulsed and washed macrophages pretreated with cell culture medium and 1 µM GDC-0623, respectively. Representative CFSE dilution blots. (F) Quantification of total OT-I numbers per well. Mean±SEM, n=3. Unpaired two-tailed t-test. (G) Representative IHC stainings of human PDAC tissue specimen sections for the indicated markers and quantification of samples from mutiple PDAC patients. n = 90-100 . Scale bar: 100 µm. (H) Flow cytometric analysis of macrophages in freshly dissociated human PDAC sample upon gating on the live CD45+, CD11b+ subset. (I) Dose–response curves of monocyte-derived human macrophages. M1 (50 ng mL −1 M-CSF+50 ng mL−1 IFNγ+20 ng mL−1 LPS) and M2 (50 ng mL−1 M-CSF+20 ng mL−1 IL-4 or 20 ng mL−1 TGFβ+10 ng mL−1 IL-10) macrophages were treated with GDC-0623. Mean±SEM, n=3. Numbers indicate best-fit IC50 values. (J) Flow cytometric analysis of surface markers and costimulatory molecules on human macrophage cultures as described in (I). Mean±SEM, n=3. Two-way ANOVA with post hoc Sidak test. Significance levels are indicated by asterisks (*p≤0.05; **p≤0.01; ***p≤0.001: ****p≤0.0001). ANOVA, analysis of variance; FACS, fluorescence-activated cell sorting; IFNγ, interferon-γ; IHC, immunohistochemistry; IL, interleukin; LPS, lipopolysaccharide; M-CSF, macrophage-colony stimulating factor; MEK, mitogen-activated protein kinase kinase; PDAC, pancreatic ductal adenocarcinoma; TGFβ, transforming growth factor-β.
As mentioned above, human pancreatic tumors are rich in M2-like macrophages, which in the human setting are characterized by expression of the markers CD163 and CD68,12 as well as the marker CD206 (figure 2G,H; online supplemental figure S2A). To assess whether human M1 and M2 macrophages display differential sensitivity to MEK inhibition, we generated monocyte-derived human macrophage cultures and polarized these with, respectively, IFNγ+LPS, IL-4 or TGFβ+IL-10. As previously reported, CD206 can be induced by IL-4 in in vitro macrophages cultures13 (online supplemental figure S2B). As shown in figure 2I,J, also human M2-like macrophages were significantly more sensitive to MEKi, whereas M1 macrophages largely survived this treatment without losing their proinflammatory phenotype. Taken together, these data suggested that the MAPK/ERK pathway is critical for M2 macrophage survival while being not essential for the sustenance of M1 macrophages.
Supplemental material
p38 MAPK signaling rescues M1 macrophages from death under MEK inhibition
