Article Text

Abstract
Background Probody® therapeutics are antibody prodrugs that are activated in the tumor microenvironment by tumor-associated proteases, thereby restricting the activity to the tumor microenvironment and minimizing ‘off-tumor’ toxicity. We report dose-escalation and single-agent expansion phase data from the first-in-human study of CX-072 (pacmilimab), a Probody checkpoint inhibitor directed against programmed death-ligand 1 (PD-L1).
Methods In the dose-escalation phase of this multicenter, open-label study (NCT03013491), adults with advanced solid tumors (naive to programmed-death-1/PD-L1 or cytotoxic T-lymphocyte-associated antigen 4 inhibitors) were enrolled into one of seven dose-escalation cohorts, with pacmilimab administered intravenously every 14 days. The primary endpoints were safety and determination of the maximum tolerated dose (MTD). In the expansion phase, patients with one of six prespecified malignancies (triple-negative breast cancer [TNBC]; anal squamous cell carcinoma [aSCC]; cutaneous SCC [cSCC]; undifferentiated pleomorphic sarcoma [UPS]; small bowel adenocarcinoma [SBA]; and thymic epithelial tumor [TET]); or high tumor mutational burden (hTMB) tumors were enrolled. The primary endpoint was objective response (Response Evaluation Criteria In Solid Tumors v.1.1).
Results An MTD was not reached with doses up to 30 mg/kg. A recommended phase 2 dose (RP2D) of 10 mg/kg was chosen based on pharmacokinetic and pharmacodynamic findings in the expansion phase. Ninety-eight patients enrolled in the expansion phase: TNBC (n=14), aSCC (n=14), cSCC (n=14), UPS (n=20), SBA (n=14), TET (n=8), and hTMB tumors (n=14). Of 114 patients receiving pacmilimab at the RP2D, grade ≥3 treatment-related adverse events (TRAEs) were reported in 10 patients (9%), serious TRAEs in six patients (5%), and treatment discontinuation due to TRAEs in two patients (2%). Grade ≥3 immune-related AEs occurred in two patients (rash, myocarditis). High PD-L1 expression (ie, >50% Tumor Proportion Score) was observed in 22/144 (19%) patients. Confirmed objective responses were observed in patients with cSCC (n=5, including one complete response), hTMB (n=4, including one complete response), aSCC (n=2), TNBC (n=1), UPS (n=1), and anaplastic thyroid cancer (n=1).
Conclusions Pacmilimab can be administered safely at the RP2D of 10 mg/kg every 14 days. At this dose, pacmilimab had a low rate of immune-mediated toxicity and showed signs of antitumor activity in patients not selected for high PD-L1 expression.
Trial registration number NCT03013491.
- B7-H1 antigen
- immunotherapy
- therapies
- investigational
Data availability statement
All data relevant to the study are included in the article or uploaded as online supplemental information. The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
The emergence of monoclonal antibodies directed against programmed-death-1 (PD-1) and programmed-death ligand-1 (PD-L1) has resulted in substantial clinical benefit, including improved overall survival in patients with an increasing range of solid tumors.1–6 Despite these considerable advances, dose-limiting—and occasionally fatal—immune-related adverse events (irAEs) reflecting non-tumor-specific immune system activation have been frequently reported. In a systematic review and meta-analysis of 13 randomized controlled trials involving more than 6000 patients receiving anti-PD-1/PD-L1 therapies, increased risk compared with standard therapies was reported for a range of irAEs, including hypothyroidism (odds ratio [OR]=6.92), pneumonitis (OR=5.37), colitis (OR=2.88), and hypophysitis (OR=3.88).7 While not common, despite appropriate management, irAEs can lead to death.8 Moreover, irAEs often necessitate treatment interruptions or discontinuation and prolonged use of steroids, conceivably reducing anti-tumor efficacy.9 Therefore PD-1/PD-L1-targeted therapies with improved tolerability and safety are needed.
Probody® therapeutics are novel therapeutics designed to improve the safety and tolerability profile of monoclonal antibodies, antibody–drug conjugates, and other antibody-based therapies. These fully recombinant monoclonal antibody prodrugs comprise an active antibody (or antibody fragment) component, a masking peptide, and a protease-cleavable substrate linker peptide (figure 1). Exploiting the dysregulated protease activity in cancer cells, Probody therapeutics remain largely intact—and thus inactive—in the circulation, whereas in the tumor microenvironment, the masking peptide linker is cleaved by proteases allowing antibody binding to the target antigen expressed on the tumor.10–12

(A) Probody antibodies like pacmilimab comprise an anti-cancer antibody, a cleavable linker, and a masking peptide. (B) The Probody antibody enters the tumor microenvironment where tumor-associated proteases remove the masking peptide by cleaving the linker. (C) The unmasked antibody then selectively binds its target.
