Article Text

Abstract
Background Probody® therapeutics are antibody prodrugs designed to be activated by tumor-associated proteases. This conditional activation restricts antibody binding to the tumor microenvironment, thereby minimizing ‘off-tumor’ toxicity. Here, we report the phase 1 data from the first-in-human study of CX-072 (pacmilimab), a Probody immune checkpoint inhibitor directed against programmed death-ligand 1 (PD-L1), in combination with the anti-cytotoxic T-lymphocyte-associated protein 4 (anti-CTLA-4) antibody ipilimumab.
Methods Adults (n=27) with advanced solid tumors (naive to PD-L1/programmed cell death protein 1 or CTLA-4 inhibitors) were enrolled in the phase 1 combination therapy dose-escalation portion of this multicenter, open-label, phase 1/2 study (NCT03013491). Dose-escalation pacmilimab/ipilimumab followed a standard 3+3 design and continued until the maximum tolerated dose (MTD) was determined. Pacmilimab+ipilimumab was administered intravenously every 3 weeks for four cycles, followed by pacmilimab administered every 2 weeks as monotherapy. The primary objective was identification of dose-limiting toxicities and determination of the MTD. Other endpoints included the rate of objective response (Response Evaluation Criteria In Solid Tumors v.1.1).
Results Twenty-seven patients were enrolled in pacmilimab (mg/kg)+ipilimumab (mg/kg) dose-escalation cohorts: 0.3+3 (n=6); 1+3 (n=3); 3+3 (n=3); 10+3 (n=8); 10+6 (n=6); and 10+10 (n=1). Dose-limiting toxicities occurred in three patients, one at the 0.3+3 dose level (grade 3 dyspnea/pneumonitis) and two at the 10+6 dose level (grade 3 colitis, grade 3 increased aspartate aminotransferase). The MTD and recommended phase 2 dose was pacmilimab 10 mg/kg+ipilimumab 3 mg/kg administered every 3 weeks. Pacmilimab-related grade 3–4 adverse events (AEs) and grade 3–4 immune-related AEs were reported in nine (33%) and six (22%) patients, respectively. Three patients (11%) discontinued treatment because of AEs. The overall response rate was 19% (95% CI 6.3 to 38.1), with one complete (anal squamous cell carcinoma) and four partial responses (cancer of unknown primary, leiomyosarcoma, mesothelioma, testicular cancer). Responses lasted for >12 months in four patients.
Conclusions The MTD and recommended phase 2 dose of pacmilimab (10 mg/kg)+ipilimumab (3 mg/kg) every 3 weeks is active and has a favorable tolerability profile.
- B7-H1 antigen
- immunotherapy
- CTLA-4 antigen
- therapies
- investigational
Data availability statement
All data relevant to the study are included in the article or uploaded as online supplemental information. The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Immune checkpoint inhibitor (ICI)-targeted therapies have transformed the landscape of cancer treatment. Patients with a wide array of solid tumors have attained marked improvement in outcomes, with antibodies targeting programmed cell death protein 1 (PD-1), its ligand programmed death-ligand 1 (PD-L1), and cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4).1 2 Combination therapy with these agents has greater efficacy,3 4 but is associated with higher toxicity.4–6 The combination of nivolumab 1 mg/kg and ipilimumab 3 mg/kg improved progression-free survival compared with single-agent therapy but grade 3–4 treatment-related adverse events (AEs) were observed in 55% of patients treated with the combination compared with 16% with nivolumab monotherapy and 27% with ipilimumab monotherapy.7 Multiple different nivolumab plus ipilimumab regimens with reduced dosing for either nivolumab or ipilimumab are approved by the US Food and Drug Administration for the treatment of four types of solid tumors.8 Full dose (ie, 3 mg/kg for both agents) combination therapy was not tolerable.9
ICIs can result in potentially serious organ-specific immune-related AEs (irAEs)6 10 11 including grade 5 toxicity from colitis, myocarditis, pneumonitis, or hepatitis.5 6 These toxicities necessitate dose reductions or permanent treatment discontinuation. A dose-response relationship has been demonstrated with ipilimumab in patients with melanoma, with a survival advantage observed with increased exposure.12 It is therefore possible that ipilimumab dose reduction or discontinuation compromises clinical efficacy. Optimization of ICI treatment combinations may provide better antitumor activity without dose-limiting toxicity (DLT).
