Article Text

Abstract
While immune checkpoint inhibitors (ICIs) have ushered in major changes in standards of care for many solid tumor malignancies, primary and acquired resistance is common. Insufficient antitumor T cells, inadequate function of these cells, and impaired formation of memory T cells all contribute to resistance mechanisms to ICI. Adoptive cellular therapy (ACT) is a form of immunotherapy that is rapidly growing in clinical investigation and has the potential to overcome these limitations by its ability to augment the number, specificity, and reactivity of T cells against tumor tissue. ACT has revolutionized the treatment of hematologic malignancies, though the use of ACT in solid tumor malignancies is still in its early stages. There are currently three major modalities of ACT: tumor-infiltrating lymphocytes (TILs), genetically engineered T-cell receptors (TCRs), and chimeric antigen receptor (CAR) T cells. TIL therapy involves expansion of a heterogeneous population of endogenous T cells found in a harvested tumor, while TCRs and CAR T cells involve expansion of a genetically engineered T-cell directed toward specific antigen targets. In this review, we explore the potential of ACT as a treatment modality against solid tumors, discuss their advantages and limitations against solid tumor malignancies, discuss the promising therapies under active investigation, and examine future directions for this rapidly growing field.
- immunotherapy
- adoptive
- immunotherapy
- receptors
- chimeric antigen
- lymphocytes
- tumor-infiltrating
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Immune checkpoints refer to a collection of pathways that normally serve to prevent the immune system from destroying healthy cells. Cancer cells take advantage of these innate mechanisms and subsequently evade T-cell responses which would otherwise result in tumor destruction.1 It is now well known that blockade of these immune checkpoints can result in significant tumor regression.2 Consequently, the immune checkpoint inhibitor (ICI) revolution has ushered in major changes in standards of care for a variety of solid tumor malignancies including melanoma,3 lung cancer,3 4 and head and neck cancer.5 Despite the robust successes of ICI, primary and acquired resistance is common and is attributable to several factors, including insufficient antitumor T cells, inadequate function of these cells, and impaired formation of memory T cells.6
Adoptive cellular therapy (ACT) is another form of immunotherapy that is a rapidly growing area of clinical investigation which involves removing a patient’s or donor’s T cells, growing and/or modifying them in a laboratory, and reinfusing them back to the patient. In contrast to ICIs which block T-cell inhibitory receptors, ACT depends on ex vivo expansion of T cells which may allow certain inhibitory immune regulators to be bypassed. Furthermore, some forms of ACT entail genetic modification or re-engineering of T cells which can help to increase T-cell specificity. Most forms of ACT are combined with lymphodepletion chemotherapy which helps improve T-cell proliferation and persistence.7 The different forms of ACT therefore have the strong potential to address many of the limitations of ICI by its ability to augment the number, specificity, and reactivity of T cells against tumor tissue.
There are currently three major modalities of ACT: tumor-infiltrating lymphocytes (TILs), genetically engineered T-cell receptors (TCRs), and chimeric antigen receptor (CAR) T cells. TIL therapy involves expansion of a heterogeneous population of endogenous T cells found in a harvested tumor, while CAR T cells and TCRs involve expansion of a genetically engineered T-cell directed toward specific antigen targets. While successful application of ACT has been seen in hematologic malignancies,8–10 ACT in solid tumors is still in its early stages. In this review, we explore the potential of ACT as a treatment modality against solid tumors, discuss their advantages and limitations against solid tumor malignancies, discuss the promising therapies under active investigation, and examine future directions for this rapidly growing field.
Types of ACTs
Tumor-infiltrating lymphocytes
TILs refer to lymphocytes that have left the bloodstream and entered the tumor. TILs are a heterogenous population of cells that are polyclonal and have broad antigen recognition in tumor cells.11 It is believed CD8+ and CD4+ T-cell populations play a critical role in tumor control through a variety of mechanisms.12 13 Activated CD8+ T cells are able to produce proinflammatory cytokines and cause tumor cell destruction, while CD4+ T cells can promote the production of antibody-producing plasma cells and also help to activate CD8+ T-cell responses. The favorable prognostic significance of a rich T-cell infiltrate has long been reported in melanoma,14 as well as other tumors including head and neck squamous cell carcinoma (HNSCC),15 cervical cancer,15 16 and lung cancer.17 Unfortunately, these existing TILs are rendered ineffective due to tumor microenvironments filled with inflammatory infiltrates that are enriched with suppressive regulatory T cells and myeloid suppressor cells.18
As a therapeutic strategy, using adoptive cellular transfer of TILs allows for an expansion of immune cells away from this suppressive tumor microenvironment. This process involves harvesting and isolation of TILs from a resected tumor, expansion in vitro, and then reinfusion back into the patient. While several animal studies had demonstrated the potential of isolated immune cells to mediate tumor regression,19 20 the first in-human study of TILs was performed in 1988 in patients with metastatic melanoma.15 Researchers isolated extracted lymphocytes from resected melanoma specimens, expanded them in vitro, and then reinfused them back to the patients after a dose of cyclophosphamide chemotherapy. Patients in this study were also given adjuvant interleukin-2 (IL-2), a strategy based on the observation of potentiated tumor responses from immune cells with this cytokine in murine models.21 22 This study found that adoptive cellular transfer of TILs resulted in a 55% objective response rate (ORR). This landmark study was able to prove that not only was TIL therapy safe but also it could be effective in eradicating tumor cells.