CX-072 (pacmilimab) is a Probody therapeutic directed against PD-L1. In preclinical studies, pacmilimab induced antitumor responses comparable to those of the same dose of the parental antibody in mice bearing MC38 tumors.13 Pacmilimab accumulated in murine PD-L1-expressing tumors with minimal uptake in peripheral, non-cancerous lymphoid tissue, consistent with limited uptake in non-tumor PD-L1-expressing tissues and protection against autoimmunity.14 Data from translational studies evaluating on-treatment biopsies from patients with cancer showed that pacmilimab functioned as designed, with proteolytic activation of pacmilimab, expansion of intratumoral CD8+ T cells, and increased markers of T-cell activation, thereby demonstrating proof of mechanism.15
PROCLAIM-CX-072 (PRObody CLinical Assessment In Man, CX-072 Clinical Trial 001; NCT03013491), the first-in-human proof-of-concept study of pacmilimab, determined appropriate dosing and evaluated tolerability and preliminary antitumor efficacy of pacmilimab in patients with advanced, unresectable solid tumors. Here, we report safety and efficacy data of single-agent pacmilimab in a large, open-label, dose-finding proof-of-concept study in patients with selected advanced malignancies.
Methods
Study design and participants
In this open-label study, immunotherapy-naive patients with advanced, unresectable solid tumors were recruited at 32 sites in the USA, The Netherlands, Poland, Spain, Ukraine, and UK. The study included three parallel cohorts that evaluated single-agent pacmilimab: dose-escalation cohorts to evaluate safety and to determine the maximum tolerated dose (MTD) and an expansion cohort to establish preliminary antitumor activity in selected malignancies.
Eligible patients were aged ≥18 years with Eastern Cooperative Oncology Group performance status 0 or 1, with an anticipated life expectancy of ≥3 months. Patients in the dose-escalation phase were required to have metastatic or advanced unresectable solid tumors (measurable or non-measurable disease) for which no standard therapy was available and to be PD-1/PD-L1- and cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4) inhibitor-naive. Patients with treated brain metastases were eligible if the brain metastases were stable and the patient did not require radiation therapy or steroids. Patients in the biomarker dose-escalation cohort were required to have tumors suitable for biopsy. To be enrolled into this group, patients were required to have tumors with PD-L1 expression ≥1%, which was defined as Tumor Proportion Score (TPS) ≥1% membrane staining using the DAKO PD-L1 IHC 22C3 pharmDx assay (Agilent Technologies, Santa Clara, California, USA). PD-L1 expression was not required for enrollment in any other cohort.
Patients in the expansion phase were required to have measurable disease and to be PD-1/PD-L1- and CTLA-4 inhibitor-naive. Patients with one of seven tumor types were eligible: (1) advanced or metastatic triple-negative breast cancer (TNBC) with documented progression after 1–3 systemic chemotherapy regimens for metastatic disease; (2) unresectable anal squamous cell carcinoma (aSCC) with prior radiation therapy and/or chemotherapy; (3) unresectable cutaneous SCC (cSCC); (4) undifferentiated pleomorphic sarcoma (UPS) with ≥1 prior systemic therapy or unresectable, advanced disease with prior standard surgery and/or radiation therapy and TPS ≥1% membrane staining or unknown PD-L1 status; (5) small bowel adenocarcinoma (SBA) with 1–3 prior systemic chemotherapy regimens for metastatic or unresectable disease; (6) thymic epithelial tumor (TET) with stage II–IV disease and ≥1 prior chemotherapy regimen; or (7) patients whose tumors had high tumor mutational burden (hTMB), defined as ≥16 mutations/megabase in a recent sample using a validated assay and failure or refusal of standard therapy. These tumors were chosen based on a lack of available therapies to treat them at the time of study initiation and evidence in the medical literature of PD-L1 expression or preliminary responses to PD-(L)1-targeted therapies.
Exclusion criteria included prior chimeric antigen receptor T-cell therapy; prolonged QTc interval or use of medications known to cause QT prolongation; chemotherapy or immunotherapy within 2 weeks; prior or current autoimmune disease; history of a medical condition requiring >10 mg daily prednisone (or equivalent) or immunosuppressants; and unresolved grade >1 acute drug toxicity. Patients with TET who had a history of interstitial lung disease were excluded.
PD-L1 expression was determined on archived or biopsied tissue locally using the DAKO PD-L1 IHC 22C3 pharmDx assay. TMB was determined locally using a Clinical Laboratory Improvement Amendments validated assay from a recent tumor tissue or blood sample.