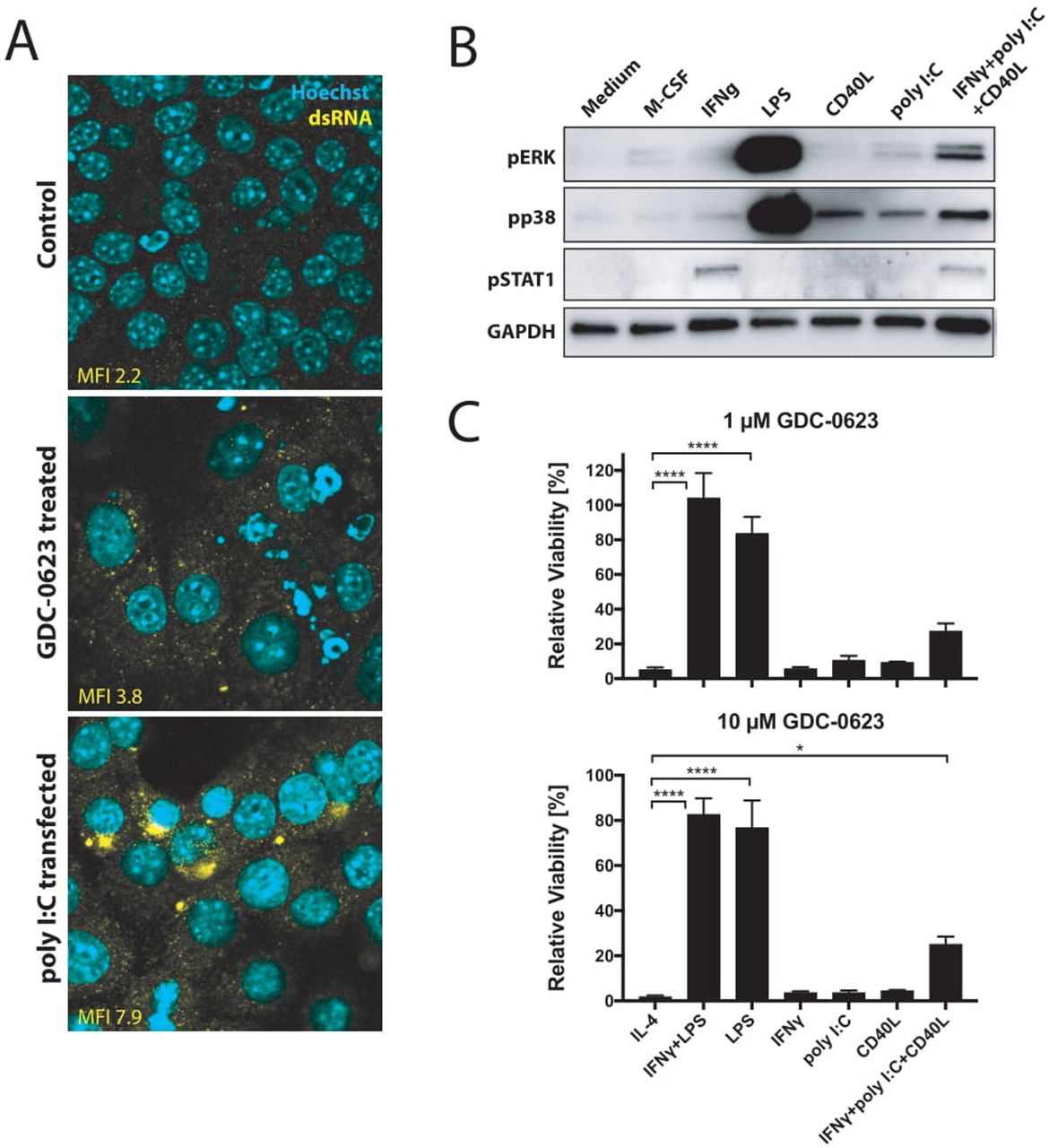
To identify differences between signaling pathways operational in M1 and M2 macrophages, we subjected lysates from these cells to DigiWest pathway activity analysis.8 As shown in figure 3A, activation of the p38 MAPK (pT180/pY182) and STAT1 (pY701) pathways was found in M1 macrophages only. This is in accordance with the fact that activation of p38 MAPK represents a prominent proinflammatory signaling pathway that is triggered by a variety of stimuli including LPS,14 while STAT1 is phosphorylated in response to IFNγ stimulation.15 We validated these data in an independent experiment by means of western blotting, confirming that the MEK/ERK and PI3K/AKT pathways are activated in both M1 and M2 macrophages in an M-CSF-dependent fashion,16 17 while M1 macrophages show M-CSF-independent activation of STAT1 and p38 MAPK (figure 3B). As expected, treatment of either macrophage subtype with MEKi results in the suppression of pERK levels, while pAKT levels are not affected. Importantly, MEK inhibition also does not suppress pSTAT1 and pp38 MAPK levels in M1 macrophages. This observation prompted us to test whether activated p38 MAPK signaling could act as survival pathway for M1 macrophages exposed to MEKi. For this, M1 and M2 cultures were incubated with BIRB 796 (doramapimod), a highly potent p38 MAPK inhibitor that is currently evaluated in clinical studies for the treatment of inflammatory diseases.18 19 Whereas BIRB 796 alone had no significant impact on M1 and M2 viability, the combination of BIRB 796 with MEK inhibition rendered M1 macrophages equally sensitive to cell death similar to M2 macrophages (figure 3C,D).

p38 MAPK signaling acts as a compensatory survival pathway in M1 macrophages. (A) DigiWest phospho-protein pathway activity analysis, log2 Fc; non-parametric Wilcoxon rank-sum test (p<0.05). (B) Western blot analysis of mouse bone marrow-derived macrophages treated for 1 hour with GDC-0623 (500 nM) or DMSO before addition of stimuli as in figure 1 for 15 min. (C) Bar graphs of treated macrophage subsets with 10 µM BIRB 796, 100 nM GDC-0623, and a combination of 2 µM BIRB 796 and 100 nM GDC-0623. (D) Same experiment as in (C); depicted 1 µM GDC-0623. Mean±SEM, n=3. Two-way ANOVA with post hoc Dunnett test. Significance levels are indicated by asterisks (*p≤0.05; **p≤0.01; ***p≤0.001: ****p≤0.0001). ANOVA, analysis of variance; DMSO, dimethyl sulfoxide; MAPK, mitogen-activated protein kinase; M-CSF, macrophage-colony stimulating factor.
