Article Text

Abstract
Background Resistance to an immune checkpoint inhibitor (ICI) is a major obstacle in cancer immunotherapy. The causes of ICI resistance include major histocompatibility complex (MHC)/histocompatibility locus antigen (HLA) class I loss, neoantigen loss, and incomplete antigen presentation. Elimination by natural killer (NK) cells would be expected to be an effective strategy for the treatment of these ICI-resistant tumors. We previously demonstrated that a lipid nanoparticle containing a stimulator of an interferon gene (STING) agonist (STING-LNP) efficiently induced antitumor activity via the activation of NK cells. Thus, we evaluated the potential of reducing ICI resistance by STING-LNPs.
Methods Lung metastasis of a B16-F10 mouse melanoma was used as an anti-programmed cell death 1 (anti-PD-1)-resistant mouse model. The mice were intravenously injected with the STING-LNP and the mechanism responsible for the improvement of anti-PD-1 resistance by the STING-LNPs was analyzed by RT-qPCR and flow cytometry. The dynamics of STING-LNP were also investigated.
Results Although anti-PD-1 monotherapy failed to induce an antitumor effect, the combination of the STING-LNP and anti-PD-1 exerted a synergistic antitumor effect. Our results indicate that the STING-LNP treatment significantly increased the expression of CD3, CD4, NK1.1, PD-1 and interferon (IFN)-γ in lung metastases. This change appears to be initiated by the type I IFN produced by liver macrophages that contain the internalized STING-LNPs, leading to the systemic activation of NK cells that express PD-1. The activated NK cells appeared to produce IFN-γ, resulting in an increase in the expression of the PD ligand 1 (PD-L1) in cancer cells, thus leading to a synergistic antitumor effect when anti-PD-1 is administered.
Conclusions We provide a demonstration to show that a STING-LNP treatment can overcome PD-1 resistance in a B16-F10 lung metastasis model. The mechanism responsible for this indicates that NK cells are activated by stimulating the STING pathway which, in turn, induced the expression of PD-L1 on cancer cells. Based on the findings reported herein, the STING-LNP represents a promising candidate for use in combination therapy with anti-PD-1-resistant tumors.
- adjuvants
- pharmaceutic
- immunotherapy
- killer cells
- natural
- melanoma
- drug therapy
- combination
Data availability statement
Data are available on reasonable request.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Background
Our immune responses against cancer involve a series of spatiotemporal events and are defined as the concept of the Cancer-Immunity Cycle.1 The Cancer-Immunity Cycle is initiated by the innate sensing of cancer cells by dendritic cells (DCs).2 3 The stimulator of the interferon gene (STING) pathway appears to be indispensable for the innate sensing of cancer cells.4 The STING pathway recognizes cytosolic DNA and cyclic dinucleotide (CDN) molecules, leading to the induction of type I interferons (IFNs) and inflammatory cytokine production associated with innate immunity.5 Therefore, agonists associated with the STING pathway (STING agonists) such as DNAs and CDNs would be expected to function as potent cancer adjuvants. Several clinical trials are currently ongoing in this area.
The intracellular transport of CDNs via SLC19A was carried out in cells that highly express SLC19A in an in vitro situation.6 However, in general, STING agonists are not able to pass through the cellular membrane and reach the cytoplasm where the STING pathway is located because they are analogous to nucleic acids. We previously reported on the development of a lipid nanoparticle (LNP) loaded with a STING agonist (STING-LNP) and succeeded in inducing effective antitumor immunity via systemic administration.7 8 The STING-LNP contained a pH-sensitive cationic lipid, YSK05 as the major component,9 and efficiently delivered the STING agonist to the cytoplasm.7 A treatment with ovalbumin (OVA) and the STING-LNP resulted in an antitumor effect against E.G7-OVA via OVA-specific cytotoxic T lymphocyte (CTL) activation.7 In addition, the intravenous injection of STING-LNP efficiently activated natural killer (NK) cells and showed an antitumor effect against lung metastatic B16-F10 cells which express only minimal amounts of the major histocompatibility complex class I (MHC-I).8 Therefore, the findings presented here indicate that the STING-LNP efficiently activated both MHC-I restricted and non-restricted antitumor immunity.
