Article Text

Abstract
Background Adoptive transfer of engineered T cells has shown remarkable success in B-cell malignancies. However, the most common strategy of targeting lineage-specific antigens can lead to undesirable side effects. Also, a substantial fraction of patients have refractory disease. Novel treatment approaches with more precise targeting may be an appealing alternative. Oncogenic somatic mutations represent ideal targets because of tumor specificity. Mutation-derived neoantigens can be recognized by T-cell receptors (TCRs) in the context of MHC–peptide presentation.
Methods Here we have generated T-cell lines from healthy donors by autologous in vitro priming, targeting a missense mutation on the adaptor protein MyD88, changing leucine at position 265 to proline (MyD88 L265P), which is one of the most common driver mutations found in B-cell lymphomas.
Results Generated T-cell lines were selectively reactive against the mutant HLA-B*07:02-restricted epitope but not against the corresponding wild-type peptide. Cloned TCRs from these cell lines led to mutation-specific and HLA-restricted reactivity with varying functional avidity. T cells engineered with a mutation-specific TCR (TCR-T cells) recognized and killed B-cell lymphoma cell lines characterized by intrinsic MyD88 L265P mutation. Furthermore, TCR-T cells showed promising therapeutic efficacy in xenograft mouse models. In addition, initial safety screening did not indicate any sign of off-target reactivity.
Conclusion Taken together, our data suggest that mutation-specific TCRs can be used to target the MyD88 L265P mutation, and hold promise for precision therapy in a significant subgroup of B-cell malignancies, possibly achieving the goal of absolute tumor specificity, a long sought-after dream of immunotherapy.
- immunotherapy
- adoptive
- hematological neoplasms
- receptors
- antigen
- cell engineering
- T lymphocytes
Data availability statement
Data are available upon reasonable request. All data relevant to the study are included in the article or uploaded as supplementary information. For further information, please contact oezcan.cinar@charite.de. Supplementary files can be found in the online version of this article. Complete length nucleic acid and amino acid sequences of T-cell receptors can be found in the published patent application: WO 2020/152161 A1.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Recently, clinical studies of adoptive T-cell therapy (ATT) using chimeric antigen receptor T (CAR-T) cell therapy against the B-cell antigen CD19 have achieved remarkable success and have shown response in around 50% of refractory and relapsed patients with diffuse large B-cell lymphoma (DLBCL).1 2 Similar strategies have been developed targeting other B-cell lineage antigens such as CD20, CD22 and BCMA (B-cell maturation antigen).3–5 However, tumor escape by modulating surface expression of the target antigen is a limitation of this strategy, leading to relapse in up to 50% of patients treated with CD19 CAR-T cells.6 7 Furthermore, although being much more specific than standard chemotherapy, CD19 CAR-based ATT is lineage-specific rather than truly cancer-specific, as major parts of the B-cell compartment including normal B lymphocytes are eliminated. This frequently leads to severe B-cell aplasia that may cause morbidity or requirement for long-term immunoglobulin substitution and/or antibiotic prophylaxis.8
Surface antigens druggable by antibodies or antibody derivatives are only rarely tumor-specific, as oncogenic mutations occur mostly in intracellular proteins regulating cell proliferation and survival.9 In contrast to CAR-based strategies, T-cell receptor (TCR)-based ATT relies on classical TCR recognition of processed antigen-derived epitopes presented in the context of MHC (major histocompatibility complex) molecules. The ability to target any protein independent of cellular localization greatly widens the spectrum of target antigens including truly cancer-specific mutant antigens, so called ‘neoantigens’, derived from somatic mutations that are acquired in the course of tumor development.10
Among all cancer-associated somatic mutations, oncogenic driver mutations are obviously the most attractive targets for TCR gene therapy. A prerequisite for TCR-based therapies is that the mutation leads to generation of peptides presented on MHC molecules with high affinity.11 12 Selection of antigen-loss variants within the tumor is unlikely, if a driver mutation is targeted that is crucial for oncogenic transformation. However, many driver mutations such as common p53 mutations occur at many variable positions of a given protein, creating a large number of potential target epitopes, which would need to be addressed individually. In contrast, a missense mutation almost consistently changing leucine in position 265 to proline (L265P) in the MyD88 adaptor protein is one of the most common driver mutations found in about one-fifth of all lymphoid malignancies. MyD88 L265P is the hallmark mutation in Waldenström macroglobulinemia, but it is also frequently found in aggressive B-cell lymphomas, for example, 30% of activated B cell (ABC)-like DLBCL, 45%–60% of central nervous system lymphomas and testicular lymphomas,13–16 diseases with a huge need for novel specific and well-tolerated therapies.
In this study, we developed high-affinity TCRs recognizing the mutant peptide sequence in MyD88 L265P with specific killing capacity towards the mutant malignant B-cell population. Targeting cancer-specific genetic alterations with highly specific immunotherapies represents a huge step towards precision cellular therapy in oncology.
