Article Text

Abstract
Chimeric antigen receptor (CAR)-modified T-cells targeting CD19 represent a promising therapy for relapsed or refractory (r/r) lymphoma and leukemia. The most common adverse events are immune related and include cytokine release syndrome and neurotoxicity. However, early and late hematological toxicity has emerged as a substantial clinical hurdle leading among others to an increased risk for infections or bleeding. The underlying pathophysiology remains elusive and supportive measures comprise stem cell support or the use of growth factors. Here, we report a 66-year-old woman with r/r diffuse large B-cell lymphoma that received anti-CD19 CAR-T-cells achieving a complete metabolic remission. At month 3 after adoptive cell transfer, the patient still exhibited a grade 3 anemia and a grade 4 thrombocytopenia. The latter required regular platelet transfusions. Bone marrow smear revealed hypocellularity without dysplasia. Despite reduced megakaryopoiesis, immature platelet fraction was elevated indicating an at least partially consumptive underlying component. Based on the successful use of Romiplostim, a thrombopoietin receptor-agonist, in aplastic anemia and immune thrombocytopenia, we treated our patient accordingly. Platelet count (and hemoglobin levels) increased and the patient remains transfusion-free. Taken together, our therapeutic approach could represent a novel strategy for managing CAR-T-cell-related hematotoxicity but, self-evidently, requires further controlled clinical studies.
- immunotherapy
- adoptive
- receptors
- chimeric antigen
- hematologic neoplasms
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Background
To date, CD19-directed chimeric antigen receptor (CAR)-T-cell therapies have revolutionized the treatment of relapsed or refractory (r/r) B-cell derived non-Hodgkin's lymphoma and acute lymphoblastic leukemia producing durable responses in diseases not responsive to standard therapy. Initially, most practitioners focused on cytokine release syndrome (CRS) and CAR-T-cell related encephalopathy syndrome (CRES) as the most common toxicities, which are also seen in alternative immunotherapies redirecting T-cells such as bispecific T-cell-engaging antibodies.1 With increasing experience in treating patients with CAR-T-cells, partially persistent hematological toxicities including neutropenia, anemia, and thrombocytopenia have emerged as a substantial clinical obstacle.2 However, underlying mechanisms remain to be elucidated. Obviously, conditioning chemotherapy that precedes CAR-T-cell infusion contributes to (early) myelosuppression but cannot explain long-lasting (late) cytopenia as well as cytopenia occurring even in patients that have not received chemotherapy.3 Early cytopenia affects 80%, late cytopenia occurs in 30%–40% of the patients. A recent study described cytopenia persisting >90 days on CAR-T-cell infusion in 16% of the cases, and disease entities do not appear to play a role.4 As of to date, no preventive measures against late cytopenia exist and treatment includes the application of granulocytes-colony stimulating factor (G-CSF) or transfusion of blood or platelets. Here, we describe our first use of a thrombopoietin (TPO) receptor agonist (ie, Romiplostim) in a patient with persisting anemia and in particular transfusion-dependent thrombocytopenia upon successful CAR-T-cell therapy against r/r diffuse large B-cell lymphoma (DLBCL).