Procedures
In the dose-escalation phase, patients received single-agent pacmilimab (0.03, 0.1, 0.3, 1, 3, 10, and 30 mg/kg) intravenously over 60 min (3–5 min for the 0.03 mg/kg dose; 90 min for the 30 mg/kg dose) every 2 weeks. The dose-limiting toxicity (DLT) assessment period was 28 days. Initially, one patient each was enrolled into the 0.03, 0.1, and 0.3 mg/kg groups, and these patients were eligible for intra-patient dose escalation. Subsequent dose levels followed a 3+3 design. In the biomarker group, six additional patients with PD-L1–positive solid tumors received intravenous single-agent pacmilimab at doses of 0.3 mg/kg, 1 mg/kg, 3 mg/kg, and 10 mg/kg every 2 weeks. Patients in the expansion phase received intravenous pacmilimab at a dose of 10 mg/kg every 2 weeks, until confirmed disease progression or unacceptable toxicity.
Outcomes
AEs were assessed every 2 weeks and were graded using the National Cancer Institute’s Common Terminology Criteria for Adverse Events (v.4.03). Biopsies (if performed) occurred during the 28-day screening period and on study day 29. A DLT was defined as grade 2 pneumonitis or ocular toxicity necessitating discontinuation of pacmilimab, any grade 3 central nervous system event, treatment-related grade ≥3 AEs (except grade 4 lymphopenia, non-febrile neutropenia, nausea, diarrhea, vomiting, asthenia, or constipation lasting ≤48 hours; correctable, asymptomatic electrolyte imbalances; clinically manageable endocrinopathy; or tumor flare). irAEs were predefined as AEs with no clear alternate etiology that required systemic corticosteroid or immunosuppressant treatment within 30 days of onset or that required the use of systemic hormonal supplementation (online supplemental table 1).
Supplemental material
The primary efficacy endpoint was objective response rate (ORR) assessed by Response Evaluation Criteria In Solid Tumors (RECIST) v.1.1. Stable disease was defined according to RECIST v.1.1 and duration of at least 8 weeks was required to be included in the disease control rate. Duration of response (DOR), defined as the time from first documentation of objective response to first documented disease progression by RECIST v.1.1 or death from any cause, was a secondary efficacy endpoint. CT or MRI scans were performed every 8 weeks for 12 months and every 12 weeks thereafter until disease progression. Treatment beyond the first instance of RECIST progressive disease was allowed according to the modified immune-related RECIST criteria.16 Patients with TNBC with skin lesions had cutaneous lesions photographed at all time points used for tumor assessment. After completing study treatment, patients were followed up every 3 months for survival (patients with progressive disease) or DOR.
Statistical analyses
To observe a sufficient number of objective responses in the dose-expansion phase with 95% probability, 20 patients with UPS and 14 patients with each other cancer type were required. For most tumor types (aSCC, SBA, TET, TNBC, and hTMB), one patient with a confirmed objective response supported further study of single-agent pacmilimab in that tumor type. For UPS and cSCC, a confirmed objective response was required in two and three patients, respectively, for further study. Data from the dose-escalation phase were pooled by dose with data from patients who received ≤1 mg/kg combined into a single category. Data from the expansion phase were analyzed by tumor type; statistical summaries were descriptive. Data from the 16 patients in the dose-escalation 10 mg/kg group were included in the dose expansion analyses, for a total of 114 patients. Because patients with TET may be at high risk of irAEs,17–19 a separate post hoc safety analysis was conducted for these patients.
Safety data were summarized using the safety population (all patients who received ≥1 dose of study drug). The efficacy population comprised all patients who received ≥1 dose of study drug and who had measurable disease at baseline. Tumor response was analyzed per RECIST v.1.1, using exact two-sided 95% CI for all proportion estimates. Kaplan-Meier analyses were used to estimate DOR, with median duration and 95% CI based on the Brookmeyer and Crowley method.20
Results
Patients were enrolled between January 19, 2017, and September 16, 2019; data are reported as of August 28, 2020. In the dose-escalation phase, a total of 53 patients enrolled: ≤1 mg/kg (n=21), 3 mg/kg (n=13), 10 mg/kg (n=16), and 30 mg/kg (n=3). Reasons for treatment discontinuation in the dose-escalation phase were symptomatic deterioration (defined as clinical signs of disease progression in the absence of confirmed radiographic evidence; 29 [54.7%]), disease progression (13 [24.5%]), treatment-related AEs (6 [11.3%]), investigator decision (2 [3.8%]), and patient decision (2 [3.8%]). As of the cut-off date, one patient from this study phase remained on study on active treatment. A further 98 patients were enrolled in the expansion phase: 14 patients with TNBC, 14 with aSCC, 14 with cSCC, 20 with UPS, 14 with SBA, 8 with TET, and 14 with hTMB. Reasons for treatment discontinuation in these patients included symptomatic deterioration not confirmed as disease progression (46 [46.9%]), disease progression (28 [28.6%]), patient decision (7 [7.1%]), AE (2 [2.0%]), death due to disease progression (2 [2.0%]), pregnancy (1 [1.0%]), and discontinuing the parent study to enroll in the long-term extension study (1 [1.0%]). As of the cut-off date, 11 of these patients remained on study on active treatment.
