Article Text

Abstract
Background Advanced cancer therapy is targeted at primary tumors and also recurrent or metastatic cancers. Combinational cancer treatment has recently shown high efficiency against recurrent and metastatic cancers. In this study, we synthesized a thermal responsive hybrid nanoparticle (TRH) containing FimH, an immune stimulatory recombinant protein, for the induction of a combination of photothermal therapy (PTT) and immunotherapy against cancer and its metastasis.
Methods The hybrid nanoparticle was incorporated with a near-infrared (NIR) absorbent, indocyanine green, and decorated with FimH on its surface to form F-TRH. F-TRH was evaluated for its anticancer and antimetastatic effects against CT-26 carcinoma in mice by combining PTT and immunotherapy.
Results NIR laser irradiation elicited an elevation of temperature in F-TRH, which induced apoptosis in CT-26 carcinoma cells in vitro. In addition, F-TRH and NIR laser irradiation promoted photothermal-mediated therapeutic effects against CT-26 and 4T1 tumors in mice. The release of FimH from F-TRH in response to elevated temperature and apoptotic bodies of cancer cells via PTT elicited dendritic cell-mediated cancer antigen-specific T-cell responses, which subsequently inhibited the second challenge of CT-26 and 4T1 cell growth in the lung.
Conclusions These data demonstrate the potential use of F-TRH for immuno-photothermal therapy against cancer and its recurrence and metastasis.
- adjuvants
- immunologic
- dendritic cells
- immunotherapy
Data availability statement
All data relevant to the study are included in the article or uploaded as online supplemental information. J-OJ: https://orcid.org/0000-0003-4216-8111.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Recurrence and metastasis of cancer occur in 90% of all cancer-related deaths.1 If patients with cancer are not properly treated at the cancer site, regardless of the site of the primary cancer, the cancer cells spread through the blood vessels, causing cancer recurrence or metastasis.2 Therefore, local therapy, including surgery, radiotherapy, and photothermal therapy (PTT), has limitations in the eradication of cancer recurrence and metastasis.3 Compared with other approaches, PTT has received increasing attention owing to its low side effects and ability to specifically target tumor cells.4 Recent studies have demonstrated apoptotic and necrotic cell death using PTT.5 6 Although PTT has sufficient therapeutic efficacy at the primary cancer site, it has limitations in the elimination of distant metastatic tumors and the prevention of tumor recurrence. Apoptosis-mediated release of cancer antigens (Ags) via PTT is poorly immunogenic, as Ags originate from somatic cells.7 To improve the therapeutic efficiency in cancer metastasis and recurrence, PTT, combined with chemotherapy or immunotherapy, has shown promising outcomes in animal studies.8
Photothermal therapeutic nanoparticles (PTNs) effectively absorb near-infrared (NIR) lasers and convert them into heat. PTNs have been delivered to tumor cells, followed by thermal ablation of tumor cells under NIR laser irradiation. Various PTNs such as black phosphorus quantum dots, Cu5FeS4 nanoparticles, and bismuth selenide nanocrystals have recently been reported and shown effective therapeutic results against tumors.9–14 However, these PTNs exhibit instability in air-exposed water environments, as well as body coagulation and potential long-term toxicity and nongradability in vivo.15 16
Compared with inorganic PTNs, indocyanine green (ICG), which exhibits better solubility, degradability, and lower long-term toxicity, has been approved by the Food and Drug Administration (FDA) for human use.4 As ICG features a stronger NIR absorbance and high thermal conversion yield than other PTNs, it can augment the theranostic effect in cancer cells.17–19 However, ICG is not suitable in biological environments because it is easily degradable in aqueous solutions and decomposed by light and temperature. Thus, encapsulation of ICG in nanoparticles has been widely used to improve stability in aqueous solutions and to control the release time.
Among various lipid-based nanoparticles, liposomes have been widely used owing to their good biocompatibility and convenient modification of the surface.20 However, their use is limited by poor drug-loading ability, unsatisfactory aqueous stability, and rapid release.21 To overcome these limitations, several therapeutics based on lipid–polymer hybrid nanoparticles (HNPs) have been developed.22 HNPs can be easily fabricated by self-assembly with a biodegradable polymer and lipid; they comprise a polymeric core encapsulated by hydrophobic material such as ICG, and an outer lipid layer.22
Immunotherapy approaches, such as cancer vaccines, blockade of immune checkpoints, or chimeric receptor T-cell transfer, have shown efficient results in the treatment of cancer in animals and humans.23 These immunotherapies activate cytotoxic T lymphocytes (CTLs), which are the most powerful immune cells for killing cancer cells.5 Blockade of immune checkpoints has shown effective therapeutic results; however, it induces undesirable side effects, such as inflammation, in healthy tissues.24 Unlike immune checkpoint blockade, the induction of Ag-specific T-cell activation targets only Ag-expressing cells, which reduces undesired side effects. Ag-specific T-cell activation is mediated by macrophages and dendritic cells (DCs).25 Among them, DCs have the most powerful function of Ag presentation and T-cell activation.26
Stimulation of pattern recognition receptors (PRRs) promotes the activation of DCs, which upregulates co-stimulator expression.27 Moreover, activated DCs present Ags on their surface via class I or class II major histocompatibility complex (MHC).28 In addition, DCs release pro-inflammatory cytokines to induce T-cell differentiation. Conventional murine DCs contain two main subsets: CD8α+ and CD8α– DCs. CD8α+ DCs present intracellular Ag on MHC class I, which induces the activation of CD8-expressing CTLs. CD8α− DCs are involved in the induction of helper T (Th)-cell activation by the presentation of extracellular Ags on MHC class II.29 Since CTL and Th immune responses are required for induction of anticancer immunity, activation of both CD8α+ and CD8α− DCs is essential for cancer vaccines and immunotherapy.30
PRR ligands contain various types of macromolecules, including DNA, RNA, lipids, and proteins.31–33 The Escherichia coli type 1 fimbriae adhesion portion, FimH, functions as an immune stimulator in humans and mice.34 As FimH can easily be used to decorate the surface of the nanoparticles through nickel–nitrilotriacetic acid (Ni-NTA) and histidine (His) interactions, in this study, we developed an FimH-conjugated dual-functional thermal response hybrid nanoparticle (F-TRH). The TRH was based on poly lactic-co-glycolic acid (PLGA)–lipid HNP, comprising polymeric cores, such as PLGA and lipids, including lecithin and 1,2-dioleoyl-sn-glycero-3-[(N-(5-amino-1-carboxypentyl)iminodiacetic acid)succinyl] (nickel salt) [DGS-NTA(Ni)], as the shell and TRH incorporated with ICG as the NIR absorbent. Finally, FimH was decorated on the shell of the TRH. We hypothesized that F-TRH would treat primary tumors through PTT and prevent second challenged cancer growth via immunotherapy.
Materials and methods
Preparation of F-TRH
Soybean lecithin (90%) was obtained from Alfa Aesar (Ward Hill, MA, USA). DGS-NTA(Ni) was purchased from Avanti Polar Lipids (Birmingham, AL, USA). PLGA (lactide/glycolide (50:50), molecular weight=30,000–60,000) was purchased from Sigma Aldrich (St. Louis, MO, USA). ICG was acquired from Tokyo Chemical Industry (Tokyo, Japan).