Since then, many studies have been done with TILs in melanoma. More recent data presented in 2020 at the American Society of Clinical Oncology annual meeting assessed melanoma patients whose cancers had progressed after either PD-1 blockade or a BRAF±MEK inhibitor for BRAF V600-positive disease. Of the 66 patients treated, there was an ORR of 36.4% and at a median follow-up of 8.8 months, the median duration of response was not reached.23 Furthermore, the disease control rate was 80%. The therapy was well tolerated with a majority of the toxicities being related to lymphodepleting chemotherapy and/or IL-2. A majority of the patients who responded to the TILs had progressed on ICI, suggesting that the mechanisms between the two are not redundant. This is especially important to consider given that the early TILs studies were done prior to the checkpoint inhibitor era. How to adequately generate and when to sequence TILs with checkpoint inhibitors will need to be elucidated with future studies. There are already data to suggest that patients whose cancers have progressed on ICI still have a significant amount of tumor-reactive T cells.24 Novel strategies to reinvigorate these T cells in this patient population remains difficult but may help to further delineate the role of TILs in the ICI era.
Some of the differences seen between the most recent studies on TIL therapy in melanoma and the studies done in the 1980s are due to a better understanding of the importance of lymphodepletion. Lymphodepletion is believed to contribute to enhanced antitumor efficacy of TILs not only by the removal of regulatory T cells which hinder CD8+ T-cell immunity25 but also by removing homeostatic cytokine sinks that otherwise would weaken effector cell proliferation.26 While the initial studies in the 1980s used a single dose of cyclophosphamide for lymphodepletion, the more recent studies used more intensive regimens either through the addition of fludarabine or total body irradiation. Although not definitive, some data suggest that ORRs are higher for TIL populations that have more intensive lymphodepletion regimens.7
TIL therapy has been most extensively studied in melanoma, though there are emerging data to suggest a potential therapeutic role of TILs in a variety of other solid tumors as well.27 The more recent observations that these other tumor types are also responsive to ICI provided the proof of principle that endogenous T cells are capable of mediating tumor regression in these solid cancers. For these tumor types, TILs could potentially have several advantages to ICI. ICI by its very nature relies on tumor-reactive T cells since its primary mechanism of action is to remove the breaks on the immune system. However, since it is speculated that many of these T cells may be dysfunctional28 (or absent due to low tumor immunogenicity in some cancers),29 this strategy does not work for a relevant proportion of patients. The generation of TILs relies on the creation of a population of T cells that may be able to overcome this dysfunction either through coculture with IL-2 and/or selection for tumor antigen specific T cells.
There are now several multi-institutional trials exploring TIL therapy for HNSCC, lung cancer, and cervical cancer (table 1). While previously HNSCC were thought to lack immune infiltration, a recent analysis of The Cancer Genome Atlas found that transcriptomes from HNSCC were some of the most highly immune infiltrated.30 Furthermore, CD8+ T-cell infiltrates in HNSCC are associated with improved survival and favorable clinical outcomes.31 These studies suggest that the therapeutic approach using TILs could be very promising.
Selected adopted cellular therapy trials in solid tumor malignancies
While some of these trials have shown significant successes in TILs as a treatment modality, widespread adoption has not yet been seen. The traditional manufacturing process takes approximately 5–7 weeks from tumor resection to TIL production32 which may be too long for some patients with metastatic disease. Furthermore, many times isolated TILs are unable to grow or demonstrate strong effector responses,33 which may reflect tumor tissue with poor immune infiltration that existing processes are unable to overcome. Combination approaches such as the addition of checkpoint blockade to TIL products (NCT02621021; NCT03645928; NCT03215810) aim to improve TIL efficacy and to help overcome some of these issues. For example, recently preliminary data from a TIL therapy in lung cancer with ICI were presented at the American Association of Cancer Research (AACR) meeting in 2020. In this study, patients with metastatic non-small cell lung cancer (NSCLC) whose cancers had progressed on nivolumab were given TILs and then resumed nivolumab after TIL infusion. Of the 20 enrolled patients, most of the patients had initial tumor regression with a median best overall change in their tumor of approximately 38%.34 Two patients in the study had complete regression of their tumor which was ongoing for nearly a year. Further studies will help to elucidate the potential benefits of this combination therapy in lung as well as other tumors.
Genetically engineered TCR
TCR-engineered T-cell technologies are a natural extension of the rationale for TIL therapy. Since TILs are ostensibly equipped with acquired tumor recognition, this raised the possibility of genetically modifying naive lymphocytes to also recognize tumor antigen. In the 1980s, Dembic et al identified that TCR α and β chains are the critical components that determine antigen specificity of T cells.35 These TCR ɑ and β heterodimers form TCR–CD3 signaling complexes with specificities for target antigens presented to it by human leukocyte antigen (HLA)-dependent mechanisms.36 Kessels et al used retroviral vectors to transduce specific TCRs into T cells via gene transfer into murine models.37 In the late 1990s at the National Institutes of Health (NIH), researchers were able to successfully transduce human peripheral blood lymphocytes with genes encoding a MART-1-specific TCR, found in a majority of melanoma tumors.38 With this established framework in mind, Morgan et al tested an anti-MART-1 TCR on 15 patients with melanoma.39 The study demonstrated clinical responses in 2 of 15 patients with metastatic melanoma providing proof of principle of TCRs as a feasible strategy to affect tumor regression.