Patient and disease characteristics are shown in table 1. Tumor types in the dose-escalation phase included TET (n=10); castration-resistant prostate cancer (n=4), cervical carcinoma (n=4), esophageal carcinoma (n=4), colon carcinoma (n=3), pancreatic carcinoma (n=3), uterine carcinoma (n=3), rectal carcinoma (n=3), aSCC (n=2), TNBC (n=2), ovarian carcinoma (n=2), uterine sarcoma (n=2), and other (n=11). In the dose-escalation phase (n=53), patients had received a median of two prior cancer regimens (range: 0–11). PD-L1 expression was high (defined as >50% TPS) in 23%, low (≥1% and ≤50% TPS) in 28%, absent (<1% TPS) in 34%, and unknown in 15% of patients. In patients treated at the recommended phase 2 dose (n=114), PD-L1 expression was high in 19%, low in 31%, absent in 43%, and unknown in 7% of patients. The median number of prior regimens ranged from 0.5 (cSCC) to 4.5 (hTMB tumors) among tumor types, with the median number of prior regimens being two to three for most tumor types. PD-L1 expression varied by tumor type, with the highest expression in TET (38% with high PD-L1 expression) and the lowest expression in TNBC and SBA (no expression in 93% and 79%, respectively).
Baseline patient and disease characteristics
In the dose-escalation phase, the median duration of pacmilimab treatment was 2.3 months (range: 0.5–28.0 months). Two patients experienced DLTs (one patient with grade 4 infusion-related reaction treated with 1 mg/kg; one patient with grade 3 febrile neutropenia treated with 3 mg/kg), but an MTD was not reached. In the absence of an MTD, the 10 mg/kg dose level was selected as the recommended phase 2 dose based on overall tolerability in addition to pharmacokinetic and pharmacodynamic considerations.21 The AE profile in the dose-escalation phase is summarized in table 2 and detailed in online supplemental tables 2 and 3. Grade 3–4 treatment-related AEs were observed in eight patients (15%), with no AE occurring in more than one patient. All-grade irAEs occurred in six patients (11%). Three of these patients had grade 3–4 irAEs: febrile neutropenia and thrombocytopenia in one patient (3 mg/kg group), alanine aminotransferase increase and aspartate aminotransferase increase in one patient (30 mg/kg group), and pneumonitis in one patient (3 mg/kg group). Serious AEs observed in >1 patient were diarrhea (10 mg/kg; n=2, 1 was grade 2 and 1 was grade 3), infusion-related reaction (≤1 mg/kg and 10 mg/kg; n=2, 1 was grade 2 and 1 was grade 4), and lower respiratory tract infection (≤1 mg/kg and 3 mg/kg; n=2, both were grade 3). In the dose-escalation phase, seven patients (13%) experienced AEs leading to treatment discontinuation; no AEs led to death.
AEs in dose-escalation phase
Among all patients who received pacmilimab 10 mg/kg (n=114), the median duration of exposure was 3.0 months (range: 0.5–28.7 months). The safety profile is shown in table 3 and detailed in online supplemental tables 4 and 5. Grade 3–4 treatment-related AEs were observed in 10 (9%) patients: increased gamma glutamyl transferase (2%), increased lipase (2%), fatigue (1%), increased aspartate aminotransferase (1%), maculopapular rash (1%), enterocutaneous fistula (1%), hypertension (1%), and myocarditis (1%). All-grade irAEs occurred in 17 patients (15%). Only two events were grade 3–4: maculopapular rash (n=1) and myocarditis (n=1). Serious AEs were observed in 40 patients (35%); those observed in >1 patient are listed in table 3. Grade 3–4 serious AEs occurring in more than two patients were pneumonia (n=5, 4%), pericardial effusion (n=3, 3%) and sepsis (n=3, 3%). Six serious AEs were considered related to pacmilimab, including two that were grade 3–4 serious AEs (grade 3 myocarditis and grade 3 enterocutaneous fistula). Four patients (4%) had AEs leading to treatment discontinuation, and in two patients they were treatment-related (myocarditis and gamma-glutamyltransferase increased). No patients had AEs resulting in death.
AEs at pacmilimab 10 mg/kg in dose-escalation and expansion phases
Compared with other tumor types, patients with TET had a greater incidence of all-grade (70% vs 57%) and grade 3–4 treatment-related AEs (30% vs 9%). The incidence of irAEs in the TET subgroup was similar to that in other tumor types (10% vs 15%).