First, TRHs were prepared via self-assembly of PLGA, lecithin, and DGS-NTA(Ni) through a single-step nanoprecipitation method. Briefly, PLGA was dissolved in chloroform; then lecithin/DGS-NTA(Ni) (8.5:1.5 molar ratio), containing a 15% wt ratio of PLGA, was dissolved in 4% aqueous ethanol solution; and ICG was dissolved in a mixture of chloroform/methanol (4:1 v/v%). The lecithin/DGS-NTA(Ni) solution was heated to 70°C to ensure clarity. The PLGA and ICG (20 w/w% of total lipids) mixture was added dropwise to the preheated aqueous solution under gentle stirring. The solution was vortexed for 3 min and then stirred gently for 4 hours at room temperature to produce TRH (10 mg/mL). HNPs were formulated without ICG. To prepare F-TRH, the FimH proteins (0.5 mg) were dispersed in the TRH solution with slight stirring for 2 hours to allow NTA(Ni) to conjugate with the His-tagged FimH proteins. Finally, the remaining free lipids, polymers, and FimH proteins were removed using an ultracentrifuge (10,000×g, 60 min). The encapsulation efficiency and loading capacity of ICG in the F-TRHs were determined to be 74.07%±0.51% and 4.3%±0.18%, respectively.
Characterization of F-TRHs
Transmission electron microscopy (TEM) images were obtained using an H-7600 transmission electron microscope (Hitachi, Tokyo, Japan). Fourier-transform infrared spectroscopy (FT-IR, Spectrum 100; Perkin Elmer, Waltham, MA, USA), conducted at the Core Research Support Center for Natural Products and Medical Materials in Yeungnam University, was used to observe the IR spectra of the lyophilized TRHs and F-TRHs. The loaded concentration of ICG in the TRHs was determined using a UV–vis spectrophotometer (Cary 100 Bio; Varian, Palo Alto, CA, USA). A fiber-coupled continuous-wave diode laser (808 nm, 10 W) was obtained from Changchun New Industries Optoelectronics Technology Co., Ltd (Jilin, China). Thermographic images were obtained, and temperature changes were measured using an FLIR ONE imaging system (FLIR Systems, Wilsonville, OR, USA).
Cell lines
The murine colon carcinoma cell line CT-26 (ATCC, CRL-2638; Korean Cell Line Bank, Seoul, Korea) and CT26.WT-iRFP-Neo cells (Imanis Life Sciences, CL091, Rochester, USA) were cultured in RPMI-1640 medium, supplemented with 1× penicillin/streptomycin and 10% fetal bovine serum (FBS) in a 5% CO2 cell incubator at 37°C. Murine mammary carcinoma 4T1-Fluc-Neo/iRFP-Puro cells (Imanis Life Sciences, CL078) were cultured in RPMI-1640 medium, supplemented with 1× penicillin/streptomycin, 10% FBS, 0.1% G418, and 2 µg/mL puromycin, in a 5% CO2 cell incubator at 37°C.
Apoptosis assay
CT-26 cells (1×105 cells) were cultured in 24-well plates. Six hours later, the cells were treated with phosphate-buffered saline (PBS), FimH, HNPs, TRHs, or F-TRHs and irradiated with an NIR laser (2 W/cm2 for 5 min). The cells were stained with annexin V-FITC and 4′,6-diamidino-2-phenylindole (DAPI, Sigma-Aldrich) for 15 min at room temperature, 24 hours after treatment. The apoptotic cells were analyzed using flow cytometry (NovoCyte; ACEA Biosciences, San Diego, CA, USA).
Mice
BALB/c mice (female, 5–8 weeks old) were obtained from Hyochang Science (Daegu, Korea). The mice were kept under pathogen-free conditions at 20°C–24°C and 40%–60% humidity. In all experiments, efforts were made to minimize the suffering of the mice, and the mice were euthanized via CO2 inhalation.
Primary tumor challenge and PTT
BALB/c mice were subcutaneously injected with 1×106 CT-26 or CT-26-iRFP cells. Once the tumor size at the longest dimension reached approximately 5.0 mm on day 7, the mice were randomly separated into 10 groups: PBS, FimH, HNP, TRH, and F-TRH with or without NIR laser irradiation. After intratumorally injecting into the mice, the tumor site was irradiated with an NIR laser at 2 W/cm2 power for 5 min. The elevated temperature was imaged using the FLIR One Thermal Imaging System (FLIR Systems). The tumor volume was calculated using the formula V ¼ 1/2 (longest dimension/2 shortest dimension).
In vivo fluorescence imaging
iRFP expression in the mice was measured using a fluorescence in vivo imaging system, FOBI (Cellgentek, Cheongju, Republic of Korea), on day 10 after the first CT-26-iRFP challenge or day 12 after the second CT-26-iRFP challenge.
Antibodies
Mouse antibodies (Abs) and isotype control Abs (IgG1, IgG2a, and IgG2b), anti-CD3 (17A2), CD4 (GK1.5), CD8α (53–6.7), tumor necrosis factor (TNF)-α (MP6-XT22), perforin (S16009A), CD11c (N418), CD40 (3/23), CD80 (16-10A1), CD86 (GL-1), anti-interferon (IFN)-γ (B27), MHC class I (H2Kd, 28-8-6), and MHC class II (I-A/I-E, M5/114.15.2) were purchased from BioLegend (San Diego, CA, USA).
Enzyme-linked immunosorbent assay
Mouse interleukin (IL)−6, IL-12p40, IFN-γ, and TNF-α ELISA kits were obtained from BioLegend. A mouse perforin/pore-forming protein (PF/PFP) ELISA kit was purchased from ABBKINE (Wuhan, China). A Mouse Granzyme B ELISA kit was obtained from Abcam (Cambridge, UK). The concentrations of TNF-α, IFN-γ, and perforin were measured in triplicate using ELISA kits according to the manufacturer’s protocol.
Secondary tumor challenge
BALB/c mice were injected intravenously with CT-26 carcinoma cells (0.5×106/100 µL of PBS). The survival of the mice was monitored for 25 days after cancer cell injection.
H&E staining
Lung, colon, and liver tissue samples were fixed in 4% formaldehyde and embedded in paraffin. The tissue paraffin blocks were then sectioned at 5 µm thickness and stained with H&E.
ELISPOT assay
IFN-γ production in response to cancer Ag was measured using ELISPOTs according to the manufacturer’s protocol (BioLegend). In brief, capture Abs were coated on the plate and 5×104 splenocytes were seeded in the wells. The cells were then treated with 10 µg/mL of cancer Ag and incubated at 37°C for 24 hours. The wells were stained with detection Abs, and the plates were counted automatically using a CTL ELISPOT reader (CTL Europe GmbH, Bonn, Germany).