There is no sign that the breakthrough of cancer immunotherapy, led by the success of immune checkpoint inhibitors (ICIs), will stop. As of January 2021, anti-cytotoxic T lymphocyte-associated protein-4 (CTLA-4) antibody, anti-programmed cell death-1 (PD-1) antibodies and anti-programmed cell death ligand-1 (PD-L1) antibodies have been approved. However, only a small fraction of patients can receive the benefit of ICIs and a large portion do not respond to ICIs. Resistance to ICIs, especially anti-PD-1/PD-L1 therapy, is a major obstacle in the current status of cancer immunotherapy.10 The resistance can roughly be classified as primary resistance (primary escape) and acquired resistance (secondary escape) based on the time axis of occurrence, whereas the resistant mechanisms overlap.11 12 One of the resistant mechanisms involves the absence of antigen presentation due to the loss of MHC/HLA class I, neoantigen loss and incomplete antigen presentation.13 14 In cases of ICI-resistant tumors which are due to the MHC/HLA class I loss, neoantigen loss, and incomplete antigen presentation, the elimination by NK cells should be an effective strategy. Because the cytotoxicity of NK cells is independent of antigens, it would therefore appear that the activation of NK cells would be a useful strategy for overcoming ICI resistance.
Recent reports revealed that NK cells activated by stimulation of the STING pathway can be promising effector for antitumor responses.15 16 Although various types of STING agonist loaded nanoparticles have been used for enhancing T cell–mediated antitumor immunity,17–22 little attention has been given to activating NK cells by STING agonist loaded nanoparticles in cancer immunotherapy.8 The effect of NK cell activation by STING agonist loaded nanoparticles against anti-PD-1-resistant tumor has not been studied so far. In this study, we investigated the potential of activating NK cells by a STING-LNP to overcome the resistance to anti-PD-1 therapy. We developed a new type of STING-LNP which was mainly composed of YSK12-C4. YSK12-C4 is a synthesized cationic lipid and shows a high affinity for immune cells and can be used to effectively deliver nucleic acids to the cytosol of cells.23–26 B16-F10 cells were used as the tumor model in which antigen presentation is absent.27 Although monotherapy by anti-PD-1 showed no antitumor effect in a B16-F10 lung metastatic model mouse, interestingly, a therapy involving the STING-LNP combined with anti-PD-1 showed a synergistic antitumor effect. The findings suggest that the immune status in the B16-F10 lung metastasis model had undergone a change to an immunologically hot tumor as the result of NK cell activation derived from liver macrophages with the incorporated STING-LNP. The activated NK cells appeared to then produce IFN-γ, resulting in an increase in the expression of PD-L1 by cancer cells and immunosuppression via PD-1/PD-L1 interaction. The findings reported herein indicate that the STING-LNP could be a promising adjuvant system for overcoming PD-1 resistance and provide new insights regarding the innate immunity mediated by intravenously administered STING-LNP and the reduced anti-PD-1 resistance by NK cells.
Methods
Reagents, cell lines and mice
YSK12-C4 (6Z, 9Z, 28Z, 31Z)−19-(4-(dimethylamino)butyl) heptatriaconta-6,9,28,31-tetraen-19-ol was synthesized as previously described.23 Cholesterol was purchased from Avanti Polar Lipids (Alabaster, Alabama, USA). 1,2-dimirystoyl-sn-glycerol methoxyethylene glycol 2000 ether (PEG2000-DMG) was obtained from the NOF Corporation (Tokyo, Japan). STING agonist, cyclic di-GMP (c-di-GMP), was obtained from Yamasa Corporation (Chiba, Japan). Anti-mouse PD-1 (clone: RMP1-14) and the isotype control (clone: 2A3), anti-mouse NK1.1 (clone: PK136), anti-mouse CD8a (clone: 2.43) and anti-mouse IFN alpha/beta receptor subunit 1 (IFNAR1) (clone: MAR1-5A3) were purchased from Bio X Cell (West Lebanon, New Hampshire, USA). FITC anti-mouse CD3 (clone: 17A2), PE anti-mouse NK1.1 (clone: PK136), APC anti-mouse CD279 (PD-1) (clone: 29F.1A12), PE anti-mouse CD274 (PD-L1) (clone: 10F.9G2), PE/Cy7 anti-mouse CD69 (clone: H1.2F3), APC anti-mouse CD314 (NKG2D) (clone: CX5), FITC anti-mouse I-Ab (clone: AF6-120.1), PE anti-mouse CD11c (clone: N418), PE anti-mouse F4/80 (clone: BM8), purified anti-mouse CD16/32 (clone: 93), 7-AAD Viability Staining Solution, each isotype control and recombinant mouse IFN-β1 were purchased from BioLegend (San Diego, California, USA). Lycopersicon esculentum lectin, FITC conjugate tomato lectin was obtained from VECTOR Laboratories (Burlingame, California, USA). Clophosome-A (clodronate liposomes) was purchased from FormuMax Scientific (Sunnyvale, California, USA). Collagenase D from Clostridium histolyticum was obtained from Sigma-Aldrich (St. Louis, Missouri, USA). Recombinant murine IFN-γ was purchased from PeproTech (Rocky Hill, New Jersey, USA).