Response data for patients treated with pacmilimab 10 mg/kg are summarized in table 4. The overall ORR across tumor types was 12%. The highest ORRs were 36% observed in patients with cSCC and 29% observed in patients with hTMB. The disease control rate in all patients treated at 10 mg/kg (defined as stable disease or better) was 42%. The disease control rates ranged from 14% to 71%, depending on the tumor type. There were two complete responses, one in a patient with cSCC and one in a patient with an hTMB tumor (aSCC). One partial response was observed in the dose-escalation phase (n=53) in a patient with anaplastic thyroid cancer who received pacmilimab 10 mg/kg; no responses were observed in patients in the other dose-level groups. Among the 14 patients with objective responses, 12 remained on study as of the data cut-off, and 10 patients had responses lasting more than 12 months. Pseudoprogression defined by disease response (irRECIST criteria) following the first instance of RECIST v.1.1 disease progression was also observed in two patients treated with pacmilimab at 10 mg/kg. One of these patients (rectal cancer) later achieved a disease response allowing the patient to undergo successful surgical resection of the tumor; the patient was disease-free at the time of data cut-off.
Best tumor response per RECIST v.1.1 in evaluable patients* who had at least one post-baseline disease assessment (pacmilimab 10 mg/kg dose-escalation and expansion phases)
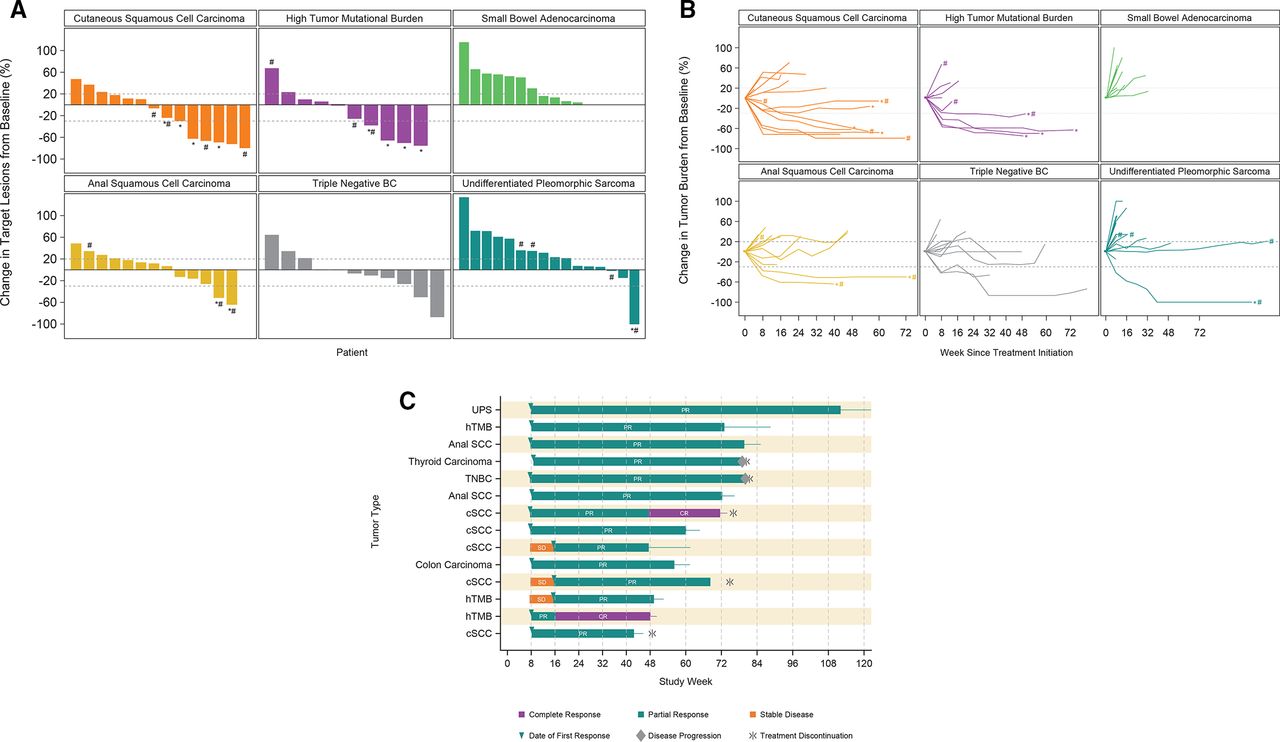
Changes in target lesions from baseline over time are shown in figure 2 by tumor type. Among responders, decreases in tumor size from baseline were observed in the first 8–16 weeks (figure 2B) and responses were durable (figure 2C).