Antibody-based therapies that demonstrate high-affinity, high-specificity tumor antigen binding cause off-tumor toxicity due to the presence of target antigen in healthy tissues.13 Probody therapeutics are antibody prodrugs that minimize off-tumor toxicity by leveraging aberrant upregulation of proteases in the tumor microenvironment to achieve preferential local activation.14–16 They consist of the antibody backbone and a masking peptide held in place by a protease-cleavable linker peptide. The linker peptide is cleaved by tumor-associated proteases allowing the antibody to bind to its target.14 15 17 Over 90% of tumors across different indications, stages, and treatment histories demonstrated sufficient protease activity to enable cleavage of the linker peptide, ensuring that Probody therapeutics would be active in various tumor microenvironments (data not shown).
CX-072 (pacmilimab) is a Probody antibody directed against PD-L1. In MC38 tumor-bearing mice, a surrogate anti-PD-L1 Probody antibody demonstrated comparable antitumor responses with lower PD-L1 occupancy on peripheral T cells compared with the parent antibody and protected against autoimmune diabetes observed with the parent drug at the same dose.18 Further, only limited uptake of pacmilimab was observed in PD-L1-expressing non-tumor murine lymphoid tissues.19 Taken together, these data suggest pacmilimab is active and functions as designed in preclinical models. Preliminary translational studies confirmed proteolytic activation of pacmilimab and biological activity in tumor biopsy specimens of patients with cancer, supporting proof of mechanism.20
We hypothesized that combining pacmilimab with ipilimumab would improve tolerability of combined ICI therapy, allowing combination with optimal therapeutic doses of both agents and consequently an increase in treatment efficacy. PROCLAIM-CX-072 (PRObody CLinical Assessment In Man, CX-072 Clinical Trial 001) is a first-in-human study evaluating the tolerability, and preliminary antitumor activity of pacmilimab in patients with advanced, unresectable solid tumors (NCT03013491). The study consisted of multiple monotherapy and combination therapy cohorts. Here, we present data from patients receiving pacmilimab in combination with ipilimumab.
Methods
Study design and participants
In this phase 1/2, open-label study, immunotherapy-naive patients with metastatic/locally advanced, unresectable solid tumors were recruited at 32 sites in the USA, Netherlands, Poland, Spain, Ukraine, and UK. The study included one cohort that evaluated dose escalation of the combination of pacmilimab and ipilimumab. Enrolment into this cohort was initiated after the corresponding dose cohort for pacmilimab monotherapy successfully passed the DLT period. Dose escalation of the pacmilimab/ipilimumab combination followed a standard 3+3 design and continued until the maximum tolerated dose (MTD) was determined.
Eligible patients were aged ≥18 years with Eastern Cooperative Oncology Group performance status 0 or 1 and had an anticipated life expectancy of ≥3 months. Patients were required to have metastatic or advanced unresectable solid tumors (excluding thymic epithelial tumor, thymoma, and thymic carcinoma) naive to prior PD-1/PD-L1 and CTLA-4 inhibitor therapy and to be ineligible for standard-of-care therapy. Patients were eligible regardless of PD-L1 status. Exclusion criteria included prior chimeric antigen receptor T-cell therapy; chemotherapy or immunotherapy within 14 days, live vaccine within 30 days, or radiation therapy within 3 months prior to study drug; HIV-related illness, hepatitis B or C; age-related macular degeneration; solid organ or bone marrow transplant; prior or current autoimmune disease; history of a medical condition requiring treatment with >10 mg daily prednisone equivalents or immunosuppressants; and women who were pregnant or breastfeeding. Patients with treated stable brain metastases were eligible for the study if their treatment did not require radiation therapy or steroids. Active screening for brain metastases was not required.