Case
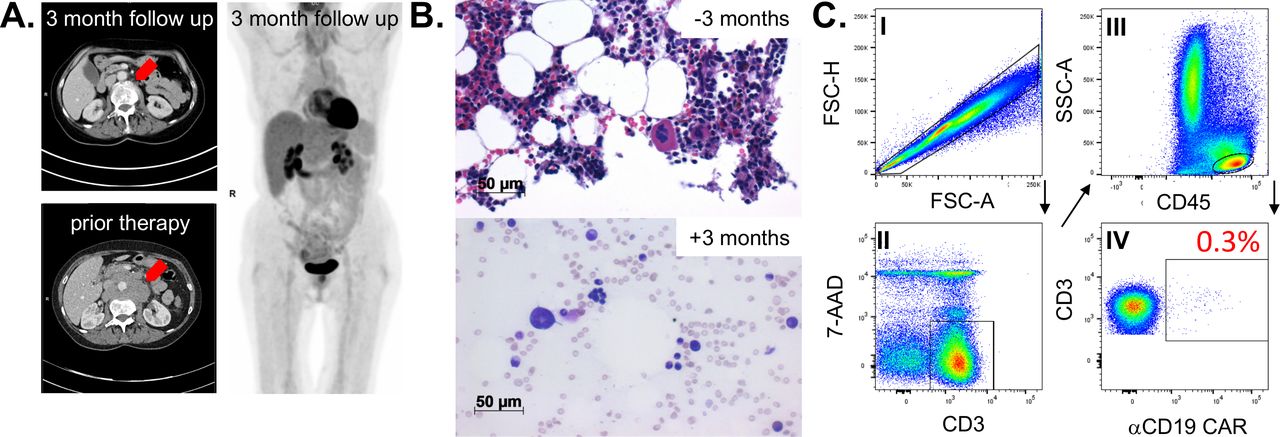
A 66-year-old female patient was diagnosed with a c-myc expressing DLBCL in March 2019. A CT scan revealed cervical, thoracic, and abdominal lymph node enlargement, pulmonary involvement, affection of autochthonous back muscles, and splenomegaly. Lactate dehydrogenase was increased 1.5-fold above normal and no bone marrow involvement could be detected following bone marrow puncture. According to the Ann Arbor classification system the patient suffered from advanced stage IV disease with an additional high-intermediate age-adjusted International Prognostic Index (IPI) and with a high-risk central nervous system (CNS) IPI. The latter one estimates the risk for CNS relapse or progression in DLBCL patients receiving first-line treatment with curative intent. In line with the current established practice,5 our interdisciplinary lymphoma board decided to suggest treatment with six cycles chemoimmunotherapy consisting of rituximab (375 mg/m2), cyclophosphamide (750 mg/m2), doxorubicin (50 mg/m2), vincristine (2 mg), and prednisone (40 mg/m2, day 1–5 of each cycle) (R-CHOP) and two cycles rituximab (375 mg/m2) in combination with high-dose methotrexate (1.5 g/m2) serving as CNS prophylaxis. Eight months upon completion of the therapy the patient developed an extended CD19+ relapse of the initial DLBCL and a salvage approach with rituximab (375 mg/m2), high-dose cytarabine (2×1000 mg/m2), cisplatin (100 mg/m2), and dexamethasone (40 mg, day 1–4) (R-DHAP) was immediately initiated. Treatment was poorly tolerated with delayed hematological recovery and renal dysfunction while not reaching satisfactory efficacy (ie, maximum stable disease). Therefore, it was decided to not further pursue a high-dose chemotherapy concept, followed by autologous stem cell transplantation. The patient received one cycle of rituximab (375 mg/m2), gemcitabine (1000 mg/m2), and oxaliplatin (100 mg/m2) but CT scans revealed progressive disease. Based on the previous disease course, we anticipated an overall chemorefractory biology and decided to switch towards an anti-CD19 CAR-T-cell-based concept with axicabtagene ciloleucel (Axi-cel) that are commercially available (for r/r DLBCL6) second-generation CAR-T-cells containing a CD28 signaling domain. The bridging therapy consisted of a combination of lenalidomide (15 mg, d1-21) and rituximab (375 mg/m2) (R2) and lymphocytes could be successfully collected. Platelet count was found reduced at 66x10ˆ9/L (160–400x10ˆ9/L) upon discontinuation of (hematotoxic) R2. Twenty-five days after initiating R2 Axi-cel was administered, following lymphodepletion with fludarabine (25 mg/m2) and cyclophosphamide (300 mg/m2), which further lowered the platelet count to 55x10ˆ9/L (160–400x10ˆ9/L). On day +2 the patient fulfilled the criteria for CRS grade 1.1 In the follow, body temperature was further rising and hypotension developed on day +4 (figure 1). After diagnosing CRS grade 2, the patient received three dosages of the interleukin-6 receptor-blocking monoclonal antibody Tocilizumab. CRS discontinued but was followed by a CRES grade 1. Neurological deficits were fully recovered under 10 mg dexamethasone per day on day +16. Before the patient was discharged, she was pancytopenic with 0.38x10ˆ9/L leucocytes (4–11x10ˆ9/L), 16x10ˆ9/L platelets (160–400x10ˆ9/L), and a hemoglobin (Hb) of 84 g/L (118–155 g/L). CAR-T-cells were detectable in the peripheral blood using the CD19 CAR