{kind=link}
{kind=link}
(A) Best percentage change in target lesion size by tumor type during the expansion phase. (B) Change in target lesion burden over time by tumor type during the expansion phase. (C) Duration of response. *Indicates that the patient is still on treatment. #Indicates tumor with high (ie, >50% tumor proportion score) PD-L1 expressionNote: One patient with a high mutational burden tumor and one patient with cutaneous squamous cell carcinoma had a confirmed complete response. Some of the target lesions were lymph nodes, accounting for reduction in target lesion values from baseline of less than 100%. One patient with undifferentiated pleomorphic sarcoma and a confirmed partial response had a complete response in target lesions and non-complete response/non-progressive disease in non-target lesions.
Discussion
This first-in-class, first-in-human study provides evidence of tolerability and clinical activity of single-agent pacmilimab in previously treated patients with advanced or metastatic solid tumors. In the dose-escalation phase, no MTD was reached. However, a recommended phase 2 dose (10 mg/kg; n=114) was identified based on the tolerability profile on chronic administration as reported here, and pharmacokinetic considerations reported separately.21 Specifically, pacmilimab 10 mg/kg had an estimated half-life of approximately 12 days, and pharmacokinetic simulations suggested that following administration every 2 weeks >95% of patients would be expected to maintain pacmilimab trough levels above the targeted level.21
Among patients treated at the recommended phase 2 dose (n=114), the discontinuation rate due to treatment-related AEs was 2%. Preliminary pharmacokinetic data suggest that pacmilimab circulates predominantly (ie, 87%) in the intact, protected form, with minimal evidence of clearance via peripheral targets (ie, target-mediated drug disposition).21 Taken together, these data suggest that Probody technology functioned as expected, with limited peripheral activation of the checkpoint inhibitor.
Anti-PD-1/PD-L1 therapies are potentially limited by the development of irAEs. The incidence of all-grade irAES varies among PD-1 and PD-L1 inhibitor therapies but have occurred in as many as 30% of patients in phase 3 trials.22 The incidence of severe irAEs requiring medical intervention or hold/discontinuation of immunotherapy is estimated to range from 0.5% to 13% of patients receiving monotherapy.23 The most common irAEs among patients receiving anti-PD-1/PD-L1 agents include hypothyroidism (5.6%), pneumonitis (2.2%), colitis (0.7%), and hypophysitis (0.3%).7 Although rare, fatal irAEs can occur with PD-1 (0.36%) and PD-L1 (0.38%) inhibitors,8 with pneumonitis and cardiac irAEs being the most common causes of death.
In the present study—the first of pacmilimab—relatively common irAEs, such as pneumonitis and colitis, were seen only once (pneumonitis) or not at all (colitis) in the 151 patients treated with single-agent pacmilimab, suggesting a low incidence of these important AEs. However, it must be noted that this is an early-phase study and is therefore limited by the number of patients exposed to pacmilimab, a heterogeneous patient population, and patients who discontinued treatment due to early disease progression.
Data from the 114 patients treated in this study with pacmilimab at 10 mg/kg suggest preliminary antitumor activity, with an ORR of 12% and a disease control rate of 42%. The prespecified efficacy criteria to justify further exploration of single-agent pacmilimab were met for TNBC, aSCC, cSCC, and hTMB. At the time of trial initiation, no PD(L)-1 inhibitor was approved for these cancer types, although responses to checkpoint inhibitor therapy had been documented in these tumor types.24–27 Of note, patients were not selected for PD-L1 expression; among patients treated with 10 mg/kg pacmilimab who had available PD-L1 expression data, 49 of 106 (46%) had no PD-L1 expression, and 35 of 106 (33%) had low expression. There was no clear relationship between response to treatment and high PD-L1 expression (figure 2A).
The clinical activity observed with pacmilimab 10 mg/kg compares favorably to historical data with other PD-L1 or PD-1 inhibitors in patient populations unselected for PD-L1 expression.28 Although none of the TNBC patients in our study had high PD-L1 expression, the ORR was 7%, which is similar to that observed with single-agent atezolizumab in patients with metastatic TNBC in the second-line setting and beyond (n=94; 6%). 29 The response rate with pacmilimab in patients with aSCC (13%) is similar to that observed with single-agent pembrolizumab in patients with recurrent, PD-L1-positive aSCC (n=24; 17%).26 Although meaningful comparisons between studies are confounded by differences in design, patient characteristics, and the non-randomized nature of the comparison, the results observed with pacmilimab suggest potentially meaningful clinical activity.