A number of trials with different antigen targets have shown clinical activity of TCRs across solid tumor types. For example, the NY-ESO-1 cancer/testis antigen is found in approximately 25% of melanomas, and one study of antigen-positive tumors found that a TCR directed toward this target resulted in 5 of 11 patients having an objective clinical response with 2 of those patients demonstrating complete regression of their disease.40 NY-ESO-1 is also expressed in approximately 18% of NSCLCs41 42 and 80% of synovial sarcomas.43 44 A TCR for NY-ESO-1 in solid tumors (NCT02650986) is ongoing and will hopefully clarify the effectiveness of this TCR for these patient populations. Another therapy being actively studied is a MAGE-A3/A6 TCR (NCT03139370). Similar to NY-ESO-1, MAGE is a cancer-germline antigen that is expressed in multiple cancer types.45 Proof of concept of this as a possible therapy was explored in a study of nine patients with cancer treated with an anti-MAGE-A3 TCR in 2013,46 though several of the patients experienced a life-threatening neurologic toxicity due to cross-reactivity with the MAGE-A12 protein expressed on a subset of neurons. Optimization of these TCRs to avoid activation in areas of the body without tumor is needed to avoid potentially life-threatening side effects.
Identification of TCR targets for virally mediated cancers such as HPV or Epstein-Barr virus may also be an especially promising strategy. Viral antigens which are presented in HLA-dependent mechanisms are constitutively expressed by tumor cells and expression of these antigens is unlikely to be found on normal tissue. In one study at the NIH of an HPV E7 TCR in HLA-A2*01-positive patients, 6 of 12 patients demonstrated objective clinical responses with no cytokine release syndrome (CRS).47 48 Of the patients who did not have clinical responses, defects were found in HLA-A2*01 or β-2 microglobulin, both of which are considered necessary for peptide antigen recognition of this specific TCR. Ongoing trials are testing TCRs in HPV-related cancers (NCT03912831).
However, virally mediated malignancies represent a small proportion of the cancer epidemiologic burden. Attempts to broaden the TCR strategy may largely depend on the identification and targeting of neoantigens, epitopes that arise from somatic mutation and are not present on normal cells. Neoantigens are generally a result of non-synonymous somatic mutations. Tumor neoantigens are presented by major histocompatibility complex (MHC), have the potential for garnering an adaptive immune response, and represent ideal targets for ACT since by definition they are not present on normal cells.49 Some data even suggest that objective responses to ICI are mediated by neoantigen-specific T cells.50–52 Identification of neoantigen T cells would allow for the creation of TCR cell therapy products that are highly individualized and specific. Next-generation sequencing, mass spectrometry, whole-exome/transcriptome sequencing, and high-throughput T-cell-based assays are all approaches being used to identify neoantigen T cells currently.53 One study using neoantigen TCRs in combination with ICI in patients with incurable solid tumor malignancies is ongoing (NCT03970382). The key to the neoantigen strategy will heavily rely on the ability to identify and development high-affinity and immunogenic TCRs. Enhancing genetically re-engineered TCR on-target cytotoxicity and outcomes will depend on a variety of factors including, but not limited to, improving the affinity of the peptide MHC–TCR interaction and reducing competition from native TCRs which would otherwise reduce transgenic TCR concentrations.54
Nevertheless, identifying individual patient neoantigens is laborious and costly, so targeting a shared mutational antigen that is specific to tumors and clonally conserved may prove more practical. Targeting neoantigens derived from potential driver oncogenes such as BRAF V600E or PIK3CA55 may prove to be a more fruitful strategy since these neoantigens may be more likely to appear in a significant portion of patients within the same tumor type.
These strategies no matter how successful may prove ultimately are ineffectual if the re-engineered TCRs are unable to meaningfully traffic to tumor sites. To overcome this barrier, multiple tumor types may benefit from intratumoral or locoregional administration of TCRs. For example, in light of the accessibility of most local lesions for HNSCC, there are clinical trials studying intratumoral delivery of an E7 oncoprotein TCR for borderline resectable and unresectable HPV-associated oropharyngeal cancers (NCT04044950). An added benefit of this will be the hopeful avoidance of systemic toxicities associated with some TCR therapies. Other tumor types with accessible lesions, such as cutaneous malignancies, or those confined to a cavity, such as peritoneal or intracranial diseases, may benefit from this strategy.
It is worthy of note that genetically engineered TCRs are HLA restricted and thus must be HLA matched to be effective. Many of the TCRs in ongoing investigation are restricted to HLA-A*0201, which is under-represented in African and Asian populations while present in nearly half of Caucasian populations.56 As TCR studies become more prolific, it is imperative that non-HLA-A*02 serotypes are considered so that these novel technologies can benefit diverse patient populations. Furthermore, since HLA alterations may function as escape mechanisms for some TCRs, the ability to overcome these resistance mechanisms will be critical to future successes.