Analysis of CT-26-specific T-cell immunity
To prepare the CT-26 lysate, 1×107 CT-26 cells were washed with PBS and subjected to freezing and thawing to obtain a crude lysate. After centrifugation (10,000×g for 5 min at 4°C), the concentration of the CT-26 lysate was determined in the supernatant (Bradford assay), and the lysate was treated with 10 µg/mL of splenocytes. The CT-26 lysate was incubated with splenocytes for 6 hours. The cells were incubated with Fc block Abs and unlabeled isotype control Abs. After 20 min, the cells were incubated with surface Abs (FITC-anti-CD3, BV605-anti-CD4, and APC/Cy7-anti-CD8α) on ice for 20 min. After washing, the cells were fixed with fixing buffer and permeabilized (both from BioLegend) for 30 min. After washing again, the cells were stained with intracellular Abs (PE/Cy7-anti-IFN-γ, Alexa 647-anti-TNF-α, and PE-anti-Perforin) for 30 min. The cells were then washed and re-suspended in PBS and analyzed using NovoCyte (ACEA Biosciences) and NovoExpress software. Dead cells were excluded from the analysis using the Zombie Violet Fixable Viability Kit (BioLegend).
Ag-specific lysis of splenocytes
After labeling with 200 nM CFSE, the splenocytes were coated with 1 µg/mL CT-26 Ag. Other splenocytes were labeled with 10 mM CellTracker Orange CMTMR (Life Technologies) and coated with the control protein. The CFSE-labeled and CMTMR-labeled cells were mixed in a 1:1 ratio and the mixture was transferred into tumor-treated BALB/c mice. The spleen was harvested 6 hours after transfer, and the levels of CFSE and CMTMR in the splenocytes were analyzed using Novocyte (ACEA Biosciences).
Depletion of CD4 and CD8 cells
Anti-CD4 (GK1.5, 1 mg/mL) or anti-CD8 (YTS169.4, 1 mg/mL) Abs (both from BioXcells, West Lebanon, NH, USA) were intraperitoneally injected every 2 days in the BALB/c mice from 25 days after the first tumor challenge (3 days before the second challenge of cancer cells). Cell depletion was confirmed using a Novocyte flow cytometer.
Statistical analysis
Data are expressed as the mean±SE of the mean. The p value was analyzed using SPSS (Chicago, Illinois, USA), and p values of <0.01 were considered to represent significant differences.
Results
Synthesis of FimH-containing thermally responsive nanoparticles
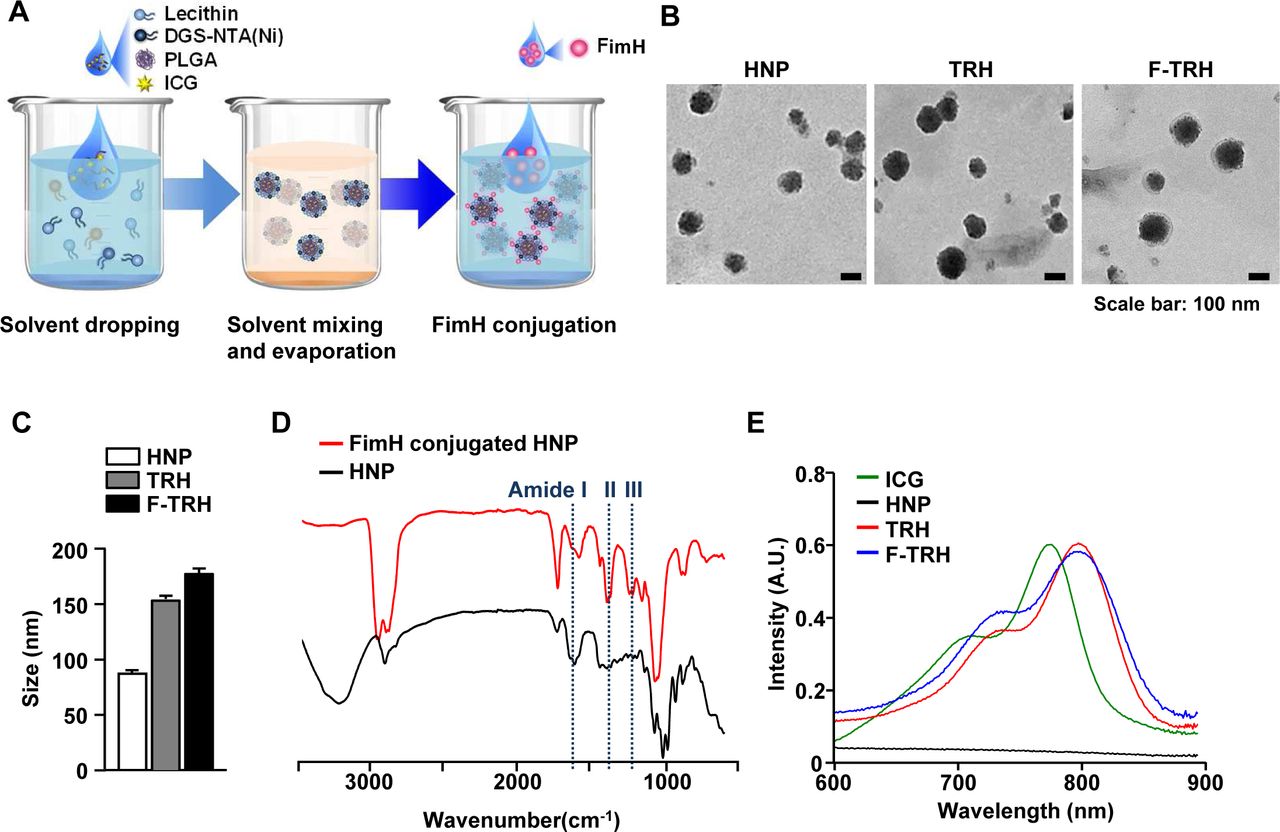
F-TRH, comprising a spherical core–shell lipid-polymeric hybrid nanoparticle with surface expression of FimH, was formulated by self-assembly and the single-step nanoprecipitation method wherein PLGA and ICG were dissolved in chloroform, and lecithin and DGS-NTA(Ni) were dispersed in an aqueous solution. The lipid mixture was added dropwise to either PLGA alone or a mixture of PLGA and ICG, under gentle constant stirring until it became clear, causing the precipitation of HNP or TRHs. During stirring, the lipids self-assembled around the PLGA core via hydrophobic interactions, resulting in the formation of TRHs. Subsequently, the His-tagged FimH proteins attached to the NTA-Ni2+ moiety of the TRHs via the NTA-Ni2+/His affinity interaction (figure 1A). Observation of a series of HNPs, TRHs, and F-TRHs, using TEM, revealed well-defined spherical structures, with the average diameters of three NPs being 89.42±0.86, 152.6±4.73, and 179.8±7.49 nm (figure 1B,C). To determine the stability of F-TRHs, they were incubated for 7 days at 4°C to examine whether ICG incorporated into the F-TRHs was released. F-TRH released approximately 26.87% of ICG by day 7 (online supplemental figure S1). The conjugation of the FimH proteins on the surface of the HNPs was analyzed using FT-IR spectroscopy. As shown in figure 1D, the spectrum of FimH conjugated to HNP presented peaks at 1596, 1406, and 1251 cm−1, which corresponded to the amide groups, compared with that of HNP. Moreover, TRH and F-TRH were encapsulated in ICG, as confirmed from the UV–vis absorbance spectra, which showed a strong peak at 800 nm and a red shift, indicating that ICG was successfully trapped in the hydrophobic moiety of the PLGA core (figure 1E). These data indicate that F-TRHs are well-defined spherical nanoparticles incorporated with FimH and ICG.