Induction of p38 MAPK signaling by therapy-induced signals in the tumor microenvironment
While the combination of LPS and IFNγ is a potent activator of p38 MAPK, we investigated whether also signals mimicking conditions in the microenvironment of MEKi-treated tumors would be able to induce this pathway. One of the signals to consider in this respect is the release of dsRNA by tumor cells as induced by small-molecule cell cycle inhibitors.20 dsRNA is known to induce p38 MAPK through the cytosolic innate receptor RIG-I.21 Indeed, culturing of PDA30364 tumor cells in the presence of MEKi induced dsRNA release, as detected by anti-dsRNA antibody in immunofluorescence stainings (figure 4A). Furthermore, incubation of bone marrow-derived macrophages with poly I:C induced phosphorylation of p38 MAPK, although to a much lesser extent than LPS (figure 4B). Notably, we observed the most prominent shift in M1/M2 ratio when combining MEK inhibition with agonist anti-CD40 antibodies, which deliver a direct trigger to macrophages through their activatory CD40 receptor as well as an indirect IFNγ signal derived from activated T cells.5 We therefore also evaluated p38 MAPK induction by CD40 ligand (CD40L), IFNγ, and their combination with poly I:C, showing that also CD40L and, to a lesser extent, IFNγ can trigger a modest activation of p38 MAPK signaling (figure 4B). Accordingly, treatment of macrophages with the combination of all three factors results in stronger p38 MAPK activation. Although the activation levels are still much less as those induced by LPS, this triple stimulation of macrophages does markedly improve the resistance of these cells to MEK inhibition (figure 4C). Taken together, these data support the notion that the p38 MAPK pathway contributes to the survival of M1 macrophages in MEKi-treated tumors, especially when applied in conjunction with agonist anti-CD40 antibodies.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
CD40 ligation and dsRNA treatment stimulate p38 signaling in macrophages. (A) Immunofluorescence analysis of dsRNA untreated or treated with 1 µM GDC-0623 for 48 hours. Poly I:C transfection served as a positive straining control. (B) Western blot of mouse BMDM stimulated with culture medium containing 20 ng mL−1 M-CSF, 10 ng mL−1 IFNγ, 1 ng mL−1 LPS, 1 µg mL−1 CD40L, 100 ng mL−1 poly I:C, or the combination of 10 ng mL−1 IFNγ+100 ng mL−1 poly I:C+1 µg mL−1 CD40L . (C) Viability of differently stimulated macrophages (20 ng mL−1 M-CSF+2.5 ng mL−1 IL-4, 10 ng mL−1 IFNγ+1 ng mL−1 LPS, 1 ng mL−1 LPS, 10 ng mL−1 IFNγ, 100 ng mL−1 poly I:C, 1 µg mL−1 CD40L, or the combination of 10 ng mL−1 IFNγ+100 ng mL−1 poly I:C+1 µg mL−1 CD40L) treated with GDC-0623 for 3 days. Mean±SEM, n=3. One-way ANOVA with post hoc Dunnett test. Significance levels are indicated by asterisks (*p≤0.05; ****p≤0.0001). ANOVA, analysis of variance; BMDM, bone marrow-derived macrophages; dsRNA, double-stranded RNA; IFNγ, interferon-γ; IL, interleukin; LPS, lipopolysaccharide; M-CSF, macrophage-colony stimulating factor; MEK, mitogen-activated protein kinase kinase.
Discussion
In addition to mediating direct antiproliferative and cytotoxic effects against tumor cells, MEKi have the capacity to increase the immunogenicity of tumor cells and to make the tumor microenvironment more permissive for immunotherapy through depletion of immunosuppressive macrophages.5 Our study demonstrates that the dependency of M2 macrophages on the MEK/ERK pathway renders these cells highly sensitive to death by MEK inhibition. In contrast, the activation of the p38 MAPK pathway in M1 macrophages rescues these cells. Consequently, MEKi treatment of tumors results in the selective elimination of M2 macrophages and thereby in an increase in the M1/M2 ratio.
