Article Text

Abstract
Background Avelumab (anti-programmed death ligand 1 (PD-L1)) is approved in multiple countries for the treatment of metastatic Merkel cell carcinoma (mMCC), a rare and aggressive skin cancer. We report efficacy and safety data and exploratory biomarker analyses from a cohort of patients with mMCC treated with first-line avelumab in a phase II trial.
Methods Patients with treatment-naive mMCC received avelumab 10 mg/kg intravenously every 2 weeks. The primary endpoint was durable response, defined as objective response (complete or partial response; assessed by independent review) lasting ≥6 months. Additional assessments included progression-free survival (PFS), overall survival (OS), safety, and biomarker analyses.
Results In 116 patients treated with avelumab, median follow-up was 21.2 months (range: 14.9–36.6). Thirty-five patients had a response lasting ≥6 months, giving a durable response rate of 30.2% (95% CI: 22.0% to 39.4%). The objective response rate was 39.7% (95% CI: 30.7% to 49.2%). Median PFS was 4.1 months (95% CI: 1.4 to 6.1) and median OS was 20.3 months (95% CI: 12.4 to not estimable). Response rates were numerically higher in patients with PD-L1+ tumors, Merkel cell polyomavirus (MCPyV)-negative tumors, and tumors with increased intratumoral CD8+ T-cell density. Exploratory analyses did not identify a biomarker that could reliably predict a response to first-line treatment with avelumab; however, a novel gene expression signature to identify the presence of MCPyV+ tumors was derived. Treatment-related adverse events (any grade) occurred in 94 (81.0%) patients, including grade 3/4 events in 21 (18.1%) patients; no treatment-related deaths occurred.
Conclusion In patients with mMCC, first-line treatment with avelumab led to responses in 40% and durable responses in 30%, and was associated with a low rate of grade 3/4 treatment-related adverse events.
- immunotherapy
- clinical trials
- phase II as topic
- gene expression profiling
- skin neoplasms
- tumor biomarkers
Data availability statement
Data are available upon reasonable request. For all new products or new indications approved in both the European Union and the United States after January 1, 2014, Merck KGaA, Darmstadt, Germany will share patient-level and study-level data after deidentification, as well as redacted study protocols and clinical study reports from clinical trials in patients. These data will be shared with qualified scientific and medical researchers, upon researcher’s request, as necessary for conducting legitimate research. Such requests must be submitted in writing to the company’s data sharing portal. More information can be found at https://www.merckgroup.com/en/research/our-approach-to-research-and-development/healthcare/clinical-trials/commitment-responsible-data-sharing.html. Where Merck KGaA has a coresearch, codevelopment, or comarketing/copromotion agreement or where the product has been out-licensed, it is recognized that the responsibility for disclosure may be dependent on the agreement between parties. Under these circumstances, Merck KGaA will endeavor to gain agreement to share data in response to requests.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
- immunotherapy
- clinical trials
- phase II as topic
- gene expression profiling
- skin neoplasms
- tumor biomarkers
Introduction
Merkel cell carcinoma (MCC) is a rare, aggressive skin cancer associated with the presence of clonally integrated Merkel cell polyomavirus (MCPyV), UV radiation exposure, increasing age, and immunosuppression.1 Before the advent of immune checkpoint inhibitors (ICIs), patients with metastatic MCC (mMCC) had a poor prognosis, with a 5-year overall survival (OS) rate of approximately 14%.2 MCC is considered chemosensitive; however, responses are seldom durable, and no specific chemotherapy regimen is recommended for mMCC in treatment guidelines.1 3 Few biomarker studies have been reported in this disease, although the presence of tumor-infiltrating CD8+ T cells has been associated with longer survival.4–6
Avelumab (anti-programmed death ligand 1 (PD-L1) antibody) was the first approved treatment for patients with mMCC and is now approved in multiple countries.7 Initial approval was based on primary analysis results from part A of the phase II JAVELIN Merkel 200 trial in patients with mMCC who received avelumab as second-line or later treatment after receiving chemotherapy,8 in addition to preliminary data from a subset of patients who received first-line treatment with avelumab in part B of the trial (n=39),9 which was initiated subsequently. Here, we report primary and biomarker analyses of part B of JAVELIN Merkel 200 after ≥15 months of follow-up in the full patient population.