Procedures
Eligible patients were enrolled in the cohort involving escalating dose combinations of pacmilimab and ipilimumab. Patients received combination therapy with pacmilimab (0.3, 1, 3, or 10 mg/kg) and ipilimumab (3 or 6 mg/kg) every 3 weeks for four doses, followed by pacmilimab monotherapy every 2 weeks. Prior to a protocol amendment mandating a 6 mg/kg maximum ipilimumab dose, one patient received ipilimumab 10 mg/kg. We did not test ipilimumab doses lower than 3 mg/kg since nivolumab combined with lower doses of ipilimumab (eg, 1 mg/kg every 3–6 weeks) is already considered tolerable. The DLT assessment period was 28 days. Pacmilimab was infused intravenously over 1 hour, followed by ipilimumab infusion over 90 min. Patients who discontinued study drug during the DLT period for reasons other than a DLT were replaced. Patients requiring a dose hold of ipilimumab were permitted to remain on pacmilimab treatment. Dose reductions were not permitted. Within the first 12 weeks after the start of treatment, permanent discontinuation was required if >10 mg/day of prednisone or equivalent was required to manage AEs. Permanent discontinuation was also required or if grade 2 or 3 treatment-related AEs did not improve to grade ≤1 or resolve within 12 weeks of the most recent dose of pacmilimab. All patients continued treatment until disease progression, unacceptable toxicity, or withdrawal of consent. Patients with evidence of disease progression who were still experiencing clinical benefit per the investigator’s judgment could be considered for continuation of treatment after discussion with the sponsor.
Tumor assessments were performed at baseline and every 8 weeks for the first 12 months and every 12 weeks thereafter. Responses were classified per Response Evaluation Criteria in Solid Tumors (RECIST) v.1.1 criteria. Treatment beyond the first instance of RECIST-defined progressive disease was allowed according to the modified immune-related RECIST criteria.21
PD-L1 expression was measured prospectively on archival tissue using the DAKO PD-L1 IHC 22C3 pharmDx assay (Agilent Technologies, Santa Clara, California, USA). High expression was defined by Tumor Proportion Score (TPS) >50% membrane staining, low expression was defined as TPS ≥1% and ≤50% membrane staining, and no expression was defined as TPS <1% membrane staining.
Outcomes
The primary objective was to evaluate the tolerability of pacmilimab in combination with ipilimumab and to determine the MTD. The MTD was defined as the highest dose at which no more than one of six patients experienced a DLT (online supplemental methods). AEs were evaluated using Medical Dictionary for Regulatory Activities terminology (v.22.0), with the National Cancer Institute’s Common Terminology Criteria for Adverse Events (v.4.03) grading. IrAEs were predefined as AEs with no clear alternate etiology that required systemic corticosteroid or immunosuppression treatment within 30 days of onset or that required the use of systemic hormonal supplementation (online supplemental table 1).
Supplemental material
Efficacy endpoints included objective response rate (ORR) as assessed by RECIST v.1.1 and duration of response (DOR). After discontinuing treatment, patients with stable disease (SD), partial response (PR), or complete response (CR) were followed every 3 months for DOR; those with progressive disease were followed for survival at the same interval.
Pharmacokinetic analyses
Blood samples were obtained at various timepoints before and after pacmilimab and ipilimumab administration for pharmacokinetic (PK) testing. Pacmilimab concentrations were assayed as reported elsewhere.22 Ipilimumab concentrations were assayed via a validated assay (online supplemental methods).
Statistical analyses
The safety evaluable population was defined as any patient who received any amount of study drug. The efficacy evaluable population was defined as any patient who had an adequate baseline tumor assessment. Exact two-sided 95% CIs were calculated for all proportion estimates. Kaplan-Meier analyses were used to estimate DOR, with median duration and its 95% CI based on the Brookmeyer and Crowley method.23
Results
Patients were enrolled between January 2017 and September 2019; data are reported as of August 28 2020. A total of 27 patients were enrolled (table 1). One patient remained on treatment and 26 had discontinued (figure 1). Reasons for treatment discontinuation were symptom deterioration (including patients with clinical signs of disease progression in the absence of confirmed radiographic evidence; n=13 (48%), of which seven had radiographic disease progression that was not confirmed with a second scan), confirmed radiographic disease progression (n=7 [26%]), AEs (n=3 [11%]), decision by the investigator or patient (n=2 [7%]), and ‘other’ (n=1 [4%]). Baseline patient and disease characteristics are shown in table 2. Patients with 21 different solid tumor types (cervical carcinoma n=3; 2 each of pancreatic and uterine carcinoma; all others of a single tumor type) were enrolled. The median patient age was 56 years (range, 28–70), and 59% and 79% of patients were female and White, respectively. The median number of prior cancer treatments in any setting was three (range, 1–10). One patient had high tumor PD-L1 expression (ie, TPS >50% membrane staining), 5 patients had low PD-L1 expression (TPS ≥1% and ≤50% membrane staining), and 15 had no tumor PD-L1 expression (6 were unknown).