CAR T cell
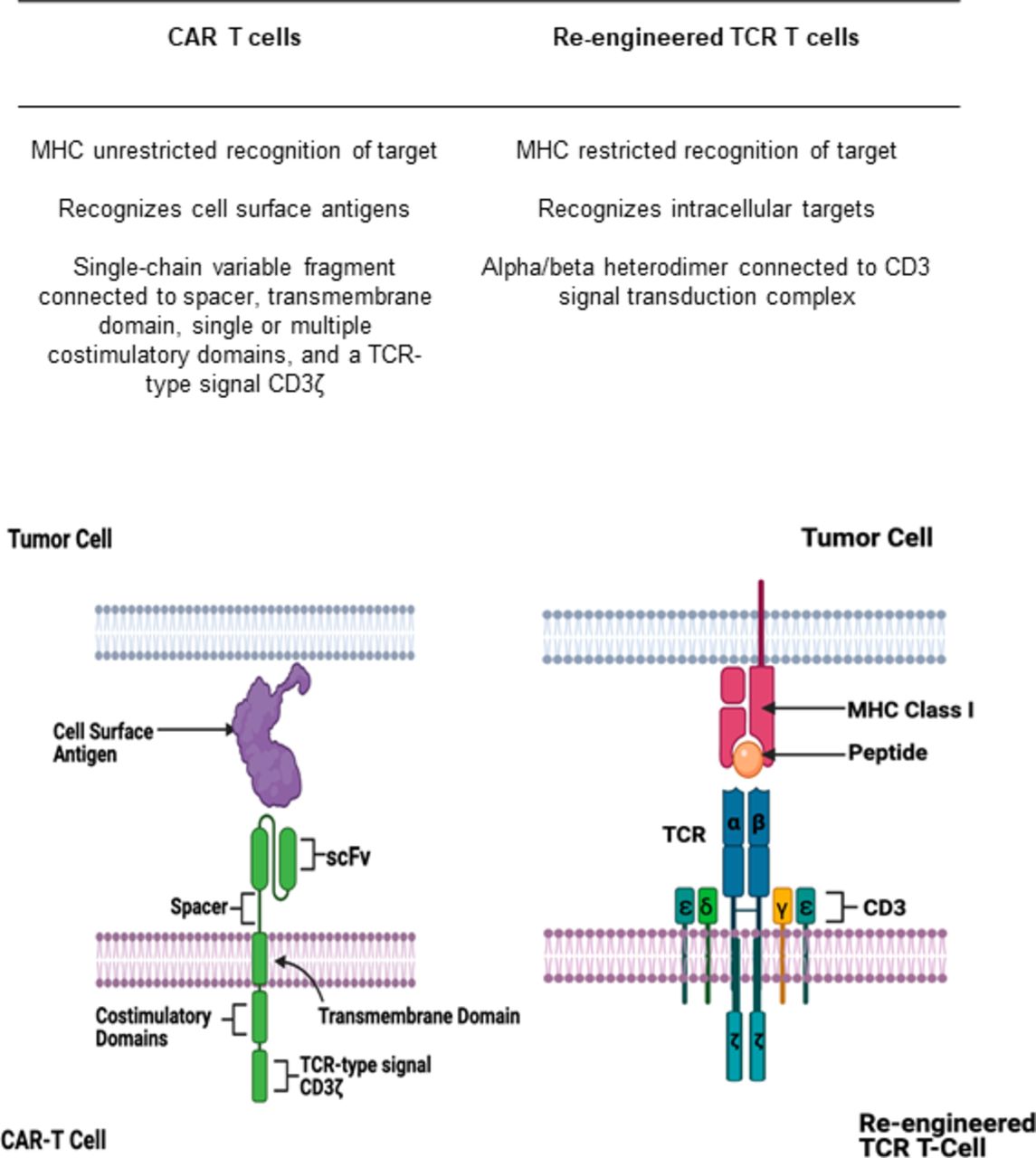
Similarly to TCRs, CAR T cells are lymphocytes that are genetically modified to have specificity for tumor antigens. In contrast to TCRs, CAR T cells recognize extracellular antigens independent of HLA presentation (figure 1).51 CAR T cells consist of a T-cell activating domain which allows for cell proliferation and a single-chain variable fragment which confers specificity for a target. While initial generations of CARs were not able to sustain T-cell responses, subsequent generations of CAR T cells include one or more costimulatory domains (usually CD28 or 4-1BB) to enhance its proliferative capacity.57–59
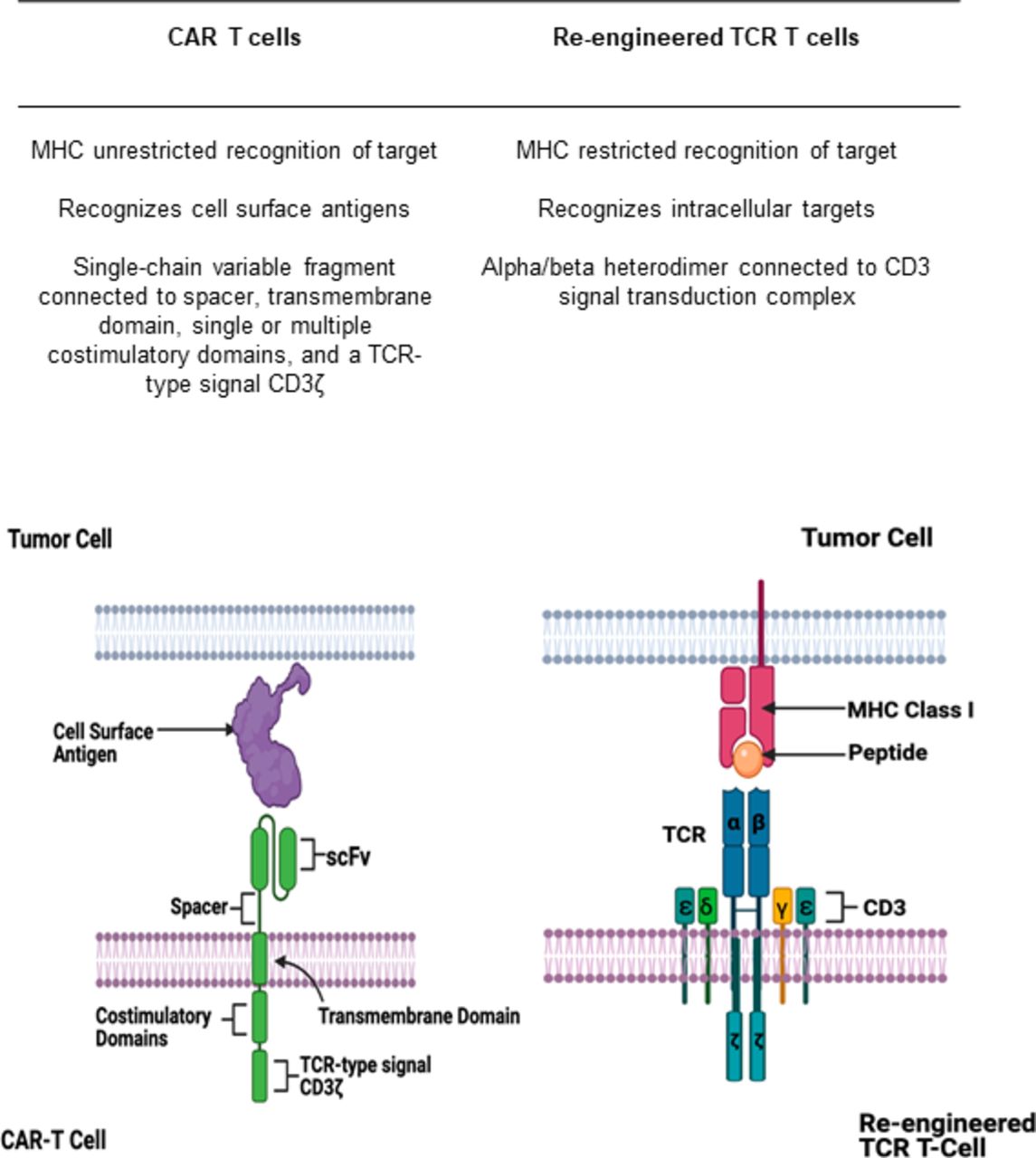
Differences between chimeric antigen receptor (CAR) T cells and re-engineered T-cell receptors (TCRs). CAR T cells recognize antigens expressed on the cell surface via an antigen-recognition domain (signal-chain variable fragment). This domain is connected to a spacer, transmembrane domain, and a single or multiple costimulatory domains which then trigger downstream T-cell activation via a signaling domain. TCRs are heterodimers with an α and β subunit which recognizes intracellular peptide antigens presented to it by the major histocompatibility complex (MHC). The heterodimer is connected to a CD3 signal transduction complex which acts to activate downstream T-cell activation. Created with http://www.biorender.com/.
The first successful CAR T cells were developed to have specificity for CD19 and were eventually FDA approved for acute lymphoblastic leukemia and refractory large B-cell lymphomas.9 60 Currently, different CAR T-cell targets in solid tumor malignancies are in active investigation.
One study used an intracranial CAR T cell directed against IL-13 receptor α 2 (IL13Rα2) in a patient with multifocal glioblastoma multiforme (GBM) and found complete regression of tumor lasting nearly 8 months.61 Another phase I trial is studying the safety and preliminary efficacy of a CAR T-cell directed against ROR1, a transmembrane tyrosine kinase receptor, found in breast and lung cancers (NCT02706392). Presentation of preliminary data from this study at the AACR demonstrated that four of five patients with breast and lung cancer had a mixed response with decreased disease burden at some of their metastatic sites.62 Another study used a regionally delivered mesothelin-targeted CAR T cell with checkpoint inhibition in patients with malignant pleural mesothelioma and metastatic lung and breast cancer.63 The mesothelin-specific CAR T cell was administered intrapleurally and then followed by ICI. Of the 14 evaluable patients treated with ICI after CAR T administration, 2 had a complete response, 5 had a partial response, and 4 had stable disease. No CAR T-cell therapy-related toxicities greater than grade 1 were observed in the study. The study is currently ongoing (NCT02414269).
A few other novel CAR T targets have shown preliminary efficacy. One phase I, open-label dose-escalation study of a HER2 CAR T cell for patients with glioblastoma found no dose-limiting toxicities and a median overall survival of 11.1 months from cell infusion. Of the 16 evaluable patients, 1 had a partial response lasting nearly 9 months, while 7 had stable disease ranging from 8 weeks to 29 months.64 Another study used a CAR T directed toward prostate-specific membrane antigen (PSMA) in patients with castrate-resistance prostate cancer.65 The PSMA–CAR was coupled to a dominant negative receptor for TGFβ in order to inhibit the immunosuppressive activity of TGFβ in the tumor microenvironment. At 12 weeks, 4 of 10 treated patients had