Materials and methods
In vitro antigen processing and mass spectrometry
Before trying to generate TCRs specific for mutated MyD88, we checked whether a peptide spanning the mutation is processed by the proteasome and potentially presented by the MHC complex. The polypeptide substrate MyD88 L265P256–281 was synthesized by the core facility of the Institute of Biochemistry using standard Fmoc (N-(9-fluorenyl) methoxycarbonyl) methodology (0.1 mmol) on an Applied Biosystems 433A automated synthesizer. The peptide was purified by high-performance liquid chromatography and analyzed by mass spectrometry (ABI Voyager DE PRO). 20S proteasomes were purified from human red blood cells, in principle following the procedure as previously described by Textoris-Taube et al.17 Proteasome digests of the synthetic MyD88 L265P256–281 polypeptide were performed in 100 µL of TEAD buffer (20 mM Tris, 1 mM EDTA, 1 mM NaN3, 1 mM Dithiothreitol, pH 7.2) over time at 37°C. For establishing a full-scale cleavage map, processing times were 48 hours. Proteasomal processing of the synthetic of polypeptides was performed at a substrate concentration of 40 µM in the presence of 4 µg 20S proteasome. Digested samples of 10 µL were loaded for 5 min onto a trap column (PepMap C18, 5 mm×300 μm×5 µm, 100 Ǻ; Thermo Fisher Scientific, Massachusetts, USA) with 2:98 (v/v) acetonitrile:water containing 0.1% (v/v) Trifluoroacetic acid at a flow rate of 20 µL/min and analyzed by nanoscale LC-MS/MS using an Ultimate 3000 and LTQ Orbitrap XL mass spectrometer (Thermo Fisher Scientific). The system comprises a 75 µm inner diameter ×250 mm nano LC column (Acclaim PepMap C18, 2 µm; 100 Å; Thermo Fisher Scientific) or a 200 mm PicoFrit analytical column (PepMap C18, 3 µm, 100 Å, 75 µm; New Objective). The mobile phase (A) is 0.1% (v/v) formic acid in water, and (B) is 80:20 (v/v) acetonitrile:water containing 0.1% (v/v) formic acid. For elution, a gradient 3%–45% B in 85 min with a flow rate of 300 nL/min was used. Full MS spectra (m/z 300–1800) were acquired on an Orbitrap instrument at a resolution of 60,000 (FWHM). At first, the most abundant precursor ion was selected for either data-dependent collision-induced dissociation (CID) fragmentation with parent list (1+, 2+ charge state included). Fragment ions were detected in an ion trap instrument. Dynamic exclusion was enabled with a repeat count of 2 and 60 s exclusion duration. Additionally, the theoretically calculated precursor ions of the expected spliced peptides were pre-elected for two Orbitrap CID (resolution 7500) and higher-energy collisional dissociation (HCD) (resolution 15,000) fragmentation scans. The maximum ion accumulation time for MS scans was set to 200 ms and that for tandem mass spectra (MS/MS) scans was set to 500 ms. Background ions at m/z 371.1000 and 445.1200 act as lock mass.
For LC-MS/MS runs using a Q Exactive Plus mass spectrometer coupled with an Ultimate 3000 RSLCnano (Thermo Fisher Scientific), samples were trapped as described previously and then analyzed by the system that comprised a 250 mm nano LC column (Acclaim PepMap C18, 2 µm; 100 Å; 75 µm Thermo Fisher Scientific). A gradient of 3%–40% B (alternatively 3%–45% B) in 85 min was used for elution. Mobile phase A was 0.1% (v/v) formic acid in water, and mobile phase B was 80% acetonitrile in water containing 0.1% (v/v) formic acid. The Q Exactive Plus instrument was operated in the data-dependent mode to automatically switch between full scan MS and MS/MS acquisition. Full MS spectra (m/z 200–2000) were acquired at a resolution of 70,000 (FWHM) followed by HCD MS/MS fragmentation of the top 10 precursor ions (resolution 17,500, 1+, 2+, 3+, charge state included, isolation window of 1.6 m/z, normalized collision energy of 27%). The ion injection time for MS scans was set to a maximum of 50 ms, automatic gain control (AGCs) target value of 1×106 ions and that for MS/MS scans was set to 100 ms, AGCs 5×104; dynamic exclusion was set to 20 s. Background ions at m/z 391.2843 and 445.1200 act as lock mass.
Peptides were identified by PD2.1 software (Thermo Fisher Scientific) based on their merged MS/MS of CID and HCD. Based on the results, further work focused on the 10 mer peptide RPIPIKYKAM (figure 1).
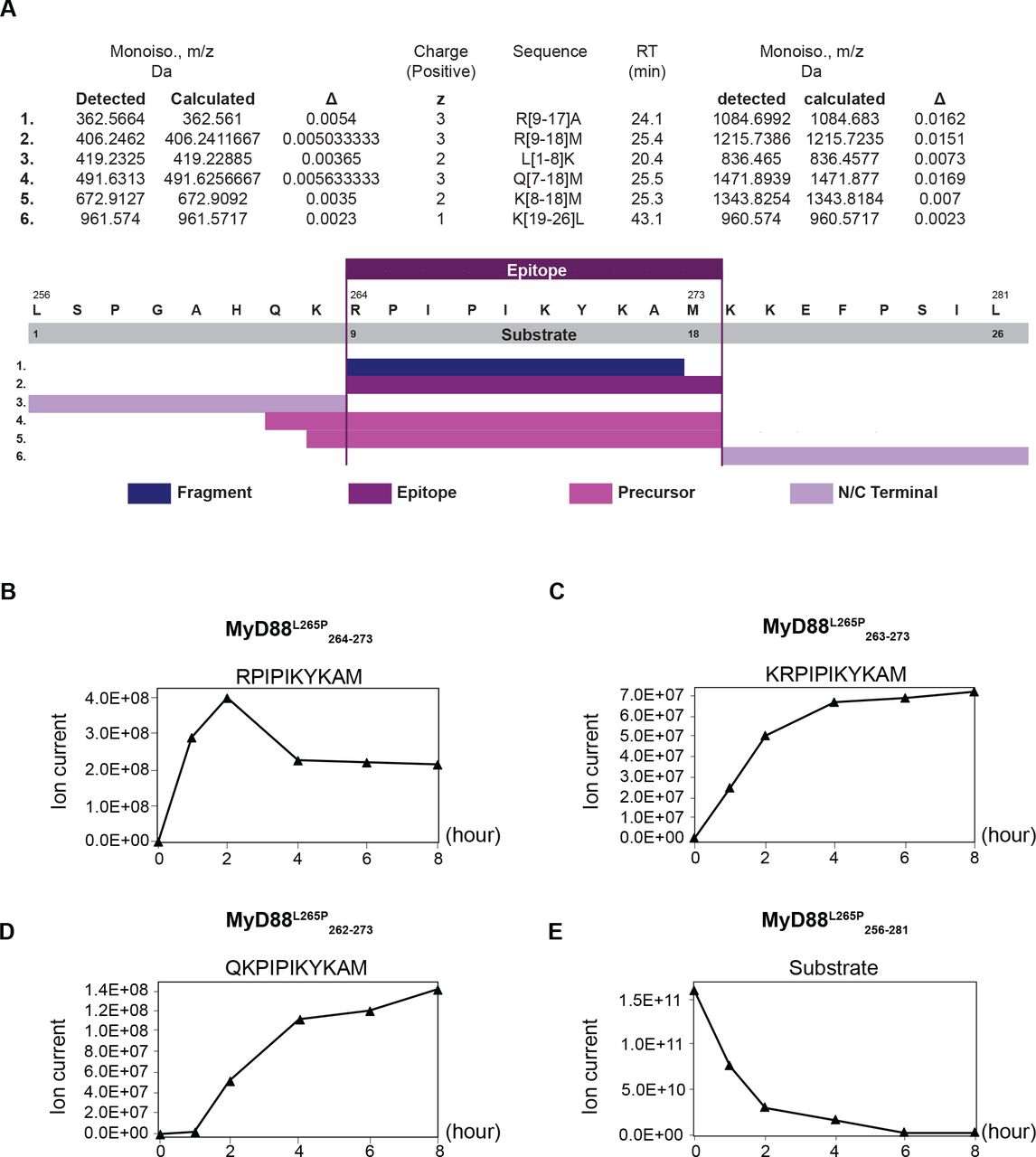
In vitro generation of the MyD88 L265P264–273 neoepitope. Kinetic proteasomal antigen processing experiments were performed using the synthetic polypeptide substrate MyD88 L265P256–281. (A) Summary of the peptides generated by the proteasome from the polypeptide substrate MyD88 L265P256–281 in an in vitro digest. The complete list can be found in online supplemental table 4. (B–D) The generation kinetics of the MyD88 L265P-derived 10 mer neoepitope and its potential N-terminally extended precursor peptides are shown. (E) Kinetics of the degradation of the MyD88 L265P-derived polypeptide substrate MyD88 L265P256–281.