To date, the most extensively explored strategy for eliminating tumor-associated macrophages involves the inhibition of CSF1R by means of blocking antibodies or small-molecule receptor tyrosine kinase inhibitors. CSF1R stimulation is essential for the differentiation and survival of M2 macrophages. Pharmacological blockade of this pathway was shown to decrease macrophage numbers in preclinical models and in human cancers.7 11 Depending on the preclinical model, this depletion was found to equally affect M2 and M1 intratumoral macrophages,22 or preferentially the M2 macrophages23 as also observed in our study. Irrespective of this difference, MEK inhibition has the advantage over CSF1R blockade that—at least in tumors dependent on the MEK/ERK pathway—it suppresses tumor cell growth and induces immunogenic tumor cell death.3–5
The presence of M1 macrophages in human cancers was found to be associated with favorable prognosis and a type I immune infiltrate,24 while selective recruitment of M1 macrophages induced by low-dose radiation was shown to facilitate infiltration of tumors by effector T cells.25 Therefore, strategies that selectively target M2 macrophages would be preferable. Exactly this is achieved by MEK inhibition. The MEK/ERK pathway is one of the two main signaling cascades triggered downstream of CFS1R engagement, the other one being the PI3K/AKT pathway.16 17 The currently prevailing view is that MEK signaling primarily drives proliferative responses in macrophages, while PI3K signaling is important for macrophage survival. Interestingly, our data show that MEKi efficiently induce M2 macrophage death, though not affecting PI3K signaling. Notably, also inhibitors of the latter pathway were found to interfere with the immunosuppressive function of tumor-associated macrophages.26 27 We therefore conclude that both CSF1R downstream pathways are important for M2 macrophage survival.
Another potential advantage of MEK inhibition over CSF1R targeting is the direct cytostatic action against tumors, at least in cancer types in which oncogenic growth is driven by an activated MEK/ERK pathway. Due to the lack of this dual function, the single-agent therapeutic efficacy of CSF1R inhibitors is limited. Consequently, many ongoing clinical studies involve combination with chemotherapy or targeted cytostatic drugs. In the majority of further clinical studies, CSF1R inhibitors are applied in conjunction with immune checkpoint inhibitory antibodies.7 11 17 Taken together, this implies that CSF1R inhibitors are ideally combined with both a cytostatic and a T-cell stimulatory drug. Due to the ‘two-in-one’ effect of MEK inhibition, one would expect that combination with an immunostimulatory antibody suffices for therapeutic efficacy, as is suggested by experimental data from preclinical tumor models reported by us and others.3–5
As we have shown, one option in this respect is the use of an agonist anti-CD40 antibody. This drug also has a dual mechanism of action, in that it indirectly enhances T-cell immunity while at the same time modulating the macrophage infiltrate, thereby synergizing with the impact of MEKi on this immune cell subset.5–7 Our data suggest that part of this synergy may be related to the capacity of CD40 agonists to activate p38 MAPK signaling in macrophages, both directly through CD40 ligation on the macrophages and indirectly through stimulation of Th1/CTL immunity, thereby rescuing M1 macrophages from MEKi-induced cell death. Notably, other studies have shown that p38 MAPK signaling is associated with repolarization of M2 macrophages toward M1 type.28 29 Furthermore, p38 MAPK signaling does not only rescue M1 macrophages from MEKi-induced cell death, but also activated T cells that receive costimulatory signals through their receptors OX40 and 4-1BB.3
In summary, our study further underpins the notion that MEKi do not merely exert a direct cytostatic effect on MEK/ERK pathway-driven tumors, but also reshape the tumor microenvironment, thereby increasing the therapeutic efficacy of immunostimulatory antibodies. Graphical abstract of the paper has been provided online supplemental file 4.
Supplemental material
Data availability statement
Data are available upon reasonable request. Antibody lists used in DigiWest analyses can be provided upon request.
Ethics statements
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors RO and DB designed the study and wrote the manuscript. DB, JD, and TH designed and performed experiments, and analyzed data. DB and GE performed statistical analyses. JD, LB, CS, and TH. performed ex vivo macrophage and OT-I experiments. DB, JD, and TH performed in vivo therapy experiments and subsequent flow cytometric analyses. DB and JD performed dsRNA staining. JD generated protein lysates and performed western Blot analysis. DigiWest analysis were performed and analyzed by PD and GE. CL and JD performed the immunohistochemistry stainings.
Funding This study was funded by the K.H. Bauerstiftung, the Helmholtz Foundation (Immunology &Inflammation Future Theme) and German Cancer Research Center PhD programme.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Author note Daniel Baumann and Jennifer Drebant are now working at Boehringer Ingelheim Pharma GmbH & Co. KG, Cancer Immunology and Immune Modulation, Biberach an der Riß, Germany.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.