at least 30% reduction of their prostate-specific antigen levels. A majority of the patients developed grade 2 or higher CRS, a constellation of inflammatory and clinical findings occurring commonly in patients with lymphoma receiving CAR T therapy. The CRS was associated with increases in IL-6, IL-15, IL-2, and IFNγ. Parallel to the PSMA CAR T, there is another promising target directed toward prostate stem cell antigen (PSCA) with its trial ongoing (NCT03873805).66 PSCA is also expressed in some gastric and pancreatic cancers and has shown preliminary safety and efficacy in this population.67 Lastly Claudin18.2, a stomach-specific isoform of Claudin-18, is overexpressed in some gastric and pancreatic adenocarcinomas. This is a new and evolving treatment option for these malignancies. One study found that a Claudin18.2 CAR T in gastric and pancreatic adenocarcinoma patients resulted in an ORR of 33%, a median PFS of 130 days, and no serious adverse events.68
Nevertheless, the development of CAR T cells for solid tumors has been difficult. While its lack of dependence on HLA-specific recognition is an advantage with regard to general applicability over TCRs, it also limits its effectiveness since it would theoretically only be effective for a unique, extracellular target which may not exist for many solid tumor malignancies. Furthermore, the limited antigen sensitivity for current CAR T cells allows for relatively easy downregulation of target antigens by tumor cells as a mechanism of acquired resistance. For example, in the seminal trial of tisagenlecleucel, a CD19 CAR T cell for patients with B-cell acute lymphoblastic leukemia,60 94% of the assessed patients who relapsed had CD19 antigen loss either through genetic mutation or maximization of CD19 variants with isoforms lacking the transmembrane domain or targeted exon.69 This proves even more problematic for solid tumor malignancies given the tremendous tumor-associated antigen heterogeneity.70 One strategy to overcome this is through ‘T cells redirected for antigen-unrestricted cytokine-initiated killing’ (TRUCKs). These fourth generation CAR T-cell products combine a CAR T cell directed toward a specific antigen with an inducible release of a transgenic protein (usually a cytokine) on activation.71 Recognition of the tumor antigen by the TRUCK leads to TCR–CD3-ζ-mediated downstream signaling which leads to eventual release of the protein. This allows for engagement with the tumor cell based on its cognate antigen with a subsequent cytokine-mediated destruction of regulatory T cells or antigen-loss tumor cells that may otherwise facilitate relapse. In one mouse model of an inducible IL-12 CAR T cell directed toward the carcinoembryonic antigen (CEA), transplanted, antigen-heterogenous tumor lesions were eliminated not only as a result of both CAR T-cell engagement with antigen-positive tumor cells but also due to IFNγ production by NK and T cells along with activation of macrophages and secreted TNF-α.71 72 Future CAR T strategies will likely include combinatorial cytokines, multitarget approaches, and novel combination immunotherapeutic strategies, which may help overcome tumor associated antigen heterogeneity and the inhibitory microenvironment of the solid tumor stroma. Table 1 summarizes relevant TCR and CAR T cells currently in active investigation.
Barriers to act for solid tumors
Target antigen heterogeneity and antigen escape