Supplemental material
Cell lines and generation of target cells
K562 leukemic cell line was cultured in RPMI with 10% fetal bovine serum (FBS) and 100 U/mL penicillin/streptomycin. Non-Hodgkin’s lymphoma cell lines SU-DHL-6, OCI-Ly3 and TMD8 were cultured in RPMI with 20% FBS and 100 U/mL penicillin/streptomycin. OCI-Ly3 and TMD8 cells were kindly supplied by groups of Professor Clemens Schmitt and Dr Martin Janz (Max-Delbruck-Center for Molecular Medicine, Berlin, Germany).
SU-DHL-6, OCI-Ly3, TMD8 and K652 cells do not naturally express HLA-B*07:02. To generate HLA-B*07:02-positive target cells, we transduced the cell lines with HLA-B*07:02-coding gamma-retroviral vector MP71.18 Moreover, K562 cells with or without HLA-B*07:02 were virally transduced to express the complete length wild-type or mutant (L265P) MYD88—coupled to the expression marker green florescent protein (GFP) via p2A—and used as artificial target cells.
Generation of mutation-specific T cells
All mutation-specific TCRs were cloned from peripheral blood mononuclear cells (PBMCs), isolated from HLA-B7-positive healthy donors. Monocytes were separated by plastic adherence for generation of dendritic cells (DCs). Following 3 days of culture with 800 IU/mL granulocyte-macrophage colony-stimulating factor (GM-CSF) and 10 ng/mL interleukin (IL)-4 in RPMI with 1% human serum, immature DCs were cultured overnight with addition of 10 ng/mL LPS and 50 ng/mL interferon gamma (IFN-γ) for maturation. Mature DCs were then loaded with mutant peptide (RPIPIKYKAM) and used for priming autologous CD8+ naive T cells. After 10 days, cells from each well were stained with a streptamer (HLA*B07:02-RPIPIKYKAM; IBA GmbH, Germany) or stained for T-cell activation markers such as CD137 (4-1BB) following a peptide restimulation. Positively stained wells were restimulated with peptide-loaded autologous PBMCs for expansion, in case it was necessary to obtain enough cells for fluorescence-activated cell sorting (FACS) isolation. The methodology for generating mutation-specific T cells is shown in figure 2A. This protocol was largely adapted from Wölfl and Greenberg.19

Generation of mutation-specific TCRs. (A) Schematic explanation of methodology for generation of mutation-specific T-cell lines. (B) Representative CD137 staining on 6 hours of restimulation with peptide, or streptamer staining for FACS isolation of peptide-specific T cells. (C) Construction of TCR gene cassettes. (D) Non-linear curve analysis of IFN-γ response by TCR-transduced CD8+ T cells from healthy donors, following coculture with K562 cells that were transduced with HLA*B07:02 and loaded with different concentrations of the mutant peptide (RPIPIKYKAM) or the corresponding WT peptide (RLIPIKYKAM). Response to the mutant peptide was detectable down to the concentration of 10−4 µg/mL with EC50 values within the nano molar (high affinity) range. Mutation-specific TCRs showed more than 10,000-fold higher affinity to the mutant peptide. IFN-γ response was measured by ELISA. FACS, fluorescence-activated cell sorting; IFN-γ, interferon gamma; TCR, T-cell receptor; WT, wild type.
Identification of mutation-specific TCRs
Viable, CD8+ and streptamer-positive cells were sorted by FACS for total RNA isolation. A representative FACS plot is shown in figure 2B. TCR alpha and beta genes were amplified via 5′-RACE PCR (SMARTER RACE cDNA Amplification Kit; Clontech, Japan) and cloned (Zero Blunt TOPO Cloning Kit; Invitrogen/Thermo Fisher Scientific, Massachusetts, USA) in competent Escherichia coli cells. Multiple bacterial clones from each TCR chain were sequenced to analyze T-cell clonality, and dominant sequences were matched to create alpha-beta (αβ)TCRs for further characterization. Online supplemental table 1 shows CDR3 sequences and gene subtypes of MyD88 L265P mutation-specific TCRs. Identified variable domains (TRBV and TRAV) were combined with murine constant domain sequences (mTRBC and mTRAC) for experimental characterization of TCRs and synthesized on codon optimization (GeneArt, Thermo Fisher Scientific) for expression in human cells. TCR gene cassettes encoding the TRBV in combination with a murine TRBC and the TRAV in combination with a murine TRAC, separated by a p2A signal, were constructed as shown in figure 2C. TCR cassettes were cloned into the vector pMP71 by restriction site cloning. CD8+ T cells from healthy donors transduced with the TCRs were again stained with streptamer to test surface expression of the TCRs and functionality of αβTCR pairing (online supplemental figure 1).