B16-F10-luc2 cells (Caliper Life Sciences, Hopkinton, Massachusetts, USA) were cultured in RPMI1640 containing 10% fetal bovine serum, 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), 1 mM sodium pyruvate and 4500 mg/L glucose.
Female C57BL/6N mice (6–9 weeks old) and female BALB/c nu/nu (6–7 weeks old) were purchased from Japan SLC (Shizuoka, Japan) and maintained under specific pathogen-free conditions. The use of mice was approved by the Ethics of Pharmaceutical Science Animal Committee of Hokkaido University (approval number: 16-0014). All experiments were carried out in accordance with the National University Corporation Hokkaido University Regulations on Animal Experimentation.
Preparation of STING-LNP
The STING-LNPs were prepared as described in previous reports.7 8 Briefly, 150 µL of 1 mM citrate buffer (pH 4.5) containing 500 nmol of c-di-GMP was added to 300 µL of 90% t-BuOH solution containing YSK12-C4 (340 nmol), cholesterol (60 nmol) and PEG2000-DMG (4 nmol) under vortex mixing. When stained with fluorescence, 2 nmol of 1,1′-dioctadecyl-3,3,3′,3′- tetramethylindodicarbocyanine (DiD) (0.5 mol%) was added to the 90% t-BuOH solution. The mixture was immediately moved to 1.6 mL of 1 mM citrate buffer (pH 4.5) under vortex mixing. After adding 4 mL of phosphate-buffered saline (PBS), the preparation was filtrated with an Amicon Ultra (molecular weight cut-off 100,000) and was replaced with PBS. The STING-LNPs were stored at 4°C. To measure the amount of c-di-GMP in the STING-LNPs, they were lysed by treatment with 5 mM deoxycholic acid and the absorbance at 252 nm (e=24,700) was measured. The recovery ratio was 11.5%±0.3%. The value is the mean±SEM (n=10).
The diameter, size distribution, polydispersity index (PDI), and zeta-potential of STING-LNP were determined with a ZETASIZER Nano (ZEN3600, Malvern Instruments, Malvern, WR, UK). The diameter, PDI, and zeta-potential of STING-LNP were 172±5 nm, 0.072±0.02 and 7.1±0.4 mV, respectively. The values are the mean±SEM (n=10).
Antitumor effect in the B16-F10-luc2 lung metastatic mouse model
The B16-F10-luc2 lung metastatic mouse model was prepared as reported previously.8 28 Mice were intravenously injected with B16-F10-luc2 cells (2×105 cells) via the tail vein. The mice were intravenously administered the STING-LNP (6 µg of c-di-GMP) or recombinant mouse IFN-β1 (14.6 ng). The dose, which was set based on the circulating blood volume of the mouse, was calculated from the body weight and the IFN-β1 concentration to be approximately 10 ng/mL. Anti-PD-1 (50 or 200 µg) was intraperitoneally injected into the mice. For the depletion of NK cells or CTLs, the mice were intraperitoneally injected with anti-NK1.1 (200 μg) or anti-CD8a (200 µg) on days 5, 9, 13 and 17. For the depletion of macrophages, the mice were intravenously injected with 200 µL of clodronate liposomes on days 5 and 11. For the inhibition of IFNAR1, the mice were intraperitoneally injected with anti-IFNAR1 (200 µg) on days 1, 5 and 9. The schedules for injection are described in the figures and the legends. After the collection of lungs, the lungs were completely homogenized and the luciferase activities were then measured using a luminometer (Luminescencer-PSN, ATTO, Japan). The values of relative light unit (RLU) per whole lung for the PBS-treated mice were set to 1.
Analysis of NK cells in the lung
The B16-F10-luc2 lung metastatic mouse model was prepared. On day 2, the mice were intravenously injected with the STING-LNP (6 µg of c-di-GMP). After 24 hours, RPMI1640 medium containing 1.25 mg/mL collagenase D was injected into the lung via trachea and the trachea was tied. The lungs were collected and incubated in RPMI1640 medium containing 1.25 mg/mL collagenase D for 30 min at 37°C. The lung cells were then dispersed using scissors and a syringe/18G and 20G needles. After washing the cells, the lymphocyte fraction was collected by Percoll density centrifugation (40% and 80% Percoll). The purified anti-mouse CD16/32 was added to the lymphocytes for blocking. The lymphocytes were then stained with FITC anti-mouse CD3, PE anti-mouse NK1.1, APC anti-mouse CD270 and the isotype controls. The lymphocytes which were stained with a 7-AAD Viability Staining Solution were analyzed by flow cytometry (Gallios, Beckman Coulter, Indianapolis, Indiana, USA). The FlowJo software was used for the data analysis.