One of the barriers to the development of effective cellular therapies in solid tumors, specifically for TCRs and CAR T cells, is target antigen heterogeneity. The successes of ACT trials in hematologic malignancies were largely dependent on three factors. First, there was a clear identifiable antigen (CD19). Second, this antigen was homogeneously expressed in the disease of interest. Third, toxicities associated with T-cell activity on other parts of the body (so-called on-target/off-tumor effects) were manageable (eg, immunoglobulin repletion for B-cell depletion)8 and expression on tumor was much higher than healthy cells allowing for a therapeutic window. In contrast, solid tumor malignancies tend to have more heterogeneity in their antigen profiles,70 especially since the formation of these cancers is not always dependent on clonal expansion of an aberrantly expressed antigen as in lymphomas or leukemias. While many of these antigens may be overexpressed in some tumor tissue, there is not consistent expression due to this intratumoral heterogeneity which generally results from genomic instability. Intratumoral heterogeneity is a concern in diffusely metastatic disease, has been found and studied in multiple cancer types, and may help to explain some forms of resistance mechanisms.73–75 Acquired resistance patterns which manifest as mixed radiographic responses may be a result of the selective pressures of immunotherapies. One study using an epidermal growth factor receptor variant III (EGFRvIII) CAR T cell in patients with recurring glioblastoma found a significant adaptive response from the tumor to avoid destruction after cell infusion.76 Analysis of tumor specimens demonstrated decreased antigen expression of EGFRvIII in five of seven infused patients. Furthermore, post-CAR T-cell tumor specimens demonstrated an increased expression of immunosuppressive molecules including indoleamine 2,3-dioxygenase (IDO1), PD-L1, and FoxP3-expressing regulatory T cells. This adaptive compensatory response adds to the difficulties in optimizing cellular therapies for the management of solid tumor malignancies.
One strategy to overcome antigen escape and heterogeneity is through the use of a multiantigen recognition circuit involving complementary antigens.77 78 One example of this is the syn-Notch receptor, which uses an engineered transmembrane receptor to induce expression of a tumor-specific CAR in response to recognition of an extracellular signal.78 79 One such study found that CD8+ T cells engineered with a syn-Notch ligand that recognizes a GBM tumor-specific but heterogeneously expressed antigen EGFRvIII and a tumor non-specific EphA2 or IL13Rα2 CAR T ligand resulted in in vitro killing of both EGFRvIII+ and EGFRvIII− cells.77 This ‘prime and kill’ strategy by Choe et al77 also hypothesized that priming with a tissue-specific antigen called myelin oligodendrocyte glycoprotein could be used in combination with the EphA2 or IL13Rα2 CAR T ligand. This resulted again in in vitro killing of GBM target cells with no effect on the priming cells. These results were confirmed in vivo in xenografts and demonstrated that the multiantigen recognition circuit resulted in cytotoxic activity in a spatially confined manner. Other strategies to overcome these barriers include bispecific CAR T cells which may optimize tumor cell engagement better than singly targeted therapies across metastatic sites and result in better tumor regression. While clinical trials with dual targeting CAR T cells are not prolific, a proof of concept with a dual-specific CAR T targeting ErbB2 and MUC1 found that engagement with the target antigen resulted in enhanced T-cell proliferation.80 Making use of these novel designs and antigen gating may help to overcome some of the barriers associated with solid tumor antigen heterogeneity and escape.