Supplemental material
T-cell activation and cytotoxicity analysis
Peripheral CD8+ T cells from HLA-B7-positive (not typed for HLA-B7 subtypes) healthy donors were transduced to express mutation-specific TCRs. Staining for murine TRBC (PE anti-mouse TCR β chain antibody; BioLegend, California, USA) was performed to check transduction efficiency before every experiment, which ranged between 25% and 65% of CD8+ T cells. Target cell lines were cocultured with TCR-transduced or control T cells for 16 hours. IFN-γ secretion was measured by ELISA (OptEIA Human IFN-γ ELISA Set; BD Biosciences, New Jersey, USA). Cytotoxicity was evaluated by flow cytometry analysis after staining of target cells for active caspase-3 (AF647 Rabbit Anti-Active Caspase-3, BD Biosciences) and fixable dead cell stain (LIVE/DEAD Fixable Violet Dead Cell Stain, Thermo Fisher Scientific). Target cells were gated on GFP-positive as reporter of wild-type or mutant MyD88 expression.
Xenograft models
All animal experiments were performed according to the institutional protocols and the national laws and regulations. Adult female NOG mice (NOD.Cg-Prkdcscid Il2rgtm1Sug/JicTac) and hIL2-NOG (NOD.Cg-Prkdcscid Il2rgtm1Sug Tg(CMV-IL2)4-2Jic/JicTac) mice, which due to their production of human IL-2 provide better support for the growth of adoptively transferred human T cells, were acquired from Taconic Biosciences (New York, USA). In short, mice were inoculated subcutaneously with 5×106 OCI-Ly3 or luciferase-expressing TMD8 cells. Once the tumors reached the predetermined volume of 100 mm3 (~2 weeks after injection), OCI-Ly3 tumor-bearing mice were treated with intravenous injection of 1×107 TCR-T cells, or mock T cells (untransduced) of the same donor, or phosphate buffered saline (PBS) as control. TMD8-bearing mice were treated similarly on day 7 after tumor cell injection. Caliper and bioluminescence measurements were used to monitor tumor growth. Tumor volume was calculated with the formula: length×width2/2. Mice were sacrificed when tumor size exceeded 1500 mm³ or signs of distress was observed as determined in the animal experimentation protocol.
Alanine scan for cross-reactivity
To define the TCR-binding motif, a list of peptides was created, in which every amino acid residue in the mutant epitope was exchanged one by one with alanine. All peptides were separately loaded on HLA-B*07:02-expressing K562 cells in the concentration of 10 µg/mL and cocultured with TCR-T cells for 16 hours. Response was measured via IFN-γ ELISA. Results were then analyzed using the online tool Expitope20 to screen for motif similarities in the human proteome. Peptides identified to have a binding-motif similarity to the original epitope were loaded on HLA-B*07:02-expressing K562 cells in the concentration of 10 µg/mL for coculture with TCR-T cells from three different healthy donors. Reactivity was measured via IFN-γ ELISA.
Lymphoblastoid cell line (LCL) scan for alloreactivity
TCR-T cells from three different healthy donors were cocultured for 16 hours with immortalized B-LCLs expressing a variety of frequent HLA haplotypes (online supplemental table 2), with or without prior peptide loading. Response was measured via IFN-γ ELISA.
Data sharing
For further information, please contact oezcan.cinar@charite.de. Supplemental files can be found in the online version of this article. Complete length nucleic acid and amino acid sequences of TCRs can be found in the published patent application: WO 2020/152161 A1.
Results
Mutation-encompassing peptide is processed by human proteasome and binds to HLA-B*07:02 with high affinity
We used NetMHC V.4.021 and NetMHCpan V.4.022 for screening of peptides spanning the mutation region on MyD88 for candidate epitopes, restricted to HLA haplotypes commonly found in the European population. A 10 mer peptide (RPIPIKYKAM) harboring a proline residue at position 2 which is highly preferred by HLA-B*07:02,23 the most common HLA-B allele found in Europe and North America, was predicted to be a strong binder, while the wild-type sequence had very low predicted HLA binding affinity (online supplemental table 3). Since it is established that the in vitro generation of epitopes by 20S proteasomes reflects the in vivo situation with high fidelity,24 we performed kinetic in vitro proteasome digestion experiments to study the generation of the predicted MyD88 L265P264–273 neoepitope from the synthetic 26 mer polypeptide substrate MyD88 L265P256–281. A summary of the different peptides generated by the proteasome in an in vitro digest is shown in figure 1A; the whole list can be found in online supplemental table 4. Giving confidence that the predicted 10 mer neoepitope is indeed generated, mass-spectrometric analysis of the proteasomal digests identified the generation of the predicted neoepitope MyD88 L265P 264–273 as well as the generation of two N-terminally extended neoepitope precursor peptides MyD88 L265P263–273 and MyD88 L265P262–273 requiring trimming by endoplasmic reticulum resident aminopeptidases. The kinetic generation of the three peptides is shown in figure 1B–D, while the degradation of the 26mer substrate is shown in figure 1E.
Isolated TCRs yielded mutation-specific and HLA-restricted T-cell activity
Next, we generated peptide-specific T-cell lines against this neoepitope by priming naive T cells with autologous peptide-loaded DCs from healthy HLA-B7-positive donors (figure 2A). These peptide-specific T-cell lines recognized the mutant peptide when restimulated as shown by CD137 expression and stained with custom MHC-peptide streptamer (figure 2B). We cloned 13 unique TCR alpha and beta genes from peptide-specific T cells of five individual donors (first two digits in TCR names indicate donor number; eg, TCR1605 and TCR1610 are cloned from the same donor) into the gamma-retroviral vector MP71, for expression on primary CD8+ T cells (figure 2C). T cells transduced with these TCRs (TCR-T) recognized HLA-B*07:02-positive K562 cells loaded with mutant peptide with varying functional avidity, while the corresponding wild-type peptide was not recognized (figure 2D). In addition, the 11 and 12 mer precursor peptides (MyD88 L265P263–273 and MyD88 L265P262–273) detected in mass-spectrometric analysis were also recognized by TCR-T cells (online supplemental figure 2).