Detection Reagent (Miltenyi Biotec, Bergisch Gladbach, Germany) and numbered 32.3 cells/µL while CD19+ B-cells were fully depleted (figure 1). At 3 months after therapy a complete metabolic remission was achieved as assessed by positron emission tomography-CT scan (figure 2A). Despite neutrophil engraftment (without any G-CSF support), Hb and in particular platelets were still not self-sustaining, constituting grades 3 and 4 toxicities, respectively. Our patients’ only potentially myelotoxic medication was low-dose cotrimoxazole as a prophylaxis against Pneumocystis jirovecii infection. However, we decided not to discontinue its intake based on our experience and published observations from patients following allogeneneic hematopoietic stem cell transplantation (allo-HSCT) suggesting a reappraisal.7 Assessment of the bone marrow smear at 3 months after CAR-T-cell infusion ruled out residual DLBCL infiltration. No signs of dysplasia typical for myelodysplastic syndrome (MDS) were detected. In contrast to the nearly normocellular bone marrow 3 months prior CAR-T-cell therapy, we noticed a rather hypoplastic bone marrow with a markedly reduced megakaryopoiesis (figure 2B). Parallel flow cytometry-based analyses confirmed a small bone marrow-resident population of persisting CAR-T-cells at 0.3% of all CD3+ T-cells (figure 2C) while CD19+ B-cells and CD45lowCD19+CD10+ hematogones were completely depleted. Cobalamin and folate levels were normal and no paroxysmal nocturnal hemoglobinuria (PNH) clone was detectable. Despite the lack of sufficient megakaryopoiesis, immature platelet fraction (IPF) calculated using a Sysmex XE2100 device (Sysmex, Kobe, Japan) based on the ratio of immature to total platelets was found elevated with 7.6% (1.2–3.7) indicating an at least partially consumptive underlying component such as immune thrombocytopenia (ITP). However, reduced megakaryopoiesis is not entirely consistent with ITP. Taken together, diagnostic criteria for aplastic anemia (AA), ITP, MDS, and PNH were all not met. The TPO receptor is expressed in many hematopoietic cells, including early progenitor cells. TPO agonists are routinely used in ITP and AA, where they have proven their capacity to not only restore thrombopoiesis but also trilineage hematopoiesis.8 Furthermore, platelet as well as Hb responses have been reported in patients receiving TPO agonists for delayed hematopoietic recovery upon allo-HSCT.9 Therefore, we decided to initiate treatment with Romiplostim (Nplate, Amgen, Thousand Oaks, California, USA) an Fc-peptide fusion protein, which is injected subcutaneously on a weekly basis and which triggers platelet production via the TPO receptor. Treatment was initiated at a dose level of 1 µg/kg (60 µg total) and gradually increased to 8.6 µg/kg (500 µg total) until a platelet count of >50 plateletsx10ˆ9/L was achieved and maintained after 2 months. In fact, no platelet or blood transfusions were necessary immediately after starting Romiplostim. The patient experienced no therapy-related adverse events and appointment frequency in our outpatient clinic could be further reduced. Six months after CAR-T-cell treatment our patients remains in complete remission, CAR-T-cells are persisting with 22/µL, B-cells are not detectable, and IPF levels are still increased with 7.4%. Hematological recovery is steadily improving with a current toxicity grade of 1 for Hb (103 g/L) and of grade 2 for platelets (60x10ˆ9/L), respectively.