Supplemental material
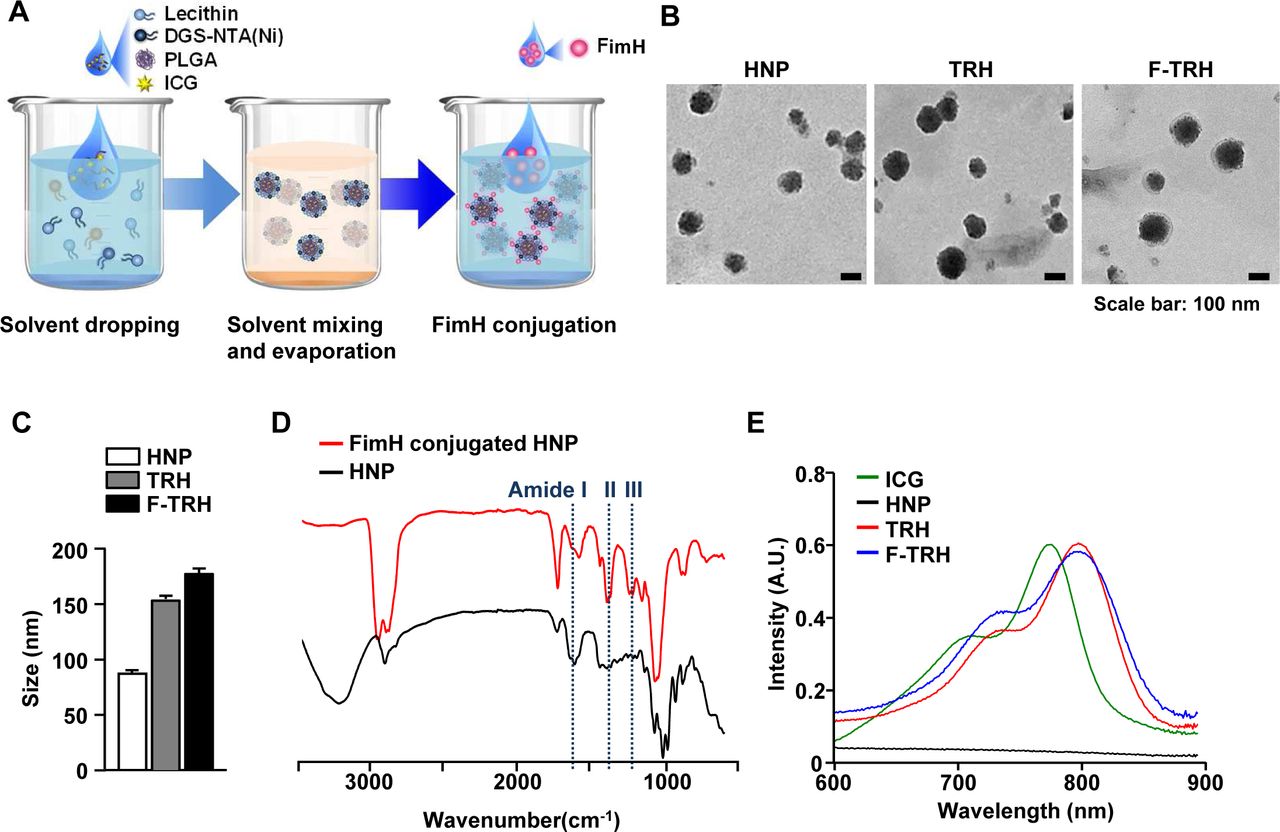
Characterization of FimH-containing thermally responsive nanoparticles (F-TRH). (A) Schematic illustration of F-TRH formation. (B) TEM images of HNP, TRH, and F-TRH (scale bars: 100 nm). (C) The average diameters of HNPs, TRHs, and F-TRHs. (D) FT-IR spectra of HNP and FimH-conjugated HNP. (E) UV–vis absorbance spectra of free ICG, HNP, TRH, and F-TRH.
Photothermal effect of F-TRH induced apoptosis in CT-26 cells
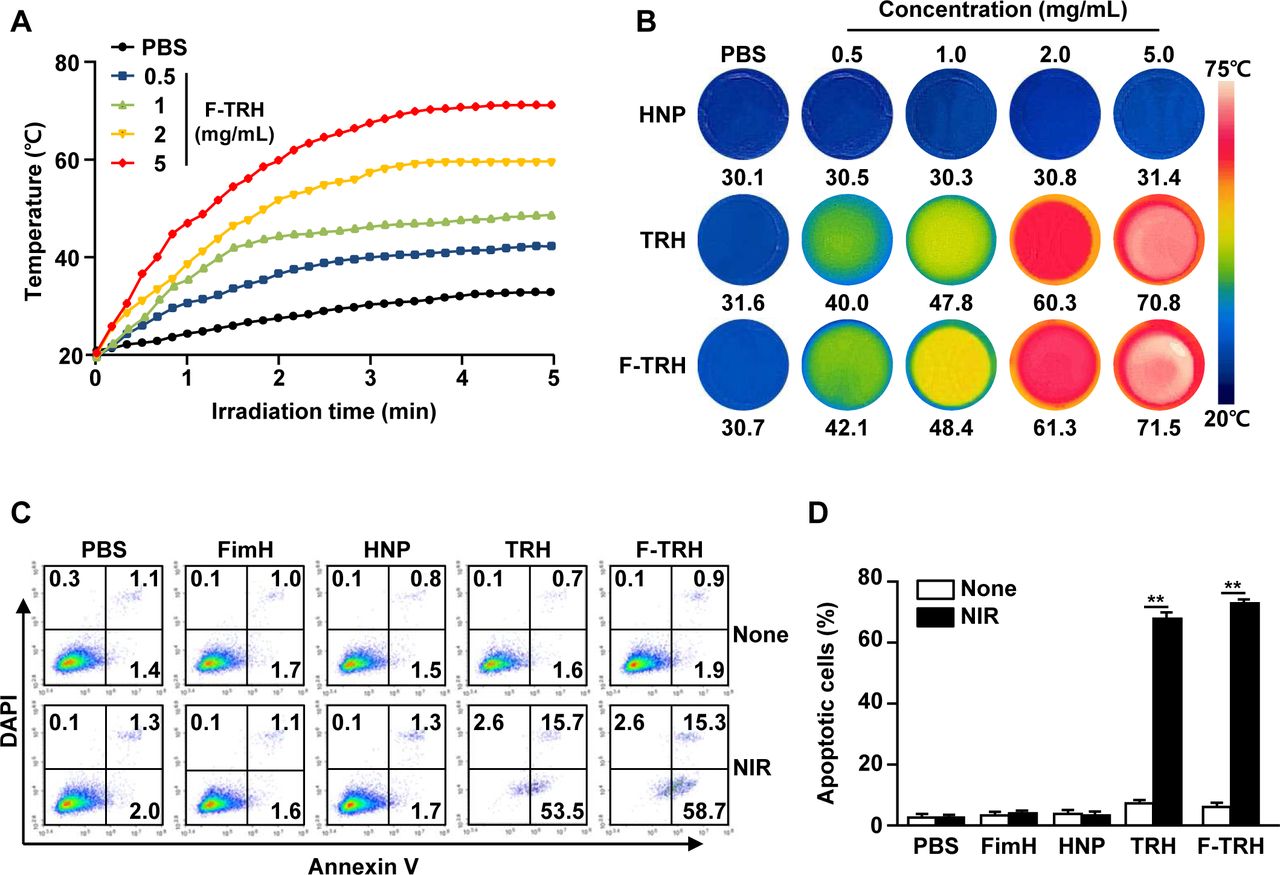
The thermal responses of F-TRH were evaluated under laser irradiation at 808 nm for 5 min. Irradiation with NIR elicited an elevation in temperature in an F-TRH dose-dependent manner (figure 2A). The temperatures of 1.0, 2.0, and 5.0 mg/mL F-TRH reached 47, 60, and 70°C, respectively, within 5 min, while HNP did not show an increase in temperature against laser irradiation (figure 2A,B). Furthermore, the photothermal conversion efficiency of F-TRH was found to be 18.7%, which was higher than that of free ICG (12.03%), using a method described in a previous study (online supplemental figure S2).35 The photothermal effects of TRH and F-TRH were then examined for the induction of apoptosis in CT-26 carcinoma cells. Treatment with PBS, FimH, or HNP did not induce apoptosis in CT-26 cells with or without laser irradiation (figure 2C,D). TRH and F-TRH induced a remarkable increase in the apoptotic death of CT-26 cells after irradiation with the NIR laser, while the cells did not die without laser irradiation (figure 2C,D). These data indicate that TRH and F-TRH can induce apoptosis in cancer cells via the photothermal effect.