Biodistribution of STING-LNP
The biodistribution of STING-LNP was performed as reported previously.29 Mice were intravenously administrated with DiD-labeled STING-LNP (total lipid amount: 320 nmol). After 1 hour, tissues (liver, spleen, lung and kidney) were collected and 20 mg of samples of tissues was homogenized with a Precellys 24 (Birthin Technologies, France) (4000–5000 rpm, 20–30 s) in a 1% sodium dodecyl sulfate solution. The supernatants were collected and the fluorescence intensity (FI) was measured using Infinite M200 (TECAN, Mannedorf, Switzerland) with λex=641 nm, λem=685 nm.
Cellular uptake of STING-LNP in the spleen
The cellular uptake of the STING-LNP in the spleen was performed following a previously reported method.30 Mice were intravenously injected with the DiD-labeled STING-LNP (total lipid amount: 320 nmol). After 1 hour, the spleens were collected and splenocytes were prepared. The purified anti-mouse CD16/32 was added to the splenocytes (1×106 cells) for blocking. The splenocytes were stained with FITC anti-mouse I-Ab, PE anti-mouse CD11c and the isotype controls. The splenocytes which were stained with a 7-AAD Viability Staining Solution were analyzed by flow cytometry (Gallios). The FlowJo software was used for the data analysis.
Distribution of STING-LNPs in the liver
Mice were intravenously injected with the DiD-labeled STING-LNP (6 µg of c-di-GMP). After 55 min, 10-time diluted FITC conjugate tomato lectin (200 µL) was intravenously injected to the mice. After 5 min, the liver was collected, counter-stained with Hoechst33342 and observed by confocal laser scanning microscopy (A1, Nikon, Tokyo, Japan).
Cellular uptake of STING-LNPs in the liver
Mice were intravenously injected with the DiD-labeled STING-LNP (6 µg of c-di-GMP). After 1 hour, the livers were collected, and the single cell suspensions were prepared. The lymphocyte fraction was collected by Percoll density centrifugation (40% and 80% Percoll). The purified anti-mouse CD16/32 was added to the lymphocytes for blocking. The lymphocytes were then stained with FITC anti-mouse I-Ab, PE anti-mouse F4/80 and the isotype controls. The lymphocytes which were stained with a 7-AAD Viability Staining Solution were analyzed by flow cytometry (Gallios). The FlowJo software was used for the data analysis.
Effect of macrophage depletion by clodronate liposomes on cytokine production
Mice were intravenously injected with 200 µL of clodronate liposomes. After 2 days, the mice were intravenously injected with the STING-LNPs (6 µg of c-di-GMP). After 2 hours, the blood and liver were collected. The concentration of IFN-β in the serum and mRNA expression in the liver were evaluated.
Statistical analysis
Comparisons between the two treatments were performed by an unpaired t-test. Comparisons between multiple treatments were made using analysis if variance (ANOVA), followed by Tukey-Kramer test. Interaction between the two factors was performed by two-factor factorial ANOVA (for synergistic effect). A p value of <0.05 was considered to be significant. The correlation was examined by Pearson’s correlation coefficient test.
Results
Preparation of the STING-LNP formulation
We used YSK12-C4 as the main component of the STING-LNPs. YSK12-C4 is a synthesized cationic lipid and shows high affinity to immune cells and can effectively deliver nucleic acids to the cytosol.23–26 A c-di-GMP was used as a STING agonist. The diameter, PDI and zeta-potential of STING-LNP were 172±5 nm, 0.072±0.02 and 7.1±0.4 nm, respectively. The size distribution showed a single narrow peak, clearly indicating that the STING-LNP was highly homogeneous (figure 1A). We also examined the 4°C storage stability of the STING-LNP for periods up to 360 days. The STING-LNP appears to be stable for 270 days at 4°C, as evidenced by the zeta-potential changing to negative on day 360 (figure 1B and online supplemental figure S1). Furthermore, we evaluated the antitumor effect of STING-LNP against mouse lung metastatic by B16-F10-luc2 cells, as previously reported.8 The STING-LNP treatment induced a significant antitumor effect (figure 1C).