Collectively, these data suggest that pacmilimab has a favorable tolerability profile with limited off-tumor toxicity. The potential for improved safety and tolerability of Probody therapeutics compared with conventional checkpoint inhibitors may yield optimal dose intensity with fewer dose interruptions or treatment discontinuations and may also allow improved tolerability of checkpoint inhibitor combinations with other checkpoint inhibitors, tyrosine kinase inhibitors, or cytotoxic chemotherapy. These preliminary findings with pacmilimab in patients with pretreated advanced solid tumors are encouraging and support further clinical evaluation. The tolerability and clinical activity of pacmilimab in combination with ipilimumab within the current study is reported separately30 .
Data availability statement
All data relevant to the study are included in the article or uploaded as online supplemental information. The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Ethics statements
Ethics approval
This study was conducted in accordance with the Institutional Review Board or independent ethics committee-approved protocol at each study site and with the International Conference on Harmonisation Good Clinical Practice Guidelines. All patients provided written informed consent.
Acknowledgments
Medical writing and editorial assistance were provided by Esther Berkowitz, MBChB, MA, and Amy Zannikos, PharmD, both with Echelon Brand Communications, an OPEN Health company, Parsippany, NJ, and by Tracy McNally, PhD, and Holly Strausbaugh, PhD, of Twist Medical, LLC. This assistance was funded by CytomX Therapeutics, Inc. The authors thank the patients and investigators who participated in the study. PROBODY is a US registered trademark of CytomX Therapeutics, Inc.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Twitter @AnaingMD, @VriesElisabeth, @ValentinaBoni7
Contributors AN, FT, EGEDV, FALME, NU, PAO, PL, JG-C, VB, JB, KAA, MR, GD, MG-M, H-TA and AS collected data. All authors interpreted the data, critically reviewed the manuscript, and provided approval to submit the manuscript for publication.
Funding This study was sponsored by CytomX Therapeutics, Inc., South San Francisco, CA.
Competing interests AN: Research funding from Amplimmune, Arcus Biosciences, ARMO BioSciences, Atterocor, BMS, Calithera Biosciences, CytomX Therapeutics, Eli Lilly, EMD, HealiosOnc, ImmuneOncia, Incyte, Karyopharm Therapeutics, Kymab, MedImmune, Merck, NCI, NeoimmuneTech, Neon Therapeutics, Novartis, Nutrition, Pfizer, PsiOxus, Regeneron, Serono, Surface Oncology, and TopAlliance Biosciences. Advisory boards for CytomX Therapeutics, Genome & Company, Kymab, Novartis, OncoSec KEYNOTE-695, and STCube Pharmaceuticals. Travel expenses from ARMO BioSciences. AN’s spouse has received research funding from Baxalta, Chao physician-scientist, Immune Deficiency Foundation, and Jeffery Modell Foundation; and has served on advisory boards for Behring, CSL, Horizon, Pharming, and Takeda. FT: Research funding and conference registration from Novartis. Consultancy/advisory role for Achilles Therapeutics, Bayer, BMS, Enara Bio, GSK, T-Knife, and Zelluna. EGEDV: Institutional financial support for clinical trials or contracted research from Amgen, AstraZeneca, Bayer, Chugai Pharma, G1 Therapeutics, Genentech, Nordic Nanovector, Radius Health, Regeneron, Roche, Servier, and Synthon. Institutional financial support for advisory boards from Daiichi Sankyo, Merck, NSABP, Pfizer, and Sanofi; all outside the submitted work. FALME: Consultancy/advisory role for Servier, Novartis, Eisai, and Ipsen. NU: Consultant for AstraZeneca, Eli Lilly, Ipsen, QED, and Taiho. Research support from EMD, Ipsen, Serono, and Taiho. Holds stock in Exact Sciences and Natera. PAO: Research funding from and advisory role for Amgen, Armo BioSciences, Array, AstraZeneca/MedImmune, Bristol-Meyers Squibb, Celldex, CytomX, Merck, Neon Therapeutics, Novartis, Pfizer, and Roche/Genentech. PL: Consultant/advisory board for AbbVie, ABL Bio, Agenus, Agios, Astellas, AstraZeneca, Black Diamond, Cybrexa, CytomX, EMD Serono, GenMab, Genentech, Glaxo-Smith Kline, ImmunoMet, IQVIA, Kineta Inc., Kyowa Kirin Pharmaceutical