Induction of apoptosis via photothermal effect of FimH-containing thermally responsive nanoparticles (F-TRH). (A) Photothermal heating curves for F-TRHs. The temperature changes in F-TRH for the different concentrations were measured during NIR laser irradiation. (B) Photothermal heating images of various concentrations of HNP, TRH, and F-TRH (2 W/cm2, 5 min). (C, D) CT-26 cells were treated with PBS, FimH, HNP, TRH, F-TRH, and NIR laser irradiation at 2 W/cm2 for 5 min. (C) Apoptotic cell death of the CT-26 cells was analyzed 24 hours after NIR laser irradiation, using Annexin V/DAPI staining. (D) The mean percentage of the apoptotic cells is shown. **p<0.01. The data are representative of the average for six independent samples (two samples for each condition per three repeated experiments).
Therapeutic effect of F-TRH through NIR laser irradiation against CT-26 tumors in mice
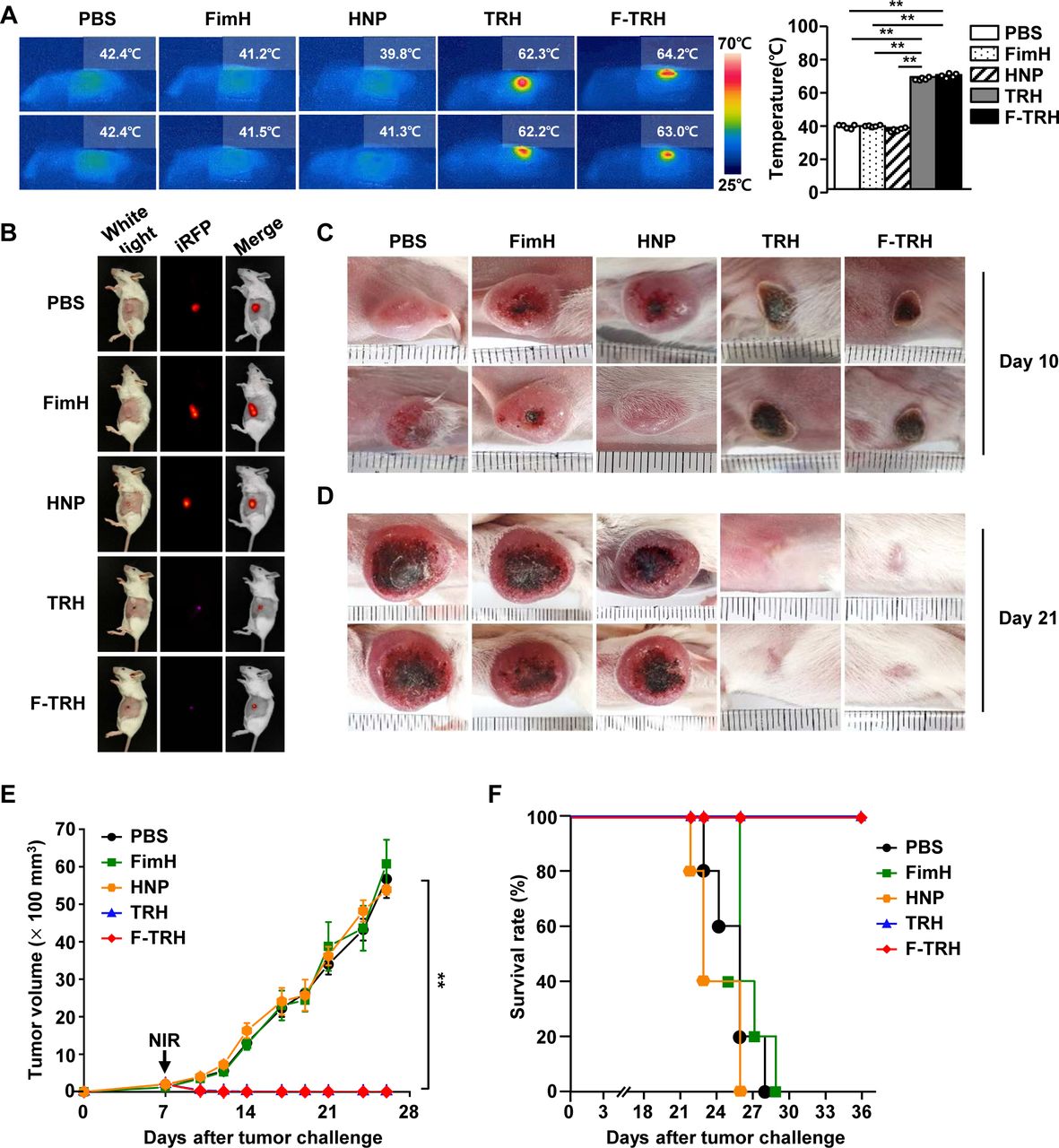
Since F-TRH could promote the apoptosis of CT-26 cells through the photothermal effect in vitro, we next examined whether F-TRH can induce anticancer effects in mice in vivo. Once the CT-26 tumor was established in the BALB/c mice, the mice were intratumorally treated with PBS, FimH, HNP, TRH, F-TRH, or NIR irradiation. As shown in figure 3A, NIR laser irradiation increased the temperature of the tumors of TRH-treated and F-TRH-treated mice, to 62°C and 64°C, respectively. However, the temperature of the tumors in mice treated with PBS, FimH, and HNP did not increase in response to NIR laser irradiation (figure 3A). Moreover, TRH-mediated PTT did not promote liver toxicity (online supplemental figure S3). On day 10 of tumor injection, the tumors in TRH-treated and F-TRH-treated mice showed a burnt tumor and the scar remained at the tumor site (figure 3B,C). On day 21, the tumor disappeared following the TRH and F-TRH treatment with laser irradiation, and the skin and hair were regenerated in the mice (figure 3D). In addition, TRH and F-TRH treatment with laser irradiation inhibited tumor growth, whereas PBS, FimH, or HNP treatment did not show any inhibitory effect on tumor growth (figure 3E). Furthermore, the mice treated with PBS, FimH, or HNP died within 28 days of tumor injection, while TRH-treated and F-TRH-treated mice survived without recurrence of the tumor (figure 3F). These findings suggest that TRH and F-TRH can treat tumors via PTT.