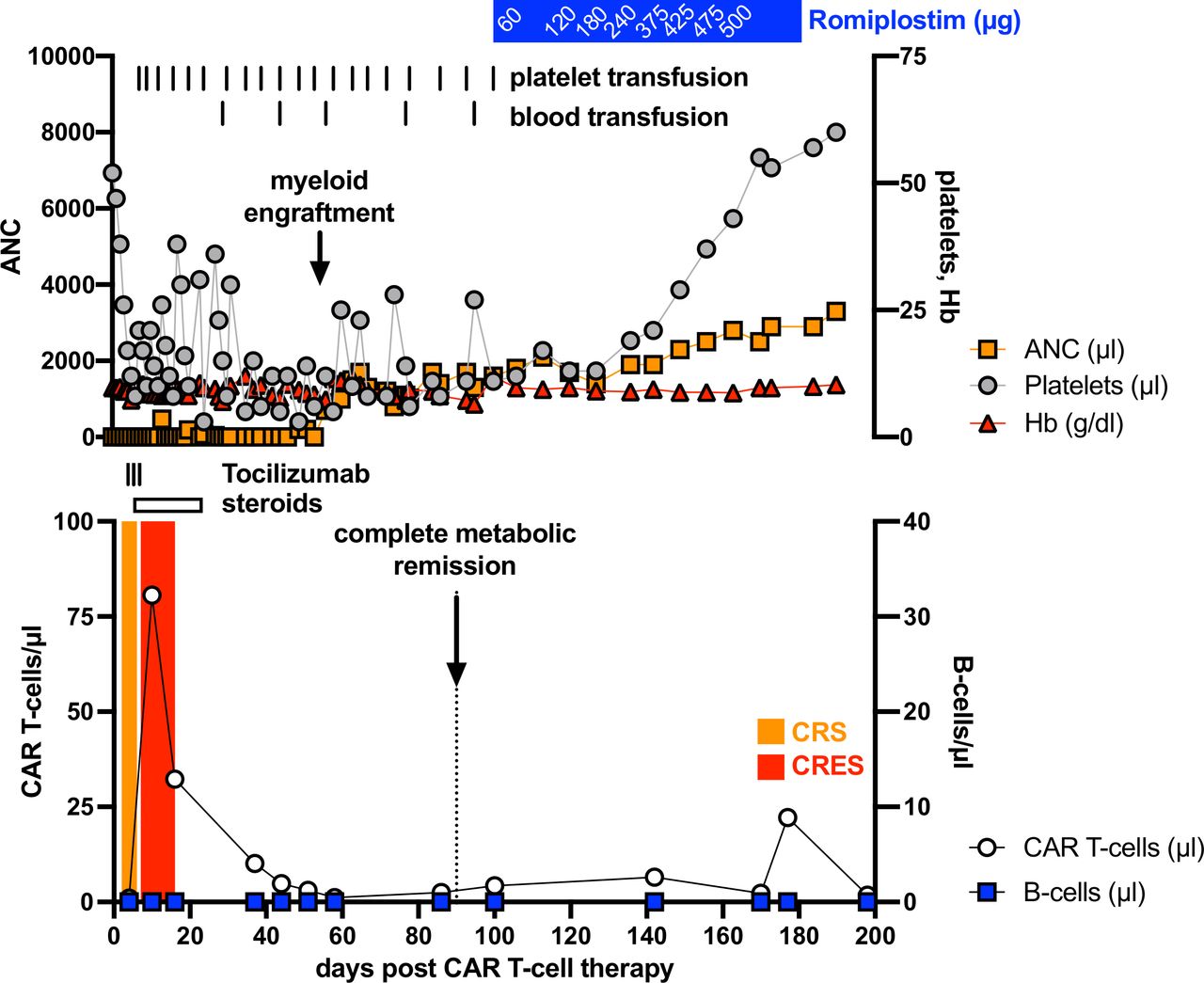
Hematological recovery and adverse events following anti-CD19 CAR-T-cell therapy for r/r DLBCL. Hematological recovery, CAR-T-cell expansion, B-cell count, CAR-T-cell therapy-related adverse event as well as the according interventions (blood and platelet transfusions, treatment with tocilizumab and steroids) were closely monitored during the first 6 months after infusion with autologous anti-CD19 CAR-T-cells. ANC, absolute neutrophil count; CAR, chimeric antigen receptor; CRES, CAR-T-cell-related encephalopathy syndrome; CRS, cytokine release syndrome; DLBCL, diffuse large B-cell lymphoma; Hb, hemoglobin; r/r, relapsed or refractory.