Patient disposition.
Patient enrolment by dose cohort
Baseline patient and disease characteristics
The median duration of follow-up was 15.4 weeks (range 4–164). The median duration of pacmilimab treatment was 1.8 months (range 0–38.5), with six patients on treatment for more than 6 months. The median number of ipilimumab doses administered was three (range 1–4). Of patients who received 3 mg/kg of ipilimumab (n=20), 7 (35%) received all four doses. DLTs occurred in three patients, one at the 0.3+3 dose level (grade 3 dyspnea, resolved without corticosteroids) and two at the 10+6 dose level (grade 3 colitis [n=1 resolved with corticosteroids] and grade 3 increased aspartate aminotransferase [n=1 resolved without corticosteroids]); the MTD was determined to be 10 mg/kg pacmilimab+3 mg/kg ipilimumab. The tolerability profile is summarized in table 3. Treatment-related grades 3–4 AEs occurred in nine patients (33%), and grades 3–4 irAEs occurred in six patients (22%). Three patients (11%) had AEs leading to discontinuation of pacmilimab (colitis, transaminases increased, and neutropenia). No grade 5 toxicity was observed. While limited by the number of patients treated, there appeared to be a trend toward higher rates of grade 3–4 treatment-related AEs and irAEs with ipilimumab 6 mg/kg compared with 3 mg/kg.
Adverse event (AE) summary
Best tumor response per RECIST v.1.1 in evaluable patients* who had at least one postbaseline disease assessment†
Response data by dose level are summarized in table 4 and figure 2A, and the change in tumor burden over time is shown in figure 2B. The ORR was 19% (95% CI 6.3% to 38.1%), with one CR in a patient with anal squamous cell carcinoma and four PRs: one patient with a cancer of unknown primary, one with leiomyosarcoma, one with mesothelioma, and one with testicular cancer. Five patients (19%) had SD; the disease control rate was 37%. Among patients with objective responses, DOR ranged from 1.9 to 32.2 months, with four patients maintaining a response for more than 1 year (figure 2B). At the time of the analysis, a patient with anal squamous cell carcinoma had maintained a CR for 24 months. Of note, this patient’s tumor did not exhibit microsatellite instability and had