Photothermal therapy via treatment with FimH-containing thermally responsive nanoparticles (F-TRH) and NIR laser irradiation protected BALB/c mice against CT-26 carcinoma. BALB/c mice were subcutaneously injected with 1×106 CT-26-iRFP or CT-26 cells. On day 7 of the tumor challenge, the mice were intratumorally treated with PBS, FimH, HNP, TRH, and F-TRH, and irradiated with NIR laser (2 W/cm2, 5 min). (A) The images show the temperature increase after laser irradiation (left panel), and the average temperature was observed during NIR laser irradiation in CT-26 tumors (right panel), n=6, **p<0.01. (B) iRFP imaging in the tumor-bearing mice on day 10, n=5. Tumor mass in mice on day 10 (C) and day 21 (D) is shown. The data are representative of the analyses of six independent samples (ie, two samples per experiment, three independent experiments). (E) The tumor growth curve is shown, n=11 per group. (F) Survival of mice was measured, n=5 for each condition.
F-TRH promoted the activation of dendritic cells in mice
Lipid-based nanoparticles melt above the melting point of lipids.21 The NIR laser irradiation in F-TRH promoted FimH-conjugated lipid release from the F-TRH by increasing the temperature (figure 4A). Since FimH is an immune stimulatory recombinant protein, we next examined whether the released FimH can induce the activation of DCs in the tumor-draining lymph node (tdLN). DCs in the tdLN were defined as lineage–CD11c+ cells, which were further divided into CD8α+ and CD8α– DCs (figure 4B). Eighteen hours after F-TRH treatment and laser irradiation, expression levels of the co-stimulator and MHC class I and II increased significantly in both CD8α+ and CD8α– DCs, which was almost similar to the results obtained with FimH treatment alone (figure 4C). In addition, F-TRH treatment induced the activation of splenic DCs, which controlled systemic immune responses (online supplemental figure S4). Furthermore, the levels of IL-6, IL-12, and TNF-α levels in the serum were substantially upregulated by F-TRH and laser irradiation (figure 4D). These results indicate that F-TRH and laser irradiation induced the activation of DCs in the tdLN and spleen of mice.

FimH-containing thermally responsive nanoparticles (F-TRH) promoted the activation of dendritic cells (DCs) in mice. (A) Illustration of the release of FimH from F-TRH in response to the elevated temperature and stimulation of DCs by the released FimH. CT-26 tumor-bearing mice were intratumorally injected with PBS, FimH, HNP, TRH, and F-TRH, and irradiated with NIR laser (2 W/cm2, 5 min). (B) Definition of DC and its subsets in the tumor-draining lymph node (tdLN) is shown. (C) The mean fluorescence intensity (MFI) of the co-stimulatory molecules and MHC class I and II in CD8α+ and CD8α– tdLN DCs was measured using flow cytometry (n=6, **p<0.01). (D) The concentrations of IL-6, IL-12p40, and TNF-α in serum were measured using ELISA (n=6, **p<0.01). All the data are representative of the average of the analyses of six independent samples (ie, three samples per experiment, two independent experiments).
Treatment with F-TRH and NIR irradiation protected mice from the second challenge of CT-26 cancer cells
Our data, showing that F-TRH and laser irradiation induced the apoptosis of tumors and activation of DCs, prompted us to examine whether F-TRH and laser irradiation can elicit tumor Ag-specific immune responses. Therefore, we next assessed the second cancer challenge in mice that survived the first tumor challenge using F-TRH and NIR laser irradiation. On day 28 of the first tumor challenge, TRH-treated and F-TRH-treated mice were injected intravenously with CT-26 cells. As controls, the mice treated with PBS, FimH, and HNP were also inoculated intravenously with CT-26 cells (online supplemental figure S5). The F-TRH and NIR laser-mediated cured mice from the first tumor were protected from the second cancer challenge (figure 5A). However, the mice that were cured of the first tumor by TRH died within 21 days of the second tumor inoculation (figure 5A). The mice treated with PBS, FimH, and HNP died within 14 days of the second tumor injection (figure 5A). In addition, the second challenge CT-26 cells did not infiltrate the lungs of the mice that were protected from the first tumor challenge by F-TRH (figure 5B–D). The cancer cells infiltrated the lungs of mice treated with PBS, FimH, and HNP on day 12 after the second cancer cell injection (figure 5B–D). Although the infiltration of cancer cells was much less than that in the mice treated with PBS, FimH, and HNP, the lungs of the TRH-mediated first tumor-protected mice were infiltrated with the second challenge CT-26 cells 12 days after the second challenge (figure 5D). The first and second tumor-challenged mice, which were treated with F-TRH and NIR laser irradiation, did not show any inflammation in the kidney, colon, and liver on day 49 after the first tumor challenge (online supplemental figure S6).

FimH-containing thermally responsive nanoparticle (F-TRH) and laser irradiation treatment prevented second challenged CT-26 cell growth in the lungs. On day 28 of the first cancer challenge, the survived mice, by TRH and F-TRH treatment, were injected intravenously with the second challenge of CT-26-iRFP. (A) The rate of survival of the mice for the first and second CT-26 challenges, n=5 per group. (B) Images of lung metastasis of CT-26 cancer. (C) iRFP expressing CT-26 was detected. (D) H&E staining of the lung on day 10 after the second CT-26 challenge. The data are representative of the average of the analyses of six independent samples (ie, two samples per experiment, three independent experiments).
Since F-TRH had a protective effect against the second challenge of CT-26 tumors, we further examined whether F-TRH can protect mice from different types of cancers, such as the 4T1 breast cancer. Consistent with CT-26 treatment, TRH and F-TRH treatment with NIR irradiation abolished the first challenge of 4T1 tumor growth in mice (online supplemental figure S7A,B). In addition, the second challenge of 4T1 cell infiltration in the lungs was also completely prevented in the mice that were cured by F-TRH treatment, whereas TRH-mediated cured mice showed 4T1 cell infiltration in the lungs (online supplemental figure S7C). These data suggest that F-TRH protects from the first tumor and also prevents the second challenge from inducing lung metastatic cancer growth in mice.