Methods
Study design and patients
The design of JAVELIN Merkel 200 (NCT02155647), a phase II, prospective, single-arm, open-label, multicenter trial, has been reported previously.8 9 In part B, eligible patients were aged ≥18 years; had histologically confirmed, distant (stage IV) mMCC; and had not received prior systemic therapy for metastatic disease. Patients who had received prior chemotherapy in the adjuvant setting were eligible if the treatment ended ≥6 months prior to study enrollment. Additional eligibility criteria included an Eastern Cooperative Oncology Group performance status of 0 or 1; life expectancy of ≥3 months; ≥1 unidimensional measurable lesion (including skin lesions) according to Response Evaluation Criteria in Solid Tumors V.1.1 (RECIST 1.1); availability of recently obtained formalin-fixed, paraffin-embedded tumor tissue (archival tumor tissue obtained ≤6 months of enrollment or fresh biopsy material obtained at enrollment); and adequate hematological, hepatic, and renal function. Previous therapy with any ICI was not permitted. Full eligibility criteria are provided in the protocol (online protocol file).
Supplemental material
Procedures
Patients received avelumab 10 mg/kg intravenously every 2 weeks until significant clinical deterioration, unacceptable toxicity, or other criterion for withdrawal occurred. Tumors were assessed radiologically and reviewed by an independent review committee (IRC) per RECIST 1.1 every 6 weeks for 12 months, then every 12 weeks thereafter. Any complete response (CR) or partial response (PR) was confirmed by a second assessment ≥5 weeks after initial documentation. Patients with a confirmed CR continued treatment for ≤12 months after confirmation; treatment beyond 12 months was allowed per investigator judgment. Treatment continuation beyond radiological progressive disease (PD) was permitted in the absence of significant clinical deterioration and based on the investigator’s assessment of potential clinical benefit. To mitigate potential infusion-related reactions (IRRs), patients received premedication of antihistamine and acetaminophen before the first four avelumab infusions.
Adverse events (AEs) were graded using the National Cancer Institute Common Terminology Criteria for AEs V.4.0. Immune-related AEs (irAEs) were identified using a prespecified list of Medical Dictionary for Regulatory Activities preferred terms, followed by comprehensive medical review using predefined criteria. IRRs were assessed using an expanded definition that included events occurring on the day of or day after infusion and signs/symptoms occurring on the day of infusion (during or after the infusion) that resolved on the day of onset or the next day, based on a prespecified list of Medical Dictionary for Regulatory Activities preferred terms. Procedures for managing AEs have been reported previously.8
PD-L1 expression was assessed using the PD-L1 73-10 immunohistochemistry (IHC) assay (Dako, Carpinteria, California, USA). PD-L1+ status was defined as PD-L1 expression in ≥1% of tumor cells. MCPyV status was determined in DNA extracted from tumor samples by real-time PCR using TaqMan (Thermo Fisher Scientific, Waltham, Massachusetts, USA) reagents and small T-antigen-specific primers and by IHC using a mouse monoclonal antibody (MCPyV large T-antigen antibody (CM2B4); Santa Cruz Biotechnology, Dallas, Texas, USA).
Exploratory biomarker analyses
In exploratory biomarker analyses, CD8 IHC was performed on tumor samples using a mouse monoclonal anti-CD8 antibody (clone C8/144B; Dako, Carpinteria, California, USA) and evaluated by digital image analysis of whole slide scan using Aperio Nuclear V.9 algorithm (Leica Biosystems, Buffalo Grove, Illinois, USA). CD8+ T-cell density was evaluated at the tumor invasive margin (IM; from 500 µm outside to 500 µm inside the leading edge of the tumor in samples with an apparent tumor/normal boundary) and at the tumor center (TC; beginning inside the inner IM border and comprising the rest of the tumor, including intervening stromal bands).
Remaining tumor samples following PD-L1, CD8+ T-cell, and MCPyV analyses were used for tumor mutational burden (TMB) analysis and whole-exome sequencing (n=52) and RNA sequencing (n=50). To assess TMB, the average number of nonsynonymous somatic variants per megabase (NSSV/Mb) was calculated using patient-matched tumor and blood whole-exome sequencing profiles. Empirical cut-offs of <2 NSSV/Mb (low) and ≥2 NSSV/Mb (high) were chosen based on the distribution of TMB values in the study population,10 to include sufficient numbers of patients per subgroup, and for consistency with analysis of part A of the trial.11
Gene expression ranks of major histocompatibility complex (MHC) class I genes (human leukocyte antigen (HLA)-A, HLA-B, and HLA-C) were calculated using RNA sequencing data from normal tissue samples (Genotype-Tissue Expression (GTEx)) and patient tumor samples. Genome-wide copy number changes and loss of heterozygosity at the HLA locus were analyzed using Sequenza12 and a modified version of OptiType.13
Gene set enrichment analysis was carried out on unselected gene signature lists using Hallmark and Reactome pathway gene sets from the Molecular Signatures Database and EMD Serono’s internal collection.14 15 A fast preranked gene set enrichment analysis package was used with ranked lists of genes between conditions. Results were filtered using a cut-off of a normalized enrichment score of 2 and a false discovery rate of <1%. Immune content deconvolution from RNA sequencing profiles was done with xCell16 and Cibersort.17 The RandomForest classification was done in R with RandomForest and Caret packages. Gene set enrichment and immune content deconvolution analysis was performed between subgroups according to clinical response, MCPyV status, PD-L1 status, and CD8+ T-cell density at the IM and TC.