To further assess specificity and functionality, we cocultured TCR-T cells with engineered HLA-B*07:02-positive or HLA-B*07:02-negative K562 target cells expressing the complete wild-type or mutant MYD88 gene. Flow cytometric analysis showed mutation-specific and HLA-restricted recognition of target cells without exogenous peptide loading, proving that the epitope can be generated from endogenously expressed MyD88 and presented by HLA-B*07:02 at the cell surface (figure 3A). Seven out of 13 peptide-specific TCRs showed strong reactivity against target cells (figure 3C). In addition, the amount of IFN-γ secreted by TCR-T cells on target recognition correlated with previously measured TCR avidity. The highly specific recognition of target cells observed here has also led to cytotoxic T-cell activity (figure 3B,D). The TCR with the highest overall activity in the comparative assessment was TCR2304. TCR2304 was thus chosen for further development. Hereafter, TCR-T cells refer to TCR2304-transduced T cells.

Mutation-specific activity of TCR-T cells. (A) Mutation-specific activation of TCR-T cells against K562 target cells—virally transduced to express complete length wild-type or mutant (L265P) MyD88 and HLA*B07:02, shown by flow cytometry analysis performed after 16 hours of coculture. (B) Viability of HLA*B07:02-positive target K562 cells that were cocultured for 16 hours with T cells expressing one of the three highest avidity TCRs, analyzed by flow cytometry. The proportions of apoptotic/dead cells are given in the upper right quadrant. (C) Comparative TCR-T cell response against target K562 cells. IFN-γ response measured by ELISA after coculture with K562 cells virally transduced to express complete length WT or mutant (L265P) MyD88 (mutation+) and/or HLA*B07:02 (HLA-B7+) as indicated. K562 cells loaded with the mutant peptide served as control (peptide+). Thirteen different TCRs were listed according to their respective EC50 values to the mutant peptide titration, from highest to the lowest. (D) Viability of target cells for cytotoxicity analysis of T cells transduced with three highest avidity TCRs. Strength of cytotoxicity as well as IFN-γ response against target cells strongly correlated with TCR avidity, previously measured by IFN-γ response to peptide titration. IFN-γ, interferon gamma; TCR, T-cell receptor; WT, wild type.
T cells engineered to express TCR2304 recognize and kill MyD88 L265P expressing lymphoma cells
To investigate the functional potential of TCR2304 when MyD88 L265P is expressed at natural expression levels, we analyzed the T-cell response against B-cell lymphoma cell lines. We used SU-DHL-6 (GCB-like DLBCL, wild-type MyD88), OCI-Ly3 (ABC-like DLBCL, homozygous L265P mutation) and TMD8 (ABC-like DLBCL, heterozygous L265P mutation) (figure 4A). Since all three cell lines originated from HLA-B7-negative patients, they were transduced to stably express the HLA*B07:02 gene. Surface expression of HLA*B07:02 measured by flow cytometry was comparable to primary peripheral blood cells and to the HLA-B*07:02-positive B-LCL STA01 (figure 4B). Overnight coculture with these lymphoma cells led to activation of TCR-T cells (figure 4C), which was accompanied by mutation-specific cytotoxicity (figure 4D,E) and T-cell proliferation (figure 4F). Specific T-cell response and cytotoxicity against naturally mutant lymphoma cells provided further evidence for the functional potential of TCR2304.

Mutation-specific activity of TCR-T cells against DLBCL cell lines with intrinsic MyD88 L265P expression. (A) Genomic sequence and allele-specific (AS) PCR analysis of lymphoma cell lines: SU-DHL-6 (GBC-like DLBCL, WT MyD88), OCI-Ly3 (ABC-like DLBCL, homozygous MyD88 L265P) and TMD8 (ABC-like DLBCL, heterozygous MyD88 L265P). (B) Side-by-side comparison of HLA*B07:02 expression of virally transduced lines versus natural expression in PBMCs isolated from an HLA*B07:02-positive donor and an HLA*B07:02-positive B-LCL line (STA01) measured by flow cytometric staining. (C) Activation analysis of TCR2304-transduced T cells via IFN-γ ELISA, after 16 hours of coculture with lymphoma cells. Cell lines virally transduced to express HLA*B07:02 are shown as ‘cell line_B7’. Results from two different blood donors plotted with error bars showing SD significance analysis by two-way ANOVA: ****p<0.0001. (D) Representative flow cytometric viability analysis of lymphoma cells expressing HLA*B07:02 after 16 hours of coculture with T cells (as explained in figure 3). (E) Viability analysis of lymphoma cells with or without HLA*B07:02 expression. T cells of three different donors were used, error bars with SD significance analysis by two-way ANOVA: ***p<0.001, ****p<0.0001. (F) Antigen-induced proliferation of TCR2304-transduced T cells following 72 hours of coculture with HLA*B07:02-positive OCI-Ly3 and TMD8 cells. T cells were labeled with CFSE to trace proliferation prior to coculture. ANOVA, analysis of variance; DLBCL, diffuse large B-cell lymphoma; IFN-γ, interferon gamma; PBMC, peripheral blood mononuclear cell; TCR, T-cell receptor; WT, wild type.