Development, MacroGenics, Molecular Templates, Pfizer, QED Therapeutics, Salarius, Shattuck, Silverback, SK Life Science, SOTIO, STCube Pharmaceuticals, Takeda, TRIGR, and Zentalis Pharmaceuticals. Data safety monitoring committee for Five Prime, Halozyme, and Tyme. Participant in imCORE Alliance (Roche/Genentech). JG-C: Speaker’s bureau for Bayer; fees to support registration and attending scientific meetings from BMS and Novartis. VB: Consulting/advisory role for CytomX Therapeutics, Guidepoint, Ideaya Biosciences, Loxo Therapeutics, Oncoart, and Puma Biotechnology. Institutional financial support for clinical trials from Abbvie, ACEO, Adaptaimmune, Amcure, AMGEN, Astellas, AstraZeneca, BMS, Boehringer Ingelheim, Boston Therapeutics, Cytomx Therapeutics, Daiichi, DebioPharm, Dynavax, GSK, Genentech/Roche, H3, Incyte, Innovio, Janssen, Kura, Lilly, Loxo, Macrogenics, Menarini, Merck, Mersana, Merus, Millenium, MSD, Nanobiotix, Nektar, Novartis, ORCA, Pfizer, PharmaMar, Principia, PsiOxus, PUMA, Regeneron, Rigontec, Sanofi, Seattle Genetics, Spectrum, Synthon, Taiho, Tesaro, Transgene, Takeda, and Zenith. JB: Institutional financial support for clinical trials or contracted research from AbbVie, Acerta Pharma, ADC, Agios, Amgen, Apexigen, Arch Oncology, Arcus Bio, ARMO, Array, Arrys, AstraZeneca, AtlasMedx, Bayer, Beigene, Bellicum, BI, Bicycle Therapeutics, Blueprint, BMS, Boston Biomedical, CALGB, Calithera, Celgene, Celldex, Cyteir Therapeutics, Cytomx, Daiichi Sankyo, Effector, Eisai, EMD Serono, Evelo, Five Prime, FORMA, Forty Seven, Foundation Bio, Genentech/Roche, Gilead, Gossamer Bio, GSK, Harpoon, Hutchinson MediPharma, IGM Biosciences, Imclone, Incyte, Innate, Innate Pharma, Ipsen, Jacobio, Koltan, LEAP, Lilly, Mabspace, Macrogenics, Marshall Edwards, MedImmune, Merck, Merrimack, Mersana, Merus, Millennium, Morphotex, Nektar, NeoImmune Tech, NGM Biopharma, Novartis, Novocare, NuMab, Oncogenex, OncoMed, Ongologie, Onyx, Pfizer, Pieris, Prelude Oncology, PureTech Health, Regeneron, Relay Therapeutics, REPARE Therapeutics, Revolution Medicines, Rgenix, Sanofi, Scholar Rock, Seattle Genetics, Shattuck Labs, Sierra, Stemcentrx, SynDevRex, Synthorx, Taiho, Takeda, Tarveda, TempestTx, TG Therapeutics, Tracon, Treadwell Therapeutics, Tyrogenex, Unum Therapeutics, Vyriad, and Zymeworks. Institutional financial support for advisory boards/consulting from Array, Agios, Amgen, Apexigen, Arch Oncology, ARMO, AstraZeneca, Bayer, Beigene, BI, Bicycle Therapeutics, BMS, Celgene, Continuum Clinical, Cyteir, Daiichi Sankyo, Evelo, Five Prime, FORMA, Fusion Therapeutics, Genentech/Roche, Gilead, GSK, Incyte, Innate, Ipsen, Janssen, LEAP, Lilly, Macrogenics, MedImmune, Merck, Merrimack, Moderna Therapeutics, Molecular Partners, Novartis, Oncogenex, OncoMed, Pfizer, Phoenix Bio, Piper Biotech, Prelude Therapeutics, Relay Therapeutics, Samsung Bioepios, Sanofi, Seattle Genetics, Taiho, Tanabe Research Laboratories, TD2 (Translational Drug Development), TG Therapeutics, Tizona, Tolero, and Torque. Food, beverages and/or travel expenses from ARMO, BI, BMS, Celgene, FORMA, Genentech/Roche, Gilead, Ipsen, Lilly, MedImmune, Merck, Novartis, Oncogenex, OncoMed, and Taiho. KAA: Advisory board for CytomX (unpaid). Institutional financial support for clinical trials or contracted research from Amgen, AstraZeneca, CytomX, GSK, Merck, Pfizer, and Tizona. MR: Honoraria from Bayer. GD: Research funding from AstraZeneca, Bristol Myers Squibb, and Merck. Honoraria from AstraZeneca. Advisory board for Curio Science. MG-M: Speaker’s bureau for Astra Zeneca, Pharmamar, and Roche. Financial support for registration and attendance at scientific meetings from MDS, Pharmamar, and Roche. MS is a former employee and ALH is a current employee of CytomX Therapeutics, Inc. Both are stockowners in CytomX Therapeutics Inc. H-TA: Advisor for Bayer, Beigene, Bicycle, Engitix, Guardant, iOnctura, Roche, and Servier. Employed by HCA Healthcare UK and Sarah Cannon Research Institute. AS: Consultant for Amgen, AstraZeneca, BMS, Merck, Mirati, and Novartis. Institutional financial support for clinical trials or contracted research from CytomX.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.