Anticancer effect of F-TRH is mediated by antigen-specific T-cell immunity
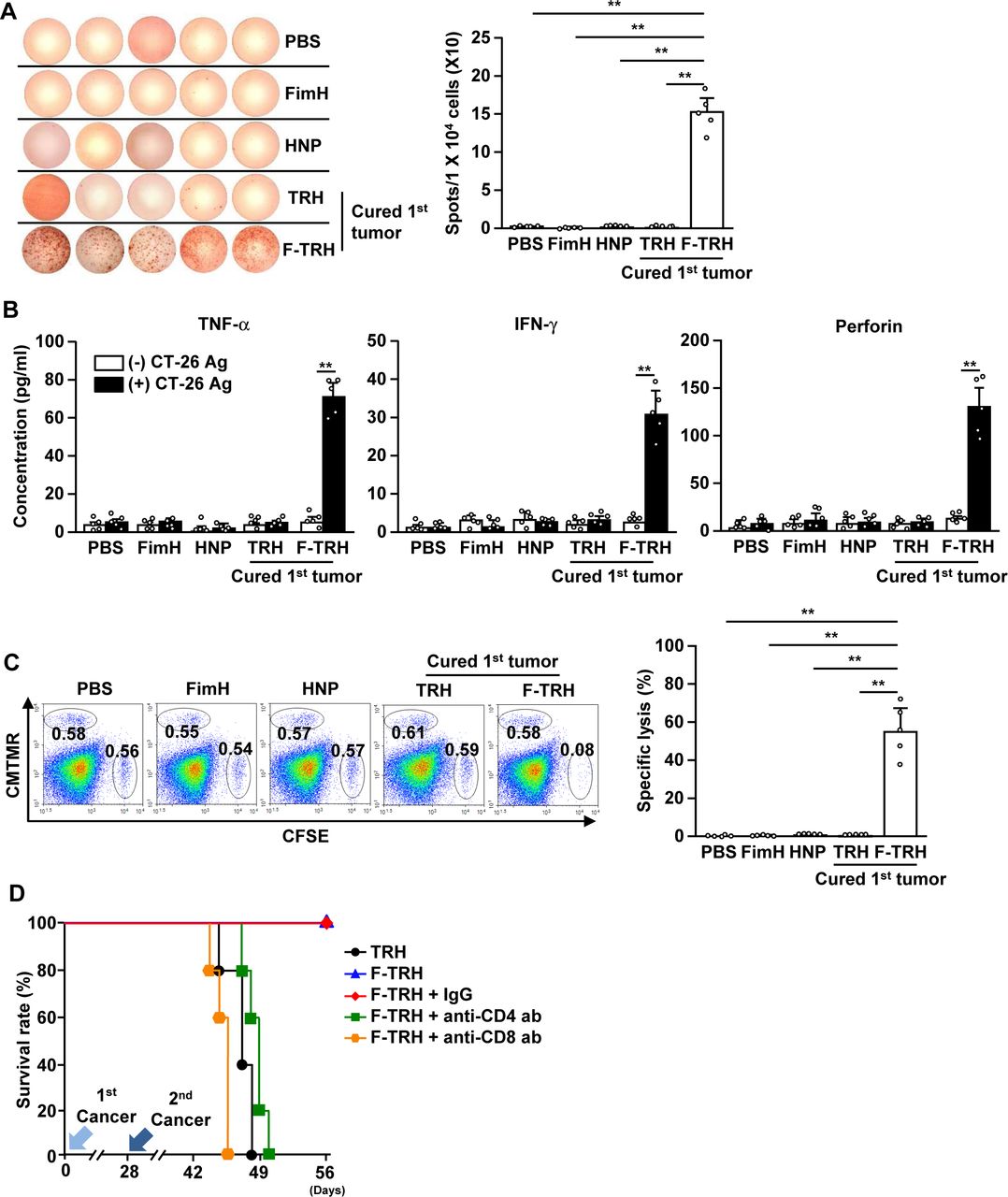
Our finding that F-TRH induces the activation of DCs and protects against second challenged cancer prompted us to examine whether F-TRH can elicit cancer Ag-specific immunity to protect mice from second challenged cancer. The splenocytes of F-TRH-treated mice showed a significant increase in the production of IFN-γ in response to CT-26 cell lysate, as shown using ELISPOT analysis, while other control splenocytes did not produce IFN-γ (figure 6A). In addition, the concentrations of TNF-α, IFN-γ, and perforin in the culture medium were significantly elevated in the splenocytes from F-TRH mice in response to CT-26 cell lysate, whereas other control splenocytes did not produce cytokines (figure 6B). Moreover, stimulation with CT-26 antigen induced intracellular production of IFN-γ, TNF-α, and perforin in CD4 and CD8 T cells in F-TRH-treated splenocytes (online supplemental figure S8). Next, we examined Ag-specific cytotoxicity and found that, in mice cured of the first tumor by F-TRH treatment, CT-26 Ag-coated splenocytes were selectively killed (figure 6C). Other controls, as well as the TRH-mediated first tumor-cured mice, failed to kill CT-26 Ag-coated splenocytes (figure 6C). Furthermore, the protective effect of F-TRH on second cancer challenge in mice diminished with the depletion of CD4 or CD8 cells (figure 6D). These findings suggest that the F-TRH-induced protective effect against the second challenge of cancer is mediated by T-cell immunity.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
F-TRH and laser irradiation induced cancer Ag-specific T-cell immunity. On day 10 after the second challenge of CT-26, the spleen was harvested and incubated with the CT-26 lysate for 24 hours. (A) IFN-γ-production levels were measured using the ELISPOT assay (n=5, **p<0.01). (B) The concentrations of IFN-γ, TNF-α, and perforin in the cultured medium are shown (n=5, **p<0.01). (C) Ag-specific cell lysis was measured by transferring either Ag or the control peptide-coated splenocytes on day 10 after the second inoculation of CT-26 in the mice (n=5, **p<0.01). (D) On day 25 after the first challenge of CT-26 cells, the mice received depletion antibodies (anti-CD4 and anti-CD8 antibodies) for every 2 days. The mice were challenged with the second CT-26 cancer by intravenous injection. The survival rates of the mice are shown, n=5 for each condition.
Discussion
In this study, we explored the use of F-TRH for PTT and immunotherapy against cancer and subsequent metastasis. The in vitro results suggested that irradiation with an NIR laser induced an increase in temperature in F-TRH-treated CT-26 cells, resulting in apoptosis of the cells. Furthermore, F-TRH and NIR laser irradiation elicited a photothermal-mediated therapeutic effect against CT-26 tumors in mice in vivo. We demonstrated that F-TRH treatment with laser irradiation induced cancer Ag-specific T-cell responses via the apoptotic bodies of CT-26 cells and released an immune stimulator, FimH. Finally, the cured mice from the first challenged CT-26 tumor by F-TRH prevented the second challenge of CT-26 cell growth in the lungs. These data demonstrate that F-TRH can be a dual-functional smart nanomaterial for the treatment of primary cancer and prevention of its recurrence or metastasis via immuno-photothermal therapy.