Myd88 L265P-redirected TCR-T cells induce durable regression of human lymphoma xenografts in mice
Having observed a strong specific T-cell response against MyD88 L265P mutant B-cell lymphoma lines in vitro, we tested TCR2304 in vivo for further assessment of its therapeutic potential. For this purpose, we generated a xenograft model by subcutaneously injecting HLA-B*07:02-expressing OCI-Ly3 cells into immunodeficient NOG mice. After tumors reached the predetermined size of 100 mm3, mice were randomly treated with a single intravenous injection of 1×107 TCR-T or mock T cells, or PBS as untreated control. All untreated or mock-treated mice showed progressive tumor growth that required sacrificing the mice between days 13 and 37 (figure 5A). Four of the six mice that received TCR2304-T cells showed complete remission of the tumor, with tumor shrinking beginning between days 18 and 25 after T-cell application, although in one of these mice the tumor started to regrow around day 40. Two mice needed to be sacrificed early in the experiment due to rapid tumor progression and severe symptoms of disease before TCR-T cells could take effect. Since human T cells grow poorly without support from human cytokines, we also used the same tumor model in hIL2-NOG (immunodeficient NOG mouse expressing human IL-2 cytokine). This model is expected to have an enhanced T-cell engraftment and has been shown to improve the outcome from T-cell therapy.25 Indeed, tumors in hIL2-NOG mice that received TCR-T cells disappeared completely after 3 weeks and did not relapse after up to 2 months following treatment (figure 5B).

Therapeutic efficacy of TCR-T cells in OCI-Ly3 xenograft mouse models. NOG and hIL2-NOG mice were subcutaneously injected with 5×106 HLA*B07:02 expressing OCI-Ly3 cells. Treatment with 1×107 T cells was given by intravenous injection after tumors reached the predetermined size of 100 mm3. (A) Change of tumor volume (significance analysis by two-way ANOVA, showing difference of tumor volume on day 31: ***p<0.001) and survival after treatment start in NOG mice. (B) Change of tumor volume after treatment start in hIL2-NOG mice. Significance analysis by two-way ANOVA, showing difference on day 30: *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. (C) Flow cytometric analysis of T cells in tissues of TCR-T cell-treated common NOG and hIL2-NOG mice, 58 days after treatment. (D) PD-1 expression in TCR-T cells in tissues of hIL2-NOG and common NOG mice on day 58 after treatment start, analyzed by flow cytometry. ANOVA, analysis of variance; IL, interleukin; TCR, T-cell receptor.
To evaluate the mechanisms leading to tumor regrowth after initial response, the NOG mouse that had shown tumor regrowth starting on day 40 was sacrificed on day 58 together with a mouse that had continuing response from the hIL2-NOG TCR-T treated group. We observed that the hIL2-NOG mouse still had a high amount of human T cells in blood, bone marrow and spleen, while the NOG mouse had only some T cells remaining in the tumor (figure 5C). TCR-T cells from tissues of the hIL2-NOG mouse did not show any significant change in the exhaustion marker PD-1 (programmed cell death protein 1), while TCR-T cells remaining in the growing tumor of the NOG mouse showed increased PD-1 staining in flow cytometric analysis (figure 5D).
However, in a second series of experiments using hIL2-NOG mice, some of the animals died independently of the size of the tumor. We had hints that these mice were developing severe graft-versus-host disease (GvHD) by proliferating human T cells, as reported by Ito et al.26 The amount of GvHD-related toxicity indeed varied significantly using different human donors; therefore, we decided to stop further experiments in this model.
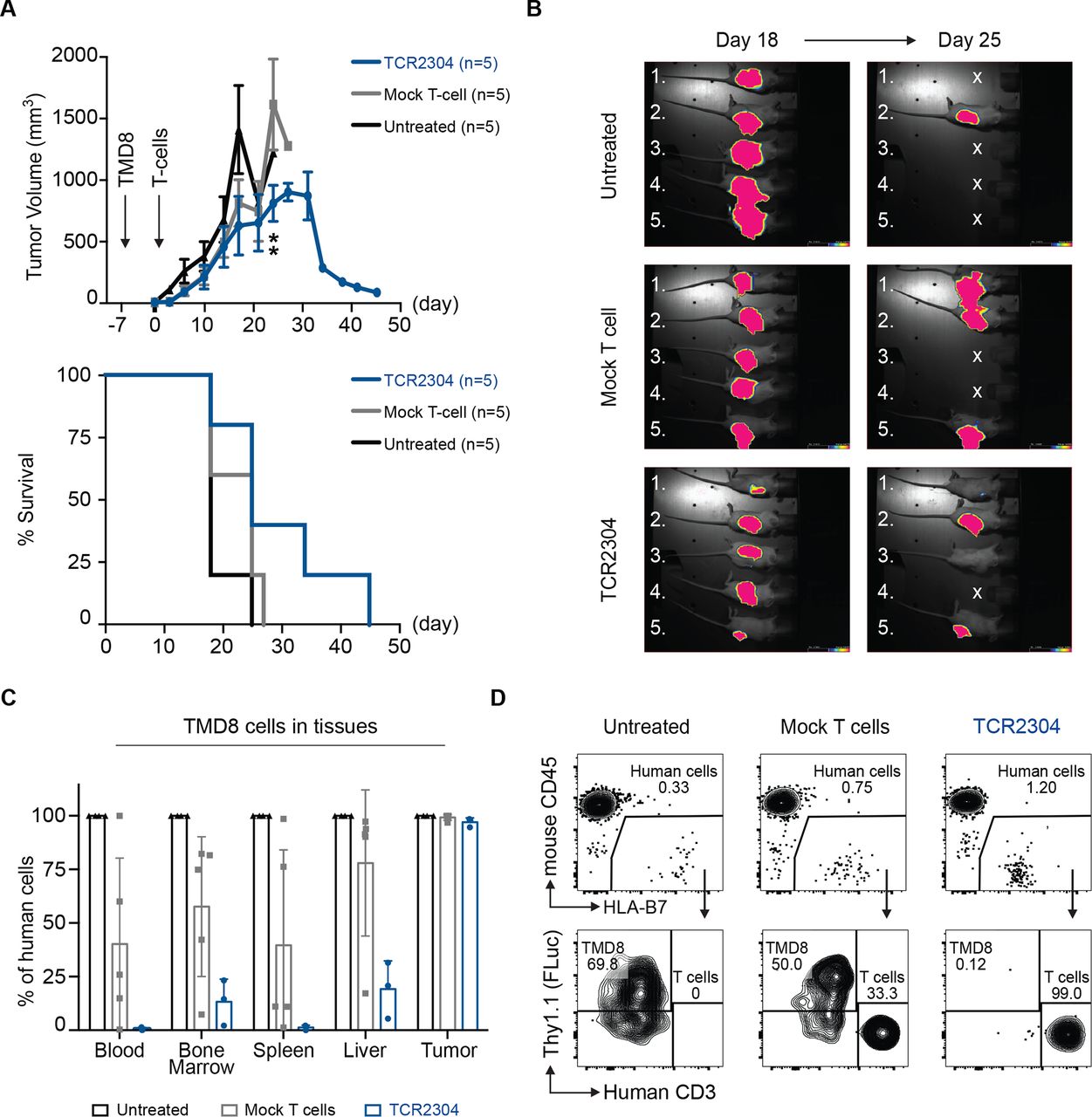
To validate our findings in a second model, luciferase-expressing HLA-B*07:02-positive TMD8 cells were subcutaneously injected into NOG mice, yet it caused some mice to die early due to strong systemic dissemination and aggressive tumor progression before treatment could be administered. Therefore, we decided to treat TMD8 bearing NOG mice 7 days after tumor inoculation. Indeed, on these experimental conditions, the T-cell treatment led to a significant reduction in tumor size starting at day 24 (figure 6A,B). Furthermore, TCR-T cell treatment prevented systemic dissemination of TMD8 cells (figure 6C,D and online supplemental figure 3).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Therapeutic efficacy of TCR2304-T cells in TMD8 xenograft mouse model. NOG mice were suncutaneously injected with 5×106 TMD8 cells that are stably transduced with HLA*B07:02 and firefly luciferase. Tumor growth was detectable via bioluminescence imaging in all mice as early as day 2. (A) Change of tumor volume over time (error bars plotted with SEM significance analysis by two-way analysis of variance, showing difference on day 24: **p<0.01) and survival of NOG mice after treatment start. (B) Bioluminescence images of tumor-bearing mice at days 18 and 25 after treatment. Exposure: 60 s. (C) Flow cytometric analysis of systemic dissemination: proportion of TMD8 cells in tissues of tumor-bearing NOG mice. Human cells meaning sum of TMD8 lymphoma cells and human T cells. (D) Representative staining of blood samples from tumor-bearing NOG mice. Shown plots were pregated on single, viable cells.