{kind=link}
{kind=link}
Remission status and bone marrow examination 3 months on anti-CD19 CAR-T-cell infusion. (A) Abdominal CT-scan of bulky lymphoma mass prior and after treatment (left panel) and full-body 18FDG-PET scan. (B) Microscopic evaluation of the patient’s bone marrow biopsy 3 months prior CAR-T-cell infusion (= −3 months) and of the bone marrow aspiration smear (May-Gruenwald- staining) 3 months after CAR-T-cell infusion (=+3 months). (C) Bone marrow-infiltrating CAR-T-cells were assessed by flow cytometry (FACS) and the according FACS-gating strategy is shown. Mononuclear cells were separated by density gradient centrifugation and stained with the indicated antibodies. Doublets were excluded by FSC-H/FSC-A, lymphocytes were determined by CD45/SSC-A, vital T-cells were gated by CD3+/7AAD− and further analyzed for CAR expression. CAR, chimeric antigen receptor; PET, positron emission tomography.
Discussion and conclusion
Hematological toxicities following CAR-T-cell treatment are common.1 2 Their course is often biphasic. Early alterations most likely result from the myelotoxic effects of the conditioning chemotherapy. The underlying pathomechanisms of the late effects (more than 1 month) after lymphodepletion remain to be elucidated. Yet, most cases of cytopenia recover spontaneously.2 Our patient suffered from persisting grade 3 anemia and grade 4 thrombocytopenia more than 3 months after adoptive CAR-T-cell transfer, which is seen in 10%–20% of the patients.4 While ITP is attributable to autoantibodies directed against platelet antigens, destruction of hematopoietic stem and progenitor cells by autoreactive T-cells is an accepted explanation for AA.10 11 Reduced megakaryopoiesis suggested a hypoproductive thrombocytopenia, whereas an elevated IPF pointed toward enhanced consumption. However, the IPF can be elevated in ITP and AA indicating a considerable overlap between both entities, which makes an unequivocal differentiation sometimes difficult.12 Practically, ITP and AA respond both to the TPO agonists Eltrombopag (Revolade, Novartis, Basel, CH) and Romiplostim. We decided to use Romiplostim based on its equally promising efficacy in Eltrombopag-naïve as well as in Eltrombopag-pretreated AA patients and its high and durable response rates in ITP.13 14 Prior such attempt, we would strongly recommend to exclude malignant infiltration of the bone marrow as well as an MDS, which actually has been reported in few patients that had received CAR-T-cells.4 Self-evidently, the responsible CAR-T-cell-associated mechanisms that lead to hematoxicity need to be identified in order to develop targeted countermeasures. Abundant proinflammatory cytokines could interfere with hematopoiesis,3 direct ‘off-target’ effects could yield a CAR-T-cell-mediated destruction of hematopoietic progenitors and/or platelets, while indirect autoimmune phenomena as observed in patients receiving immune checkpoint inhibitors15 could contribute to AA-like/ITP-like clinical pictures. Taken together, our case report implicates a role for TPO agonists in treating persistent cytopenia especially thrombocytopenia following CAR-T-cell therapy, thus reducing the need for transfusions and the patient’s burden. Further studies, especially controlled randomized trials, are required and may help to establish the use of TPO agonist in such clinical scenario that will occur more frequently in the future with emergence of CAR-T-cell therapies.
Ethics statements
Ethics approval
Blood samples were collected upon approval by the local ethics committee (number: 336_19B and 219_14B) and patient’s informed consent.
Acknowledgments
The authors would like thank the patient presented in this study.
Footnotes
Contributors RB collected and analyzed data and wrote the manuscript. RJ, SK, AS, SV, MB-H, DS, WR, and AM collected and analyzed data. DM wrote the manuscript.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.