Outcomes
The primary endpoint was durable response, defined as objective response (CR or PR) determined by IRC per RECIST 1.1, with a duration ≥6 months. Other endpoints included confirmed best overall response, duration of response (DOR; time from CR or PR until documented PD or death, whichever occurred first), and progression-free survival (PFS) determined by IRC per RECIST 1.1; OS; and safety and tolerability. Biomarkers were assessed in exploratory analyses.
Statistical analysis
Clinical activity and safety were analyzed in all patients who received ≥1 dose of avelumab. With the planned sample size of 112 patients, and assuming a true durable response rate (DRR) of 45.0%, the probability of observing a lower bound of the exact 95% CI above 20.0% was >99.0% and above 30.0% was 90.0%. DRRs and objective response rates (ORRs) were calculated with two-sided 95% CIs using the Clopper-Pearson method. Time-to-event endpoints were estimated using the Kaplan-Meier method, and 95% CIs for the median were calculated using the Brookmeyer-Crowley method.
Results
Patients
As of May 2, 2019 (primary analysis data cut-off), 116 patients were enrolled and received avelumab (online supplemental figure 1). Six (5.2%) patients had received prior anticancer drug therapy (either cisplatin or carboplatin, combined with etoposide in three patients), either for locally advanced disease (n=4; 3.4%) or as adjuvant therapy (n=2; 1.7%) (table 1). Of 108 patients evaluable for PD-L1 expression, 21 (19.4%) had PD-L1+ tumors, and 87 (80.6%) had PD-L1− tumors. Median duration of treatment was 24.0 weeks (range: 2.0–154.0), and the median number of avelumab infusions was 11.5 (range: 1–76). Median follow-up was 21.2 months (range: 14.9–36.6). At data cut-off, treatment was ongoing in 26 (22.4%) patients; the most common reasons for discontinuation were PD (n=48; 41.4%) and AE (n=23; 19.8%) (online supplemental figure 1).
Supplemental material
Patient baseline characteristics
Efficacy
Thirty-five patients had a durable response (≥6 months), resulting in a DRR of 30.2% (95% CI: 22.0% to 39.4%). In total, 46 patients had a confirmed best overall response of CR (n=19; 16.4%) or PR (n=27; 23.3%), resulting in an ORR of 39.7% (95% CI: 30.7% to 49.2%) (table 2). Responses were observed early; 43 (93.5%) of 46 patients had a response by 3 months (figure 1A) and median time to response was 6.1 weeks (range: 5–36). Median DOR was 18.2 months (95% CI: 11.3 to not estimable (NE)); 26 (56.5%) patients had an ongoing response at data cut-off (figure 1A).
Tumor response according to independent review per RECIST 1.1

Clinical activity of avelumab. (A) Time to and duration of response for patients with an objective response according to IRC per RECIST 1.1 (n=46). (B) Kaplan-Meier estimate of overall survival (N=116). (C) DRR and (D) ORR in selected patient subgroups. CR, complete response; DRR, durable response rate; ECOG PS, Eastern Cooperative Oncology Group performance status; IRC, independent review committee; MCPyV, Merkel cell polyomavirus; ORR, objective response rate; PD, progressive disease; PR, partial response; RECIST 1.1, Response Evaluation Criteria in Solid Tumors version 1.1; SLD, sum of longest diameters.
Median PFS was 4.1 months (95% CI: 1.4 to 6.1), and 6-month and 12-month PFS rates were 41% (95% CI: 32% to 50%) and 31% (95% CI: 23% to 40%), respectively (online supplemental figure 2). Median OS was 20.3 months (95% CI: 12.4 to NE), and 6-month and 12-month OS rates were 75% (95% CI: 66% to 82%) and 60% (95% CI: 50% to 68%), respectively (figure 1B).