Again, data in hIL2-NOG mice were complicated by T-cell toxicity caused by human versus mouse GvHD, which differed from experiment to experiment depending on different human donors, and although these experiments largely confirmed activity of transduced T cells in controlling tumor growth, they are not presented here.
TCR2304 is highly specific for the MyD88 L265P mutation without off-target reactivity
Even though all TCRs described in this study were isolated from healthy HLA-B7- positive donors and thus passed thymic selection, the risk of cross-reactivity against other human proteins is still potentially present. Peptides generated in the alanine scan were loaded separately on HLA-B*07:02-expressing K562 cells and cocultured with TCR-T cells to define the amino acids that are essential for the recognition (binding motif) by different TCRs (online supplemental figure 4). The proline in position 2, which reflects the amino acid substitution L265P in mutant MyD88, as well as the tyrosine in position 7, turned out to be essential for all TCRs, demonstrating the specificity of the TCRs for the mutation. Following detection of the binding motif (online supplemental figure 5A), we assessed the possibility of cross-reactivity to other human proteins, which might be caused by binding-sequence similarity, using the online tool Expitope.20 In the case of TCR2304, the screen revealed 26 human peptides exhibiting motif similarity (xPxxIxYxxx) with up to five amino acid mismatches: 12 of these had some predicted binding affinity to HLA-B*07:02 on NetMHC V.4.0 and/or NetMHCpan V.4.0 (online supplemental figure 5). We tested TCR-T cells against all 12 peptides loaded on HLA-B*07:02-positive K562 cells in high concentration (10 µg/mL) in a coculture. However, no meaningful response was observed (online supplemental figure 5B).
For assessment of alloreactivity, we cocultured TCR-T cells with a panel of 16 Epstein-Barr virus-immortalized B-LCLs, which covered a broad spectrum of HLA haplotypes and most of the frequent HLA alleles found in Europe (Online supplemental table 2). When compared with untransduced control T cells, overnight coculture only demonstrated significant TCR-T cell response when HLA-B7-positive cell lines were externally loaded with mutant peptide (online supplemental figure 5C). Thus, we did not observe any sign of alloreactivity in this setting.
Discussion
Contrary to CAR-T cell-based approaches, TCR-based cellular therapy has not yet led to breakthrough clinical results. However, because of the promise of cancer specificity, targeting tumor-specific neoepitopes by TCR gene therapy is continuing to attract attention. In solid tumors, TCRs directed against patient-individual mutations but also against a few recurrent mutations have been successfully isolated. First trials targeting mutant KRAS are currently recruiting.27 28
In this study, we present strong preclinical evidence that the lymphoma-associated L265P point mutation of MyD88 creates suitable neoepitopes for HLA-presentation, eliciting an efficient mutation-specific T-cell response. We have isolated a high-affinity TCR targeting a 10 mer neoepitope restricted to HLA-B*07:02, which mediates strong in vitro T-cell response against lymphoma cells with both homozygous and heterozygous L265P mutations. Adoptive transfer of gene-modified TCR-T cells shows promising therapeutic effect in preclinical experiments using human xenografts in immune-deficient mice. Our data indicate that the mutant epitope can indeed serve as a rejection antigen. In addition, preliminary safety screening does not indicate off-target activity of the TCR.
In a previous study by Nelde et al in 2017,29 potential peptides derived from MyD88 L265P had been analyzed and shown to generate T-cell responses restricted to HLA-B*07:02 and HLA-B*15:01. Peptide-specific T cells reported in their study showed cytotoxic reactivity against peptide-loaded target cells, and it was suggested that these peptides may be used for a peptide-based immunotherapy approach, in case the peptides are naturally processed and presented. A separate study by Nielsen et al30 in the same year confirmed that one of the mentioned peptides restricted to HLA-B*07:02 induced a mutation-specific T-cell response, and data suggested that the target epitope is endogenously processed. Peptide-specific T cells against this HLA-B*07:02-restricted epitope were only detected in one out of six donors, hinting that a peptide-based immunotherapy approach may not elicit autologous immunity in the majority of patients.