HNP was prepared with PLGA, lecithin, and DGS-NTA(Ni), which is a conjugated phospholipid containing nickel. As both PLGA and DGS-NTA(Ni) polymers have been approved by the US FDA for use in medical products and lecithin is a natural product of soybean, HNP is biodegradable, biocompatible, and safe for use in therapeutic materials.36 Moreover, PLGA, lipids, DGS-NTA(Ni), and lecithin can self-assemble in aqueous solutions. In initial studies, a typical two-step method was generally used to form HNPs, wherein preformed polymeric nanoparticles were blended with preformed lipid vesicles.37 However, HNPs are generally prepared using nanoprecipitation, which has the advantage of spontaneously forming a spherical nanostructure in a single step and directly preforming homogeneous HNPs with a relatively narrow size distribution.37 Since HNPs are synthesized from polymers and lipids, they may be able to hide from phagocytic cells owing to the presence of biomimetic surfaces. Moreover, HNPs can efficiently load therapeutic agents and show low leakage of encapsulated materials. In addition, HNPs can conjugate functional materials in outer lipids, such as proteins or antibodies. The HNPs can release conjugated biomolecules from the lipid layer at a temperature of 60°C.35 This process improves the selectivity between Ni-NTA on the surface of HNPs and His-tagged FimH via specific NTA–Ni2+–His interactions.38 In addition, the efficiency of ICG incorporation and its stability were enhanced as the HNP was coated with a lipid layer on the PLGA polymer core. Collectively, HNPs can function as multifunctional nanoparticles through the incorporation of hydrophobic drugs into the PLGA core and the decoration of proteins in the lipid shell.
Nanomaterials have recently been used to treat cancer owing to their multidrug delivery ability, combined with cancer therapy effect.39 Nanoparticles are used as delivery vehicles to load various types of biological materials, such as vaccines, Abs, nucleic acids, and chemotherapeutic agents.33 40 These nanomedicines, together with photodynamic therapy (PDT) and PTT, have advanced the targeted treatment of various types of cancers.41 Since the polymers and lipids are the main components of the HNPs, they are able to incorporate different types of therapeutic reagents against cancer.41 42 Laser-mediated therapeutic trial methods include PDT. PDT uses light to excite the photosensitizer, which then forms cytotoxic reactive oxygen species that kill tumor cells.43 In this study, we selected ICG for incorporation into the HNPs; it can be used to load photosensitizers for PDT in the HNPs.
PTT, combined with chemotherapy, has a prominent effect in the treatment of tumors.3 In addition, immunotherapy, combined with PTT, has previously shown promising effects in the treatment of metastatic cancer.8 In this study, we synthesized HNPs, through simple and easy processing, a delivery vehicle. HNPs loaded with dual molecules, such as ICG for PTT and FimH for immunotherapy, demonstrated effective therapeutic outcomes against tumors and their recurrence or metastasis.
Cancer immunotherapy is a strategy to prevent cancer by activating the immune cells. In patients with cancer, the benefit of immunotherapy prompts long-lasting responses and thus prevents metastasis and recurrence.44 Immunotherapy seeks to memorize cancer Ags and thereby activate memory T cells to combat cancer cells.45 Immune checkpoint blockade has shown promising effects in the treatment of cancer in animals and humans; however, the blockade-induced anticancer effect is not dependent on cancer cells expressing Ags.46 Therefore, immune cells are not able to memorize cancer cells expressing Ag; consequently, it may be difficult to prevent recurrence and metastasis. In contrast to immune checkpoint blockade, F-TRH induces cancer Ag-specific T-cell immunity, which effectively targets Ag-expressing cancer cells. Therefore, F-TRH is a promising therapeutic agent for the treatment of tumors and prevention of their recurrence or metastasis.
Toll-like receptor (TLR) ligands have stimulatory effects on immune cells in animals and humans. Monophosphoryl lipid A (MPLA), polyinosinic:polycytidylic acid (poly I:C), and cytosine–guanine oligodeoxynucleotides (CpG) are frequently used as immune stimulators in synthetic therapeutic materials.5 30 31 47 Since nucleic acids are easily modified during solid-phase synthesis and have good biocompatibility, CpG and polyI:C are well used to form self-assembled nanostructures. However, because CpG and polyI:C are negatively charged, it is difficult for DCs to absorb them, and they are easily degraded by nucleases in vivo.48 Moreover, MPLA is insoluble and tends to aggregate, which is a disadvantage in vivo. To enhance its solubility, it is necessary to add emulsification steps or additives.49 In a previous study, we identified a novel TLR4 ligand, the Escherichia coli adhesion protein FimH, which may have an adjuvant role in cancer treatment via immunotherapy. Compared with other TLR4 ligands, such as lipopolysaccharide, MPLA, and E. coli–derived monophosphoryl lipid A, FimH has advantages, such as water solubility, low toxicity, and ease of decoration on the surface of the nanoparticles based on His-tagged FimH.30 Therefore, we formulated dual-functional F-TRH, which could treat the first tumor therapy and also prevented second challenged tumor growth.
Previous studies have shown that PTT induces immunogenic cell death, which reduces distant tumor growth.50 We also found that cured mice from the first challenged CT-26 tumor by TRH also showed reduced second challenged CT-26 cell infiltration in the lung. However, the mice died 49 days after the first tumor injection (day 21 of the second tumor challenge). Moreover, on day 40 of the first tumor challenge, the second challenged CT-26 cells were detected in the lung, indicating that the mice were not fully protected from the second challenged CT-26 cells. In contrast to TRH, F-TRH completely prevented second tumor infiltration in the lung. The F-TRH contained FimH compared with TRH, in which FimH promoted the activation of DCs after first tumor treatment.34 These different effects in the prevention of second tumor challenge may be due to the anticancer immunity. As DCs are the main contributors to the induction of Ag-specific immune activation, F-TRH could induce cancer Ag-specific T-cell immunity; however, TRH was not promoted. Therefore, the induction of Ag-specific T-cell immunity may be essential for the long-term prevention of cancer recurrence or metastasis.
In conclusion, we developed F-TRH as a treatment to prevent cancer metastasis and recurrence. F-TRH induced the apoptosis of first challenged tumor via PTT and further prevented the second challenged metastatic lung cancer via immunotherapy. Taken together, these data demonstrate that F-TRH can be used as a dual-functional and promising nanomaterial for the treatment of first challenged tumor and also for the prevention of recurrence or metastasis of second challenged cancer.
Data availability statement
All data relevant to the study are included in the article or uploaded as online supplemental information. J-OJ: https://orcid.org/0000-0003-4216-8111.
Ethics statements
Ethics approval
The animal protocol, 2020-038, was approved by the Committee on the Ethics of Animal Experiments at the Yeungnam University Animal Facility. This study was conducted in accordance with the guidelines of the Institutional Animal Care and Use Committee at the Yeungnam University Animal Facility.
Acknowledgments
We thank the Yeungnam University animal facility for maintaining the animals used in this study and the Core Research Support Center for Natural Products and Medical Materials (CRCNM) for their technical support.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors JH and J-OJ conceived and designed the experiments. JH, H-BP, and WZ performed the experiments. JH and DY analyzed the data. J-OJ supervised the project. JH and J-OJ wrote the manuscript. YHJ and J-OJ discussed the results and commented on the manuscript.
Funding This study was supported by the National Research Foundation of Korea (NRF-2019R1C1C1003334).
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.