DRRs and ORRs in selected subgroups are shown in figure 1C,D. In preplanned biomarker analyses, a trend was seen for higher response rates in patients with PD-L1+ versus PD-L1− tumors (DRR, 47.6% (95% CI: 25.7% to 70.2%) vs 25.3% (95% CI: 16.6% to 35.7%); ORR, 61.9% (95% CI: 38.4% to 81.9%) vs 33.3% (95% CI: 23.6% to 44.3%), respectively). Median OS was not reached (95% CI: 11.3 months to NE) in patients with PD-L1+ tumors and 15.9 months (95% CI: 9.6 to NE) in patients with PD-L1− tumors; 12-month OS rates were 71% (95% CI: 47% to 86%) and 56% (95% CI: 45% to 66%), respectively. Response rates were numerically lower in patients with MCPyV+ (n=70) versus MCPyV− (n=37) tumors (assessed by IHC; DRR, 27.1% (95% CI: 17.2% to 39.1%) vs 35.1% (95% CI: 20.2% to 52.5%); ORR, 34.3% (95% CI: 23.3% to 46.6%) vs 48.6% (95% CI: 31.9% to 65.6%)). Response rates were numerically higher in patients with tumors with a CD8+ T-cell density at the IM median or higher (n=43) versus less than median (n=42; DRR, 37.2% (95% CI: 23.0% to 53.3%) vs 23.8% (95% CI: 12.1% to 39.5%); ORR, 51.2% (95% CI: 35.5% to 66.7%) vs 28.6% (95% CI: 15.7% to 44.6%)) (figure 1C,D); median OS in these subgroups was 20.3 months (95% CI: 8.4 to NE) and 15.9 months (95% CI: 6.2 to NE), respectively.
Exploratory genomic and transcriptomic analyses
Tumor mutational burden
In 52 evaluable patients, median TMB was 0.34 NSSV/Mb (range: 0.02–29.4) (online supplemental figure 3A). TMB was higher in patients with MCPyV− (n=19) versus MCPyV+ (n=31) tumors (assessed by IHC; median: 10.52 vs 0.22 NSSV/Mb) (online supplemental figure 3B), whereas TMB did not differ by PD-L1 status (online supplemental figure 3C) or by achievement of objective response (online supplemental figure 3D). Empirical cut-offs of ≥2 NSSV/Mb (high) versus <2 NSSV/Mb (low) were chosen based on the distribution of TMB values in the study population10 to include sufficient patient numbers per subgroup and for consistency with analysis of part A of the trial.11 In patients with high versus low TMB values, ORRs were 50.0% (95% CI: 26.0% to 74.0%) versus 41.2% (95% CI: 24.6% to 59.3%), and median OS was NE (95% CI: 5.2 months to NE) versus 17.2 months (95% CI: 8.4 to NE) (online supplemental figure 4).
Differential analysis of gene expression, signaling pathways, and immune signatures
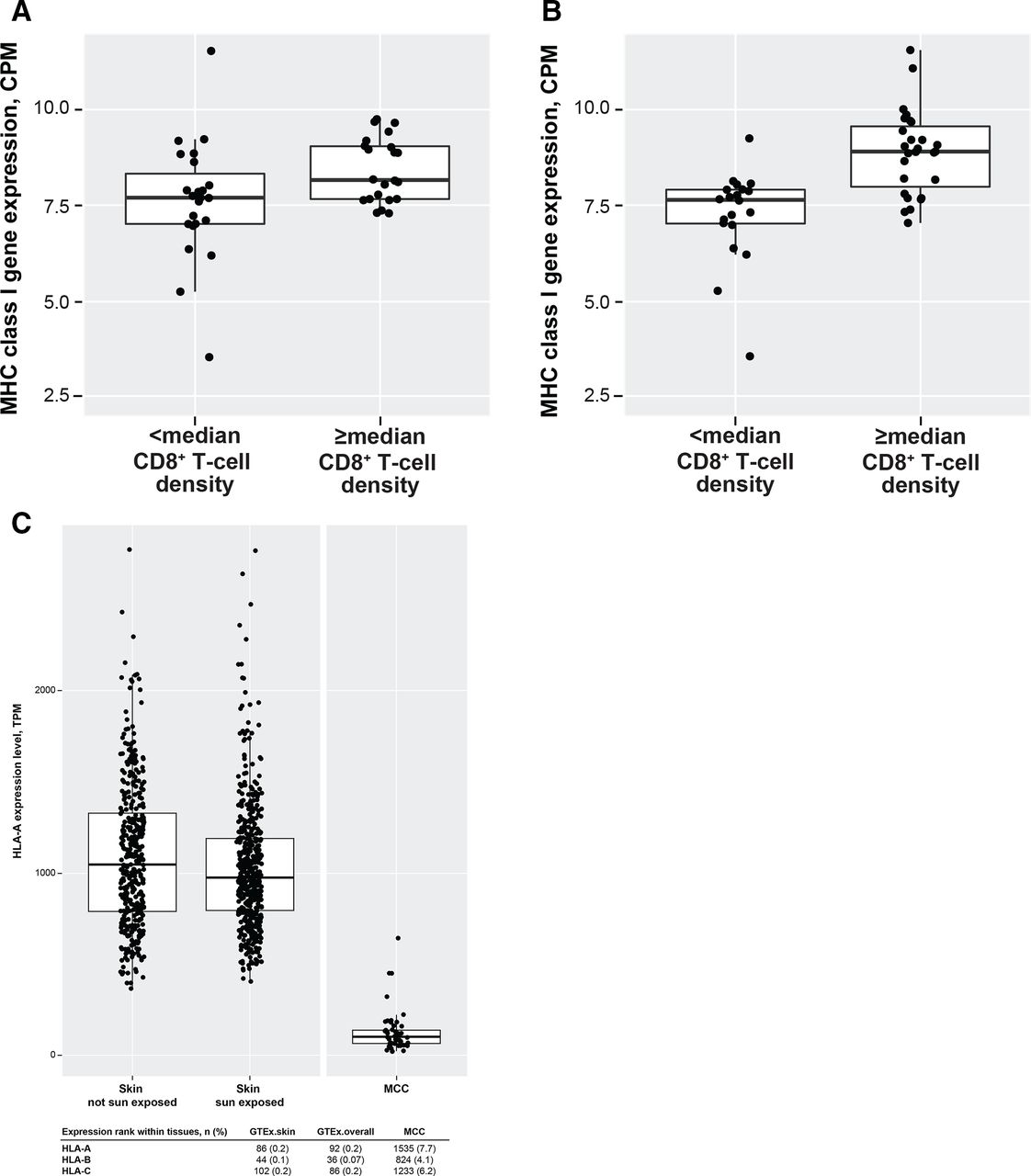
Differential gene expression analysis of RNA sequencing profiles (n=50) found that the most robust differential expression was observed in subgroups based on MCPyV status followed by CD8+ T-cell density at the TC (online supplemental table 1). As reported above, responders tended to have tumors that were PD-L1+, were MCPyV−, or had higher CD8+ T-cell density at the IM. Gene expression analysis found that gene sets associated with the inflammatory response (eg, interferon (IFN)-γ and IFN-α/β) and antigen processing and presentation were enriched in responders (online supplemental table 2). Gene sets for RNA transcription and protein synthesis pathways were enriched in nonresponders, suggesting ongoing tumor growth despite