intermediate tumor mutational burden (nine mutations/megabase). Target lesions continued to decrease during treatment with single-agent pacmilimab, eventually leading to conversion from PR to CR approximately 6 months after the last dose of ipilimumab. The patient with testicular cancer terminated treatment after 2 years, at which time the PR was maintained.
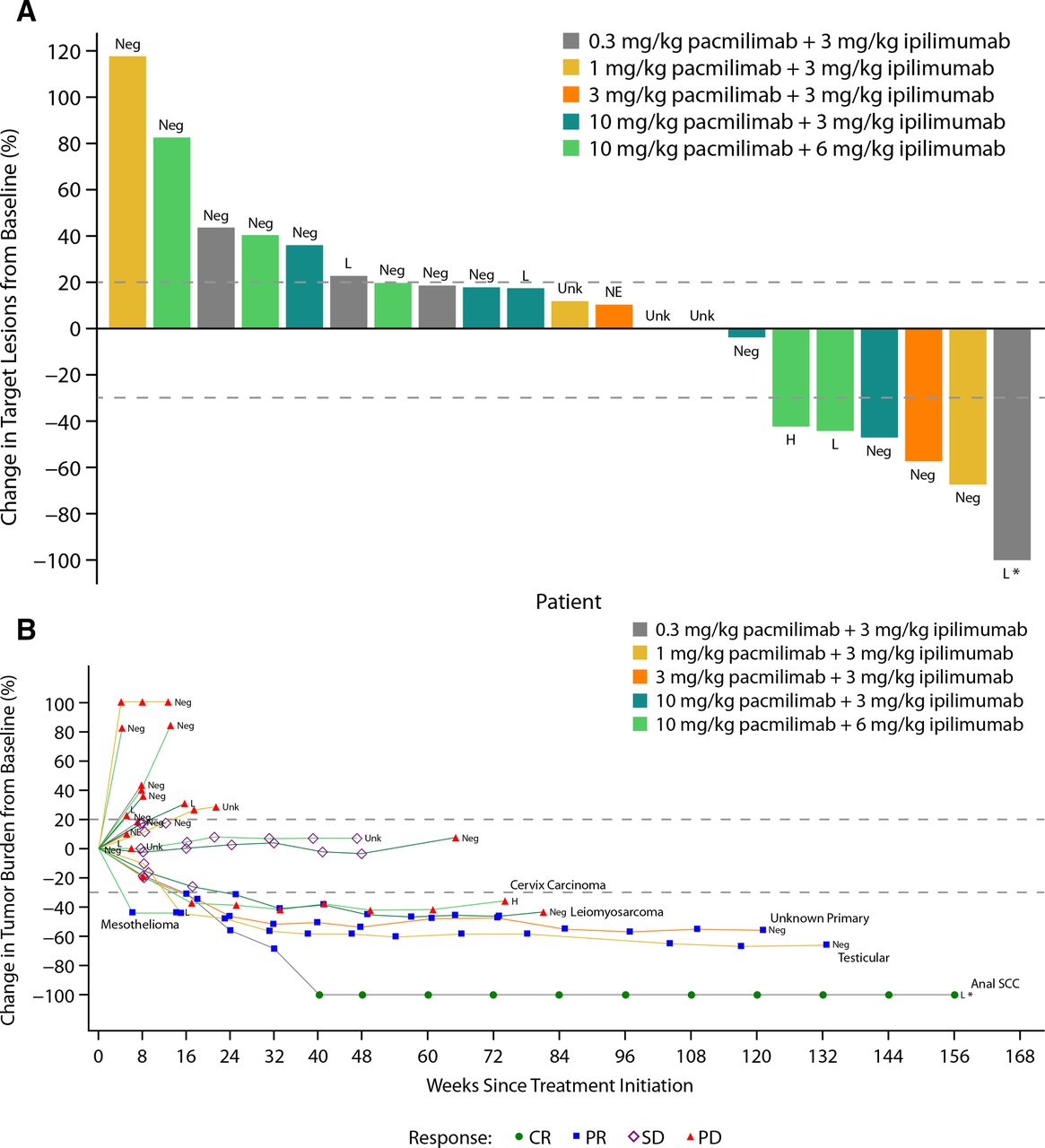
(A) Best percentage change in target lesion size from baseline and (B) percentage change in tumor burden over time. *Indicates that the patient is still on treatment. H, high PD-L1 expression; L, low PD-L1 expression; NE, not evaluable; Neg, no PD-L1 expression; Unk, unknown PD-L1 expression.
As shown in figure 3, the observed preliminary ipilimumab plasma concentrations after administration of 10 mg/kg pacmilimab followed by 3, 6, or 10 mg/kg ipilimumab administered were generally contained within the respective 90% prediction intervals of simulated ipilimumab concentrations based on the population PK model of Feng et al, suggesting pacmilimab is not a perpetrator of a PK drug–drug interaction with ipilimumab.24 As reported previously,22 patients maintained circulating plasma levels of pacmilimab following 10 mg/kg pacmilimab in combination with 3 mg/kg ipilimumab that were targeted for efficacy based on quantitative systems pharmacology modeling.

{kind=link}
{kind=link}
{kind=link}
Ipilimumab PK after (A) administration of pacmilimab followed by ipilimumab 3 mg/kg q3w×4; (B) pacmilimab followed by ipilimumab 6 mg/kg q3w×4; and (C) pacmilimab followed by ipilimumab 10 mg/kg q3w×4. Data presented are observed ipilimumab plasma concentrations (points) versus population predicted ipilimumab concentrations (line and shaded area). Line and shaded regions represent median and 90% prediction intervals of ipilimumab concentrations from a published population PK model for ipilimumab when given as monotherapy.24 PK, pharmacokinetic; q3w, once every 3 weeks.
Based in part on evidence of preliminary antitumor efficacy with similar tolerability across dose levels in those cohorts, and PK and pharmacodynamic considerations (figure 3),22 the recommended phase 2 dose was established as pacmilimab 10 mg/kg+ipilimumab 3 mg/kg.