Nevertheless, target identification that is specific to tumor cells and avoids normal human tissue has been a crucial challenge for the development of cellular therapies in solid tumor malignancies. While strategies such as syn-Notch receptors are promising, a number of studies have demonstrated dose-limiting on-target/off-tumor toxicities with currently used technologies. For example, one clinical trial studied a CEA -specific CAR T cell (CEACAM5) and found that expression of this antigen in the lung epithelium likely correlated with the dose-limiting respiratory toxicities from the target antigen being studied.81 Another study in metastatic renal cell carcinoma using autologous T cells re-engineered against carbonic anhydrase IX found a similar on-target/off-tumor toxicity when patients infused with cell product had profound liver toxicities likely mediated from expression of target antigen on bile duct epithelial cells.82 The problem of tumor antigen heterogeneity is compounded when viewed in the context of these off-tumor toxicities as even the slightest cross-reactivity could result in life-threatening toxicity.
T-cell trafficking and infiltration
For effector T-cell-mediated cell death to occur, a T cell must be able to effectively traffic to a tumor and subsequently overcome any suppressive cytokines that would prevent it from fulfilling its goal. It is well known that many metastatic lesions lack lymphocytes, thus providing evidence that effector T cells are not adequately localizing their targets.83 84 One study in melanoma found that metastatic sites with greater CD8+ T-cell infiltration also had a preferential expression of six chemokine subsets: CCL2, CCL3, CCL4, CCL5, CXCL9, and CXCL10.85 While these chemokine subsets may not be generalizable to all solid tumor malignancies, it suggests that critical chemokine receptor/ligand matches are necessary to allow for effector T-cell migration.86 Furthermore, the failure of ACTs to traffic to tumor sites as a result of chemokine/receptor mismatch and inhibitory cytokines helps to explain the lack of efficacy to date of these therapies in solid tumors, especially in light of the fact that higher and persistent CAR T levels in the blood of patients with lymphoma were associated with a higher likelihood of response.9 CAR T products in particular may have less obstacles for trafficking to hematologic tumor cells in the lymph nodes or bone marrow, since T cells are already normally trafficked to these areas.87 For solid tumor malignancies, any such T-cell product must efficiently move from the bloodstream to the tumor which generally has an overwhelming set of inhibitory cytokines which will prevent this from happening.
Immunosuppressive tumor microenvironment
The solid tumor microenvironment has optimized its oppression of active effector T cells through a variety of mechanisms including preventing T-cell infiltration and by facilitating anergy. Furthermore, even if a cell therapy product is able to effectively traffic to a tumor, it must overcome a structurally complex microenvironment and macroenvironment with altered extracellular matrices, abnormal vasculature, variable interstitial fluid pressure, and shifting hypoxic regions.88 Many tumor-associated myeloid cells produce immunosuppressive cytokines (eg, IL-10 and TGF-β) which serve to prevent T-cell-mediated destruction of tumor.89 Furthermore, deterioration of T-cell function may be a result of persistent antigen and inflammatory signal exposure.90 Some investigators claim that existing dysfunctional CD8+ T-cell infiltrates in solid tumors may be rejuvenated by checkpoint inhibitor therapy. Using single-cell RNA sequencing, one study found that two distinct tumor CD8+ T cells existed: progenitor exhausted and terminally exhausted.91 The former was able to control tumor growth, while the latter was not. This has significant developmental and therapeutic implications for ACT given that transferred T cells must be protected from becoming terminally exhausted. Innate resistance to checkpoint inhibition in patients with solid tumors may be a result of a CD8+ T-cell infiltrate ratio that favors terminal exhaustion over progenitor exhaustion.
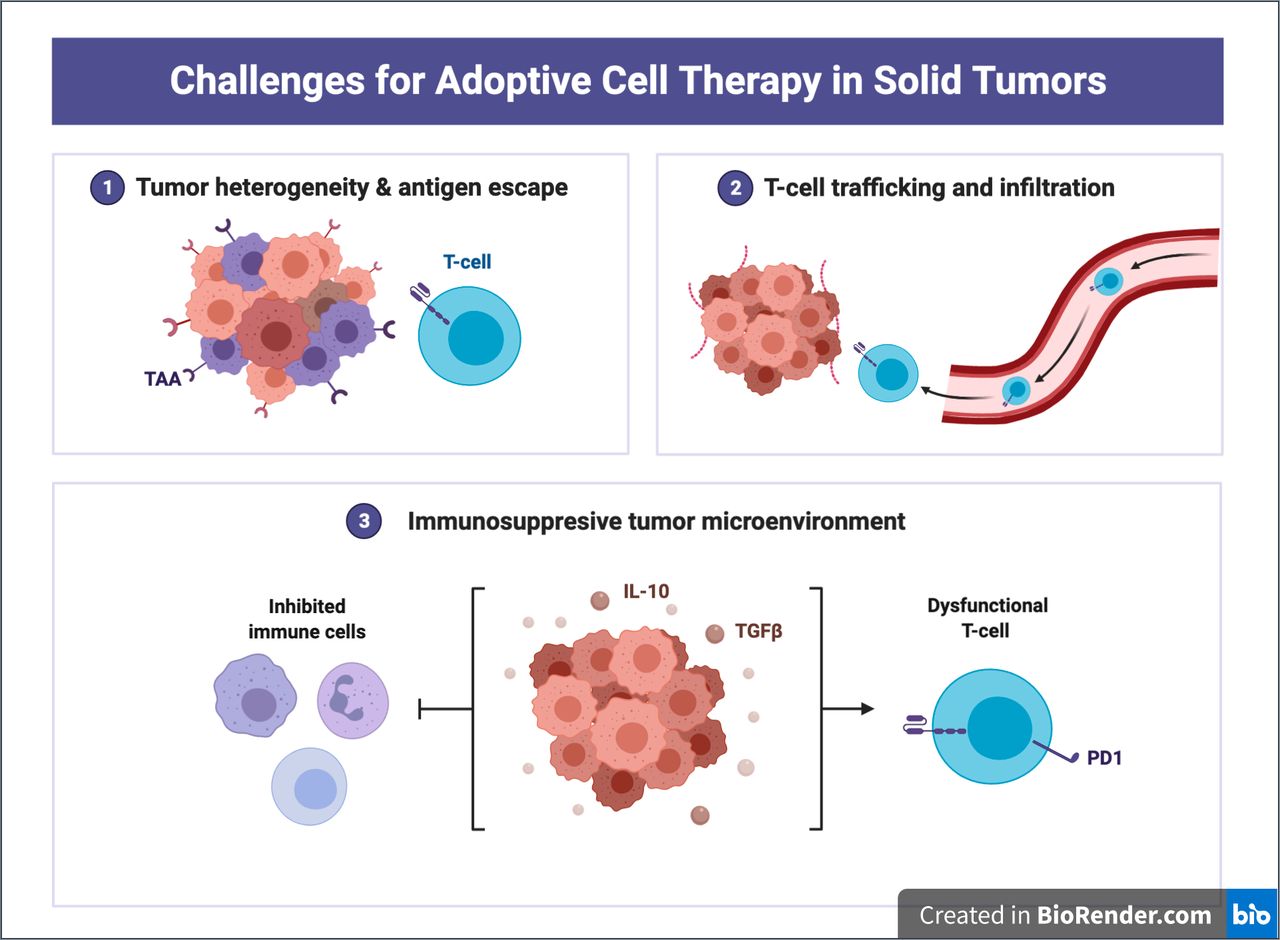