avelumab treatment. MHC class I gene expression did not correlate with response or OS; however, MHC class I gene expression was higher in patients with median or higher CD8+ T-cell density at the IM or TC (figure 2A,B). A positive correlation was observed between MHC class I gene expression and inflammatory markers associated with T-cell activation and response (eg, IFN-γ, natural killer cells, and M1 macrophages), in addition to signatures associated with T-cell exhaustion and immune suppression (online supplemental table 3).

Association of MHC class I gene expression with CD8+ T-cell density at the (A) invasive margin and (B) the tumor core and (C) MHC class I gene expression in normal tissue and MCC tumor samples (figure shows HLA-A expression). The boxes represent interquartile ranges, and the horizontal lines are medians. The whiskers denote the lower and upper quartiles, and the circles represent data points. CPM, counts per million; GTEx, Genotype-Tissue Expression; HLA, human leukocyte antigen; MCC, Merkel cell carcinoma; MHC, major histocompatibility complex; TPM, transcripts per kilobase per million.
We further evaluated differential gene expression within PD-L1, CD8+ T-cell, and MCPyV subgroups. In patients with PD-L1+ versus PD-L1− tumors, decreases in gene expression were seen in Hallmark gene sets associated with modulating PD-L1 expression (eg, interleukin-6 (IL-6)-Janus kinase-signal transducer and activator of transcription STAT-3 signaling, and tumor necrosis factor α signaling via nuclear factor-κB) (online supplemental table 4). Gene sets for CD40 pathway, B-cell antigen receptor signaling, and the inflammatory response were also decreased in patients with PD-L1+ tumors. Additionally, regulatory T-cell signatures were differentially expressed in PD-L1+ versus PD-L1− tumors. In tumors with median or higher CD8+ T-cell density at either the IM or TC, gene sets associated with the inflammatory response (eg, IFN-γ and IFN-α/β), antigen processing and presentation, cytokine-related pathways (eg, IL-12), and natural killer cell-mediated cytotoxicity were enriched (online supplemental tables 5 and 6). Gene expression signatures indicating the presence of CD8+ naive T cells were detected in samples with median or higher CD8+ T-cell density at the IM, whereas signatures of M1 macrophages and CD4+ memory T cells were detected in samples with median or higher CD8+ T-cell density at the TC, suggesting differential immune cell infiltration in different tumor regions. Patients with MCPyV+ tumors had elevated expression of gene sets for viral RNA transcription, oxidative phosphorylation, DNA repair pathways, and PD-1 signaling. Cell cycle-related pathways were enriched in patients with MCPyV− tumors. Increased expression of signatures for M2 macrophages and CD4+ T-helper cells were found in patients with MCPyV+ versus MCPyV− tumors, suggesting the presence of a more immunosuppressive tumor microenvironment consistent with our observation that patients with MCPyV+ tumors tended to have lower response rates with first-line avelumab treatment.