In vitro antigen processing experiments, generating linear non-spliced peptides, are accepted to mirror the in vivo situation with high fidelity.24 In our work, we show that the HLA-B*07:02 10 mer neoepitope is generated in vitro and that we also could generate strong T-cell responses in 5 out of 24 donors. However, only a few of 13 isolated and cloned TCRs from these five donors showed high affinity for the mutated peptide—demonstrating reactivity in the nanomolar range, which is thought to be crucial to achieve a therapeutic antitumor effect.31 Our high-affinity TCR elicited strong therapeutic activity in a preclinical mouse model, suggesting that the sequence of a high-affinity TCR could be used ‘off the shelf’ to generate a highly specific, individualized yet broadly applicable ATT product usable in all HLA-B*07:02-positive patients whose tumors carry the L265P mutation. In our opinion, such a treatment could fulfill in an ideal way the criteria for precision cancer therapy.
A main limitation to TCR gene therapy is the impaired MHC–peptide presentation in cancer cells. Mutations in the processing and presentation machinery, or downregulation of MHC molecules on the cell surface, is described in solid cancers and also in lymphomas.32 33 However, this rarely affects all malignant cells in a tumor and rarely results in a total loss of HLA antigens. IFN-γ secretion by even a few specific T cells engaging tumor might be able to transform the tumor microenvironment,34 inducing MHC upregulation and possibly enhancing the susceptibility of tumor cells to further TCR-T cells migrating into the tumor.35
In this respect, the difference we observed between hIL2-NOG and normal NOG mice is remarkable: NOG mice, in which T-cell proliferation is not supported by IL-2, showed some T-cell infiltration in tumors after adoptive transfer of MyD88 L265P-specific TCR-T cells, but tumors in some mice eventually regrew. Thus, it appears that besides lack of cytokine support for T-cell proliferation, upregulation of checkpoint inhibitory signals in T cells may have contributed to this result. Recent studies36 37 have shown that MyD88-mutant lymphomas exhibit high level expression of the immune-checkpoint mediator PD-L1, thus possibly preventing their efficient clearance by adaptive host immunity. Conversely, these mutant-specific dependencies were therapeutically exploitable by anti-PD1 checkpoint blockade. On the other hand, mice where T-cell growth is supported by IL-2 stay in remission. Experiments in human IL-2 transgenic mice, however, are complicated by strong GVHD effect of human T cells with strong variations in the severity of GVHD from donor to donor; therefore, only a limited number of experiments were performed using this model. We think that in an appropriate clinical setting where autologous transduced T cells likely encounter cytokine support—particularly if administered after lymphodepletion—escape mechanisms could be avoided. A combination of adoptive transfer of mutation-specific TCR-T cells with immune-checkpoint blockade38 might also be considered in a second phase of a pilot study.
We have tested our TCR in xenografted tumors but considered using a syngeneic lymphoma model with conditional MyD88 L265P mutation.39 However, these mice would have to be crossed to humanized HLA-B*07:02 mice,40 which is time consuming. More importantly, the information provided by such a model would be limited with respect to issues of safety and toxicity of the TCR in a clinical context, which is eventually crucial before moving to a clinical trial. This being our prior issue, we have extensively evaluated our TCR for potential toxicity. Based on our experience with generation of TCRs for ATT, we are confident that major reactivities can be excluded based on our LCL reactivity and alanine scan experiments. Moreover, while a number of TCRs are generated in transgenic mice for other targets with some of them already being approved by regulatory authorities for testing in clinical trials,41 42 TCRs described in this study were generated from healthy humans—without further modifications to enhance affinity—and had undergone thymic selection, thus at least theoretically further reducing the risk of unwanted recognition of self-proteins. Further safety testing, such as reactivity studies using larger cell line and tissue libraries or the use of amino acids other than alanine in amino acid replacements,43 has been proposed and will be discussed with safety authorities if required in preparation of a clinical trial.
We believe that besides CAR-T cell therapy, which has become a tremendous tool for the treatment of poor prognosis lymphomas and leukemias, there is a great potential in the use of TCR-T cell therapy. Our study provides a strong base for such a highly tumor-specific, molecularly defined TCR-based immunotherapy in selected hematological malignancies.
Data availability statement
Data are available upon reasonable request. All data relevant to the study are included in the article or uploaded as supplementary information. For further information, please contact oezcan.cinar@charite.de. Supplementary files can be found in the online version of this article. Complete length nucleic acid and amino acid sequences of T-cell receptors can be found in the published patent application: WO 2020/152161 A1.
Ethics statements
Acknowledgments
The authors thank Professor Dr Thomas Blankenstein (Charité–Universitätsmedizin Berlin, Germany) for constructive suggestions during the experimental phase and for reading the manuscript. The authors acknowledge Professor Dr Gerald Willimsky (Charité–Universitätsmedizin Berlin, Berlin, Germany) and Dr Armin Rehm (Max-Delbrück-Center for Molecular Medicine, Berlin, Germany) for helpful discussion through the study. The authors also thank Dr Simone Rhein (Max-Delbrück-Center for Molecular Medicine, Berlin, Germany) for technical advice and Dr Dilansu Guneykaya (Harvard Medical School, Boston, Massachusetts, USA) for experimental support and for helping the preparation of the manuscript.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Twitter @cinar_ozcan
AP and AB contributed equally.
Contributors ÖÇ designed and implemented studies, acquired, analyzed and interpreted data, and wrote the manuscript; BB designed and implemented animal studies and acquired and analyzed data; CG, CB and CAP acquired data; PMK interpreted the biochemistry data and reviewed and revised the manuscript; UK reviewed and revised the manuscript; AP designed and supervised the studies, interpreted the data, and reviewed and revised the manuscript; AB designed the studies, interpreted the data, and reviewed and revised the manuscript.
Funding This study was supported by SPARK/Validation Fund from Berlin Institute of Heath (BIH). ÖÇ was supported by fundings provided by Berlin School of Integrative Oncology (Charité–Universitätsmedizin Berlin) and The German Academic Exchange Service.
Competing interests AP and ÖÇ are inventors on a filed patent application for the T-cell receptors described in the study (WO 2020/152161 A1).
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.