Nevertheless, the cytokine interaction is multifactorial, multifaceted, and incredibly complex, and there is therefore an unmet need with regard to overcoming these inhibitory signals. More recently, CRISPR technologies have been used to identify genes that are critical to effector T-cell function.92 These technologies have the ability to create T cells resistant to multiple inhibitory pathways. For example, the generation of ablated Fas receptor CAR T cells using CRISPR allowed for a resistance to Fas ligand activation induced cell death both in vivo and in vitro when challenged with tumor cells.93 Furthermore, genome-wide CRISPR screens can help to identify potent modulators of TCR activation and effector T-cell function. One such study with CAR T and GBM stem cells using CRISPR screening found Ikaros family zinc finger protein 2 and TLE4 as critical factors in effector T-cell function, and subsequently that targeted knockouts of these genes increased CAR T-cell activity.92 CRISPR screening may prove to serve a critical role for the development of enhanced CAR T cells that can reliably reverse exhaustion and inhibitory mechanisms of the proinflammatory tumor microenvironment. Figure 2 summarizes the challenges of ACT in solid tumor malignancies.
{kind=link}
{kind=link}
Challenges for adoptive cell therapy (ACT) in solid tumors. Tumor heterogeneity, antigen escape, T-cell trafficking, and an immunosuppressive tumor microenvironment represent some of the most challenging obstacles in solid tumor ACT development. Adapted from ‘Challenges for CAR T-cell immunotherapy in solid tumors’, by BioRender.com (2021). Retrieved from https://app.biorender.com/biorender-templates. IL-10, interleukin-10.
Future directions and conclusions
The future of ACT will rely heavily on combinatorial regimens and novel structural changes to existing cellular therapy products to augment clinical efficacy and overcome resistance mechanisms. The addition of checkpoint inhibitors or cytokine modulators such as CD40 can help increase T-cell infiltration, enhance proliferation, and overcome antigen escape.94 Targeting the tumor microenvironment and/or stroma may also improve the effectiveness of ACTs. For example, adding angiogenic therapies (eg, vascular endothelial growth factor inhibitors) to ACTs may increase tumor infiltration by disrupting the tumor vasculature95 or engineering macrophages with CARs may help to overcome immunosuppressive cytokines and upregulate antigen presentation.96 The technologies of dual-specific CAR T cells, synNotch receptors, CRISPR-based modulation of the proinflammatory genes, along with inducible cytokine CAR T cells, neoantigen TCRs, and combination regimens with TILs all represent the future of enhanced ACT. Furthermore, the recent technological breakthroughs of mRNA vaccines are certain to make a strong impact on the field of cellular therapy and immunotherapies as a whole. In the USA alone, there are nearly 50 clinical trials studying ACT products for solid tumor malignancies. As we move forward to the next paradigm shift in cancer treatment, a more informed understanding of the tumor microenvironment, target antigens, resistance mechanisms, and strategies to overcome these mechanisms is critical to improving outcomes through innovation.
Ethics statements
Acknowledgments
Editorial assistance was provided by the Moffitt Cancer Center’s Office of Scientific Writing by Dr Paul Fletcher and Daley Drucker. No compensation was given beyond their regular salaries.
References
Footnotes
Twitter @kedar_kirtane
Contributors KK: conceptualization, writing of initial manuscript, and editing. HE, CHC, and DA-D: review and editing of manuscript.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests KK: Owns stock in Seattle Genetics, Oncternal Therapeutics, and Veru. HE: None. DA-D: Member of the scientific advisory board of Anixa Biosciences. He receives research funding from Intellia Therapeutics and Bluebird Bio and has been listed as inventor or coinventor in patent applications filed by Moffitt Cancer Center. CHC: Honoraria from Sanofi and Exelixis for ad hoc Scientific Advisory Board participation.
Provenance and peer review Not commissioned; externally peer reviewed.