Discussion
This first-in-human study evaluated pacmilimab in combination with ipilimumab in heavily pretreated patients with various solid tumors and established the recommended phase 2 dose of pacmilimab+ipilimumab. The pacmilimab dose range selected for this study was based on data from the pacmilimab monotherapy dose of PROCLAIM-CX-072, in which patients received pacmilimab monotherapy up to a dose of 30 mg/kg25. Based in part on evidence of preliminary antitumor efficacy with similar tolerability across dose levels in those cohorts, and PK and pharmacodynamic considerations described previously,22 10 mg/kg of pacmilimab was chosen for further investigation as monotherapy and thus was the dose level of focus in combination therapy. In the combination therapy study, the MTD/recommended phase 2 dose of pacmilimab 10 mg/kg+ipilimumab 3 mg/kg was established.
In a number of tumor types, treatment with combined ICIs has led to substantial improvements in antitumor activity including more durable ORRs and prolonged overall survival compared with chemotherapy or single-agent ICI therapy.3 4 However, combined ICI has been associated with higher risk of irAEs. Enhanced immune responses mediated by ICIs likely results in systemic activation of T-cell responses, leading to off-tumor effects manifested by irAEs. These effects are exacerbated when nivolumab and ipilimumab are combined.4–6 Thus, strategies for reducing toxicity are needed. The Probody pacmilimab is designed to restrict the inhibition of PD-L1 to the tumor microenvironment and potentially reduce off-tumor toxicity related to PD-L1 inhibition. The tolerability data from our study suggest that the Probody therapeutic platform reduces toxicity. Full monotherapy doses of both pacmilimab and ipilimumab were tolerated, in contrast to other anti-PD-1/L1 inhibitors when combined with ipilimumab.9 26 No new safety signals were identified and there were no treatment-related deaths. While some of the same toxicities observed with nivolumab plus ipilimumab were observed,4 they occurred at lower frequency and were less severe. Although only 7 of the 20 patients assigned to receive 3 mg/kg ipilimumab received all four doses, many of these patients came off trial for disease progression rather than toxicity.
Our data compare favorably to historical data for the only currently FDA-approved ICI combination nivolumab plus ipilimumab, particularly when considering the doses of ipilimumab used. Ipilimumab monotherapy is indicated for melanoma in the metastatic and adjuvant settings at doses of 3 mg/kg and 10 mg/kg, respectively.27 While monotherapy doses of nivolumab are tolerated in nivolumab ipilimumab combination regimens, ipilimumab doses of more than 1 mg/kg—which can result in greater efficacy—are associated with increased toxicity.9 28 29 Combination regimens for the treatment of colon cancer,30 non-small cell lung cancer,31 and clear cell renal carcinoma,32 are dosed at 3 mg/kg nivolumab plus 1 mg/kg ipilimumab. In these studies, rates of grade 3–4 treatment-related AEs were 32%, 33%, and 46%. In our study with a higher dose of ipilimumab (3 mg/kg), grade 3–4 treatment-related AEs occurred in 33% of patients. Similarly, the AE-related treatment discontinuation rates with 3 mg/kg nivolumab plus 1 mg/kg ipilimumab in patients with colon cancer,30 non-small cell lung cancer,31 and clear cell renal carcinoma,32 were 13%, 18%, and 22%, respectively. In our study the AE-related treatment discontinuation rate was 11%. While the conclusions that can be drawn are limited by the small sample size, our data suggest that a favorable tolerability profile can be achieved with an ipilimumab dose of 3 mg/kg, suggesting that ipilimumab doses above 1 mg/kg in combination with this Probody ICI may be feasible with higher doses of ipilimumab, potentially resulting in greater efficacy with ICI combination therapy.
The results also demonstrated preliminary evidence of clinical activity for the combination of pacmilimab and ipilimumab in heavily pretreated patients with advanced solid tumors unselected for PD-L1 expression. One CR and four PRs were observed in patients with anal squamous cell carcinoma, cancer of unknown primary, leiomyosarcoma, mesothelioma, and testicular cancer. Moreover, these responses were durable, with four of the five lasting for more than 1 year.
In conclusion, these findings add to the current body of evidence supporting the role of combination PD-1/PD-L1 and CTLA-4 inhibitor therapy in patients with advanced and/or metastatic solid tumors. The results of this study suggest that pacmilimab in combination with ipilimumab could provide a more favorable toxicity profile than traditional combination CTLA-4 and PD-1/PD-L1-targeted therapies. The tolerability of 3 mg/kg ipilimumab suggests that pacmilimab functioned as designed with preferential activation in the tumor microenvironment. This improved tolerability may allow the use of higher ipilimumab doses in combination therapy which would likely result in greater efficacy. The results support further evaluation of pacmilimab in combination with other ICIs or targeted therapies and highlight the potential value of Probody therapeutics in improving the safety and tolerability of immune therapies in patients with cancer.