We also analyzed differential gene expression in MCC compared with normal tissue. Similar to results from part A of this study,11 overall expression of MHC class I genes was lower in MCC tumors compared with normal tissues (figure 2C). Loss of heterozygosity or changes in copy number at the HLA loci did not correlate with overall MHC class I gene expression (online supplemental table 7) and is therefore unlikely to account for the relatively low MHC class I gene expression observed in MCC tumors.
A novel gene expression signature was identified that was differentially expressed in MCPyV+ versus MCPyV− tumor tissue (assessed by IHC) (figure 3 and online supplemental table 8). To derive this signature, a two-class RandomForest classifier was trained on a balanced set of 36 samples. The ability of the classifier to identify MCPyV+ samples was evaluated using an additional 12 known MCPyV+ samples. The resulting 145-gene signature identified the presence of MCPyV with 100% accuracy.

{kind=link}
{kind=link}
{kind=link}
Expression profile according to MCPyV status. IHC, immunohistochemistry; MCPyV, Merkel cell polyomavirus; NA, not applicable.
Safety
All 116 patients had an AE of any grade, of whom 70 (60.3%) had a grade ≥3 AE (table 3). Treatment-related AEs (TRAEs) of any grade occurred in 94 (81.0%) patients; the most common were fatigue (n=24; 20.7%), asthenia (n=16; 13.8%) and pruritus (n=15; 12.9%) (online supplemental table 9). Grade 3 TRAEs occurred in 20 (17.2%) patients, and 1 patient had a grade 4 TRAE (dermatitis psoriasiform). The most common grade ≥3 TRAEs were increased lipase (n=4; 3.4%) and increased amylase (n=3; 2.6%) (online supplemental table 9). Serious TRAEs occurred in 17 (14.7%) patients; IRR (single preferred term; n=3; 2.6%) was the only serious TRAE to occur in ≥1 patient. TRAEs led to treatment discontinuation in 14 (12.1%) patients. No treatment-related deaths occurred. An irAE occurred in 35 (30.2%) patients, of whom 7 (6.0%) had a grade ≥3 irAE (online supplemental table 10). An IRR (identified via an expanded definition) occurred in 34 (29.3%) patients; 1 (0.9%) patient had a grade 3 IRR, and no grade ≥4 IRRs occurred. In these 34 patients, the majority had an IRR at the time of first infusion (n=25; 73.5%), and all IRRs had onset within the first three infusions.
Key safety outcomes
Discussion
First-line avelumab treatment in the full part B cohort (N=116) of JAVELIN Merkel 200 was associated with durable responses and a clinically meaningful survival benefit in patients with mMCC. DORs in this study (median: 18.2 months) were longer than those seen in historical studies of first-line chemotherapy (range of medians: from 2.8 to 6.7 months).18 19 Median OS was 20.3 months, exceeding the median survival observed with chemotherapy (~9.0–10.5 months).3 18–20 First-line avelumab treatment resulted in mild to moderate TRAEs in most patients (81.0%) but grade 3/4 TRAEs in a low proportion (18.1%), consistent with results from previous studies of avelumab monotherapy in MCC and other tumors.21
The study population was dominated by patients with PD-L1− tumors (75.0% vs 18.1% with PD-L1+ tumors), which is in contrast to previous trials of MCC in which only 18%–48% of patients had PD-L1− tumors based on expression in tumor cells.8 22 Consistent with results from part A of this trial, responses occurred in patients with both PD-L1+ and PD-L1− tumors, with a trend for higher response rates in those with PD-L1+ tumors.11 Potential response variations were also seen between other subgroups. For example, numerically higher ORRs and DRRs were observed in patients with MCPyV− versus MCPyV+ tumors and for patients with median or higher versus less-than-median CD8+ T-cell density at the IM. Expression of MHC class I genes was lower in MCC tumors compared with normal tissues, consistent with analyses from part A of the study11; however, MHC class I expression did not correlate with response or OS. This lack of correlation with response to first-line avelumab treatment may be explained by the finding that expression of both T-cell activation and exhaustion gene signatures were correlated with higher MHC class I expression, suggesting the immune response in these tumors is primed but exhausted. Gene signature analyses also suggested that inflammatory pathways were associated with response, consistent with previous studies of other ICIs.23 24 Of note, enriched gene sets in nonresponders differed from those reported in patients who received second-line or later avelumab in part A of this trial (RNA transcription/protein synthesis in part B vs DNA replication and repair in part A).11 Additionally, cell cycle-related pathways were enriched in MCPyV− tumors, which may partly explain previous observations that these tumors are more aggressive than MCPyV+ tumors.25 The novel gene expression signature we derived predicted the presence of MCPyV+ tumors with 100% accuracy from host RNAseq profiles. To our knowledge, this is the first MCPyV-specific signature derived from host RNAseq profiles to be reported and provides a potential alternative method of determining MCPyV status to IHC/PCR assays. This signature would be useful for prioritizing RNAseq profile generation when a small amount of sample is available in a research setting. Further investigation of these biomarker findings is required.