Data availability statement
All data relevant to the study are included in the article or uploaded as online supplemental information. The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Ethics statements
Ethics approval
This study was conducted in accordance with the Institutional Review Board or independent ethics committee–approved protocol at each study site and with the International Conference on Harmonization Good Clinical Practice Guidelines. All patients provided written informed consent.
Acknowledgments
Medical writing and editorial assistance was provided by Esther Berkowitz MBChB, MA and Amy Zannikos, PharmD with Echelon Brand Communications, an OPEN Health company, Parsippany, NJ, and by Tracy McNally, PhD and Holly Strausbaugh, PhD of Twist Medical, LLC. This assistance was funded by CytomX Therapeutics, Inc. The authors thank the patients and investigators who participated in the study. PROBODY is a U.S. registered trademark of CytomX Therapeutics, Inc.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Twitter @RachelSanbornMD, @VriesElisabeth, @ValentinaBoni7
Contributors RES, OH, EGEdV, PAO, JG-C, VB, JB, KAA, DCC, RP and FT collected data. All authors interpreted the data, critically reviewed the manuscript, and provided approval to submit the manuscript for publication.
Funding This study was funded by CytomX Therapeutics, Inc. (South San Francisco, California, USA). This research was also funded, in part, through an NIH/NCI Cancer Center Support Grant to Memorial Sloan Kettering Cancer Center (P30 CA008748).
Competing interests RES: Honoraria from Amgen and AstraZeneca. Advisory boards for AstraZeneca, Blueprint Medicines, Celldex, Daiichi Sankyo, EMD Serono, Genentech/Roche, Janssen, and Seagen. Research support from AstraZeneca and Merck. OH: Consulting/advisory boards for Aduro, Akeso, Amgen, Beigene, Bioatla, BMS, Roche Genentech, GSK, lmmunocore, ldera, lncyte, Janssen, Merck, Nextcure, Novartis, Pfizer, Sanofi/Regeneron, Seattle Genetics, Tempus, and Zelluna. Speakers’ bureau for BMS, Novartis, Pfizer, and Sanofi/Regeneron. Institutional financial support for contracted research from Arcus, Aduro, Akeso, Amgen, Bioatla, BMS, CytomX, Exelixis, Roche Genentech, GSK, lmmunocore, ldera, lncyte, lovance, Merck, Moderna, Merck-Serano, NextCure, Novartis, Pfizer, Sanofi/Regeneron, Seattle Genetics, Torque, and Zelluna. EGEdV: Institutional financial support for clinical trials or contracted research from Amgen, AstraZeneca, Bayer, Chugai Pharma, G1 Therapeutics Genentech, Nordic Nanovector, Radius Health, Regeneron, Roche, Servier, and Synthon. Institutional financial support for advisory boards from Daiichi Sankyo, Merck, NSABP, Pfizer, and Sanofi; all outside the submitted work. PAO: Research funding from and has advisory role for Neon Therapeutics, Bristol-Meyers Squibb, Merck, CytomX, Pfizer, Novartis, Celldex, Amgen, Array, AstraZeneca/MedImmune, Armo BioSciences and Roche/Genentech. JG-C: Speaker’s bureau for Bayer; Fees to support registration and attending scientific meetings from BMS, Novartis. VB: Consulting/advisory role for CytomX Therapeutics, Guidepoint, Ideaya Biosciences; Loxo Therapeutics, Oncoart, and Puma Biotechnology. Institutional financial support for clinical trials from Abbvie, ACEO, Adaptaimmune, Amcure, Amgen, Astellas, AstraZeneca, BeiGene, BMS, Boehringer Ingelheim, Boston Therapeutics, CytomX