The findings reported here suggest that response rates with first-line ICI treatment for mMCC may be slightly higher than those reported for second-line or later treatment; with second-line or later avelumab treatment in part A of the JAVELIN Merkel 200 trial in 88 patients with mMCC, the ORR was 33.0% and median OS was 12.6 months after ≥3 years of follow-up.11 This suggestion is also supported by findings from the phase II KEYNOTE-017 trial of first-line treatment with pembrolizumab (anti-PD-1) in 50 patients with stage IIIB (n=7) or stage IV (n=43) MCC, which included an ORR of 56% and median OS not reached after a median follow-up of 14.9 months. In the pembrolizumab study, although PD-L1 expression did not correlate with response, there was a trend toward improved OS and PFS in patients with PD-L1+ status based on tumor cell expression.22 However, data from KEYNOTE-017 and our study cannot be compared directly due to differences between sample sizes, study designs, patient populations, and median follow-up time.22 Furthermore, in a cohort of the phase I/II CheckMate 358 trial, higher response rates have been observed with nivolumab monotherapy in treatment-naive patients with MCC compared with those who had received 1–2 prior lines of systemic treatment. A total of 25 patients with MCC received nivolumab and 22 patients were evaluable for response, of which 14 (63.6%) patients were treatment naive and 8 (36.4%) patients had received 1–2 prior lines of systemic treatment; ORRs were 68% overall, 71% in treatment-naive patients, and 63% in previously treated patients.26
Overall, the JAVELIN Merkel 200 trial comprises the largest dataset from a prospective, global clinical trial in mMCC, and the efficacy and safety results for first-line avelumab treatment reported here provide further evidence supporting the use of avelumab as first-line treatment for mMCC.
Data availability statement
Data are available upon reasonable request. For all new products or new indications approved in both the European Union and the United States after January 1, 2014, Merck KGaA, Darmstadt, Germany will share patient-level and study-level data after deidentification, as well as redacted study protocols and clinical study reports from clinical trials in patients. These data will be shared with qualified scientific and medical researchers, upon researcher’s request, as necessary for conducting legitimate research. Such requests must be submitted in writing to the company’s data sharing portal. More information can be found at https://www.merckgroup.com/en/research/our-approach-to-research-and-development/healthcare/clinical-trials/commitment-responsible-data-sharing.html. Where Merck KGaA has a coresearch, codevelopment, or comarketing/copromotion agreement or where the product has been out-licensed, it is recognized that the responsibility for disclosure may be dependent on the agreement between parties. Under these circumstances, Merck KGaA will endeavor to gain agreement to share data in response to requests.
Ethics statements
Ethics approval
The trial was conducted in accordance with the Declaration of Helsinki and the International Council on Harmonisation Guidelines on Good Clinical Practice. The protocol was approved by the independent ethics committee or institutional review board at each participating center, and all patients provided written informed consent before enrollment.
Acknowledgments
The authors thank the patients and their families, investigators, coinvestigators, and study teams at each of the participating centers. The authors would also like to thank Manuel Silva for his work on the PD-L1, CD8, and MCPyV immunohistochemistry analyses and Naveen Jami for his work on the MCPyV+ signature.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors SPD and PN had full access to the clinical data in the study and take responsibility for the integrity and accuracy of the clinical data analysis. PS and BE-L performed statistical analysis, and PS and SG performed biomarker analysis. All authors participated in study supervision, acquisition, analysis or interpretation of data, and critical revisions of the manuscript for important intellectual content.
Funding This trial was sponsored by Merck KGaA, Darmstadt, Germany, as part of an alliance between Merck KGaA and Pfizer. Medical writing support was provided by ClinicalThinking and funded by Merck KGaA and Pfizer.
Competing interests Dr D’Angelo reports serving as a consultant or advisor for Amgen, EMD Serono (an affiliate of Merck KGaA, Darmstadt, Germany), GlaxoSmithKline, Immune Design, Incyte, Merck & Co., and Nektar; received research grants from Amgen, Bristol Myers Squibb, Deciphera, EMD Serono, Incyte, Merck & Co., and Nektar; and received reimbursement for travel and accommodation expenses from Adaptimmune, EMD Serono, and Nektar. Dr Lebbé has received honoraria from Amgen, Bristol Myers Squibb, Incyte, Merck & Co., Novartis, Pfizer, Pierre Fabre, and Roche; reports serving as a consultant or advisor for Amgen, Bristol Myers Squibb, Merck & Co., Novartis, and Roche; is a member of a speakers bureau for Amgen, Bristol Myers Squibb, Novartis, and Roche; has received research funding from Bristol Myers Squibb and Roche; has received reimbursement for travel and accommodation expenses from Bristol Myers Squibb; and has other relationships with Avantis Medical Systems. Dr Mortier has received reimbursement for travel and accommodation expenses from Bristol Myers Squibb, Novartis, and Roche/Genentech. Dr Brohl reports serving as a consultant or advisor for Bayer, Deciphera, EMD Serono, and PierianDx. Dr Fazio has received honoraria from Ipsen and Novartis; reports serving as a consultant or advisor for Advanced Accelerator Applications, Ipsen, Merck KGaA, MSD Oncology, Novartis/Ipsen, and Pfizer; and has received research funding from Merck KGaA and Novartis. Dr Grob has received honoraria from Amgen, Bristol Myers Squibb, Merck KGaA, Merck & Co., Novartis, Pfizer, Pierre Fabre, Roche, and Sanofi; reports serving as a consultant or advisor for Amgen, Bristol Myers Squibb, Merck KGaA, Merck & Co., Novartis, Pfizer, Pierre Fabre, Roche, and Sanofi; is a member of a speakers’ bureau for Novartis; and has received reimbursement for travel and accommodation expenses from Bristol Myers Squibb, Merck & Co., Novartis, and Pierre Fabre. Dr Prinzi has received reimbursement for travel and accommodation expenses from Novartis. Dr Hanna has received research funding from Bristol Myers Squibb, Exicure, GlaxoSmithKline, Kite Pharma, NantKwest/Altor BioScience, Regeneron Pharmaceuticals, Sanofi Genzyme, and Kartos Therapeutics; and reports receiving honoraria and serving as a consultant for Bio-Rad Laboratories, Bristol Myers Squibb, Kura Oncology, Maverick Therapeutics, Merck KGaA, Prelude Therapeutics, and Regeneron Pharmaceuticals/Sanofi. Dr Hassel has received honoraria from Bristol Myers Squibb, Merck & Co., Novartis, Pfizer, and Roche; reports serving as a consultant or advisor for Merck & Co. and Pierre Fabre; has received research funding from 4SC, Amgen, BioNTech, Bristol Myers Squibb, Immunocore, Novartis, Philogen, and Roche; and has received reimbursement for travel and accommodation expenses from Pierre Fabre. Dr Kiecker has received honoraria from Amgen, Bristol Myers Squibb, Merck & Co., Novartis, Pierre Fabre, and Roche; reports serving as a consultant or advisor for Amgen, Bristol Myers Squibb, Incyte, Merck & Co., Novartis, and Roche; has received research funding from Novartis; and has received reimbursement for travel and accommodation expenses from Bristol Myers Squibb and Novartis. Dr Georges reports employment at EMD Serono Research & Development Institute, Inc., an affiliate of Merck KGaA, Darmstadt, Germany. Ms Ellers-Lenz reports employment at Merck KGaA, Darmstadt, Germany. Dr Shah reports employment at EMD Serono Research & Development Institute, Inc., an affiliate of Merck KGaA, Darmstadt, Germany. Dr Güzel reports employment at Merck KGaA, Darmstadt, Germany. Dr Nghiem has received honoraria from EMD Serono and Merck & Co.; reports serving as a consultant or advisor for EMD Serono and Pfizer; has received research funding from Bristol Myers Squibb and EMD Serono; and has a pending patent for high-affinity T-cell receptors that target the Merkel cell polyomavirus.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.