Therapeutics, Daiichi, DebioPharm, Dynavax, GSK, Genentech/Roche, H3, Incyte, Innovio, Janssen, Kura, Lilly, Loxo, Mersana, Nektar, Macrogenics, Menarini, Merck, Merus, Millenium, MSD, Nanobiotix, Novartis, ORCA, Pfizer, PharmaMar, Principia, PsiOxus, PUMA, Regeneron, Rigontec, Sanofi, Seattle Genetics, Spectrum, Synthon, Taiho, Tesaro, Transgene, Takeda, and Zenith. JB: Institutional financial support for clinical trials or contracted research from AbbVie, Acerta Pharma, ADC, Agios, Amgen, Apexigen, Arch Oncology, Arcus Bio, ARMO, Array, Arrys, AstraZeneca, AtlasMedx, Bayer, Beigene, Bellicum, BI, Bicycle Therapeutics, Blueprint, BMS, Boston Biomedical, CALGB, Calithera, Celgene, Celldex, Cyteir Therapeutics, Cytomx, Daiichi Sankyo, Effector, Eisai, EMD Serono, Evelo, Five Prime, FORMA, Forty Seven, Foundation Bio, Genentech / Roche, Gilead, Gossamer Bio, GSK, Harpoon, Hutchinson MediPharma, IGM Biosciences, Imclone, Incyte, Innate, Innate Pharma, Ipsen, Jacobio, Koltan, LEAP, Lilly, Mabspace, Macrogenics, Marshall Edwards, MedImmune, Merck, Merrimack, Mersana, Merus, Millennium, Morphotex, Nektar, NeoImmune Tech, NGM Biopharma, Novartis, Novocare, NuMab, Oncogenex, OncoMed, Ongologie, Onyx, Pfizer, Pieris, Prelude Oncology, PureTech Health, Regeneron, Relay Therapeutics, REPARE Therapeutics, Revolution Medicines, Rgenix, Sanofi, Scholar Rock, Seattle Genetics, Shattuck Labs, Sierra, Stemcentrx, SynDevRex, Synthorx, Taiho, Takeda, Tarveda, TempestTx, TG Therapeutics, Tracon, Treadwell Therapeutics, Tyrogenex, Unum Therapeutics, Vyriad, and Zymeworks. Institutional financial support for advisory boards/consulting from Array, Agios, Amgen, Apexigen, Arch Oncology, ARMO, AstraZeneca, Bayer, Beigene, BI, Bicycle Therapeutics, BMS, Celgene, Continuum Clinical, Cyteir, Daiichi Sankyo, Evelo, Five Prime, FORMA, Fusion Therapeutics, Genentech / Roche, Gilead, GSK, Incyte, Innate, Ipsen, Janssen, LEAP, Lilly, Macrogenics, MedImmune, Merck, Merrimack, Moderna Therapeutics, Molecular Partners, Novartis, Oncogenex, OncoMed, Pfizer, Phoenix Bio, Piper Biotech, Prelude Therapeutics, Relay Therapeutics, Samsung Bioepios, Sanofi, Seattle Genetics, Taiho, Tanabe Research Laboratories, TD2 (Translational Drug Development), TG Therapeutics, Tizona, Tolero, and Torque. Food, beverages and/or travel expenses from ARMO, BI, BMS, Celgene, FORMA, Genentech / Roche, Gilead, Ipsen, Lilly, MedImmune, Merck, Novartis, Oncogenex, OncoMed, and Taiho. KAA: Advisory board for CytomX (unpaid). Institutional financial support for clinical trials or contracted research from Amgen, AstraZeneca, CytomX, GSK, Merck, Pfizer, and Tizona. DCC: Consultant for Nektar Therapeutics, GSK, Torque, PureTech, and HUYA. RP: Honoria for attending advisory boards from Pierre Faber, Bayer, Octimet, Clovis Oncology, Novartis, Karus Therapeutics, Biosceptre, BMS, Cybrexa, Ellipses, CV6 Therapeutics, Astex Therapeutics and Sanofi Aventis. Fees for delivery of educational talks or chairing educational meetings by AstraZeneca, Novartis, Bayer, Tesaro and BMS. Funds to support attendance at conferences from BMS and MSD. MS is a former employee and Lawrence Lu is a current employee of CytomX Therapeutics and both own stock in CytomX Therapeutics. FT: Research funding and conference registration from Novartis. Consultancy/advisory role for GSK, Achilles Therapeutics, BMS, Zelluna, Enara Bio, Bayer, T-Knife.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.
Linked Articles
- Clinical/translational cancer immunotherapy