Supplemental material
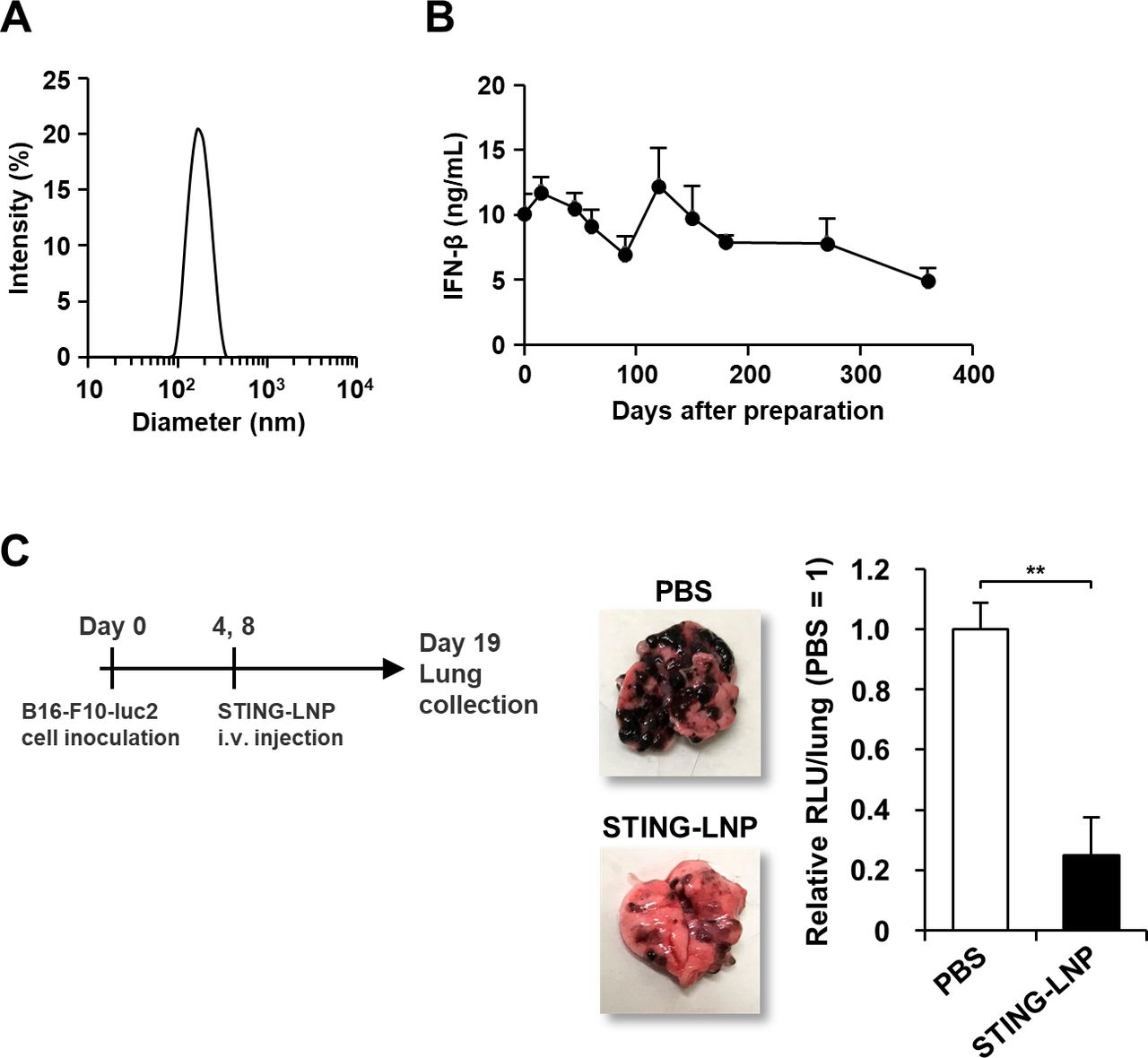
Characteristics and antitumor activity of the STING-LNP. (A) Particle size distribution of STING-LNPs based on light scattering intensity. (B) Temporal change in IFN-β production in the serum by 4°C stored STING-LNP. Mice were intravenously injected with the STING-LNP which were stored at 4°C. The x-axis shows days after the preparation of STING-LNP. Data are the mean+SEM (n=3). (C) Antitumor effect of STING-LNP against B16-F10 lung metastasis. The mice were intravenously injected with the STING-LNP (6 µg of c-di-GMP). The images represent collected lungs. The value of RLU per whole lung for the PBS-treated mice were set to 1. Data are the mean+SEM (n=4, **p<0.01). c-di-GMP, cyclic di-GMP; IFN-β, interferon-β; RLU, relative light unit; STING-LNP, lipid nanoparticle containing a stimulator of an interferon gene; PBS, phosphate-buffered saline.
A combination of the STING-LNP and anti-PD-1 shows a synergistic antitumor effect
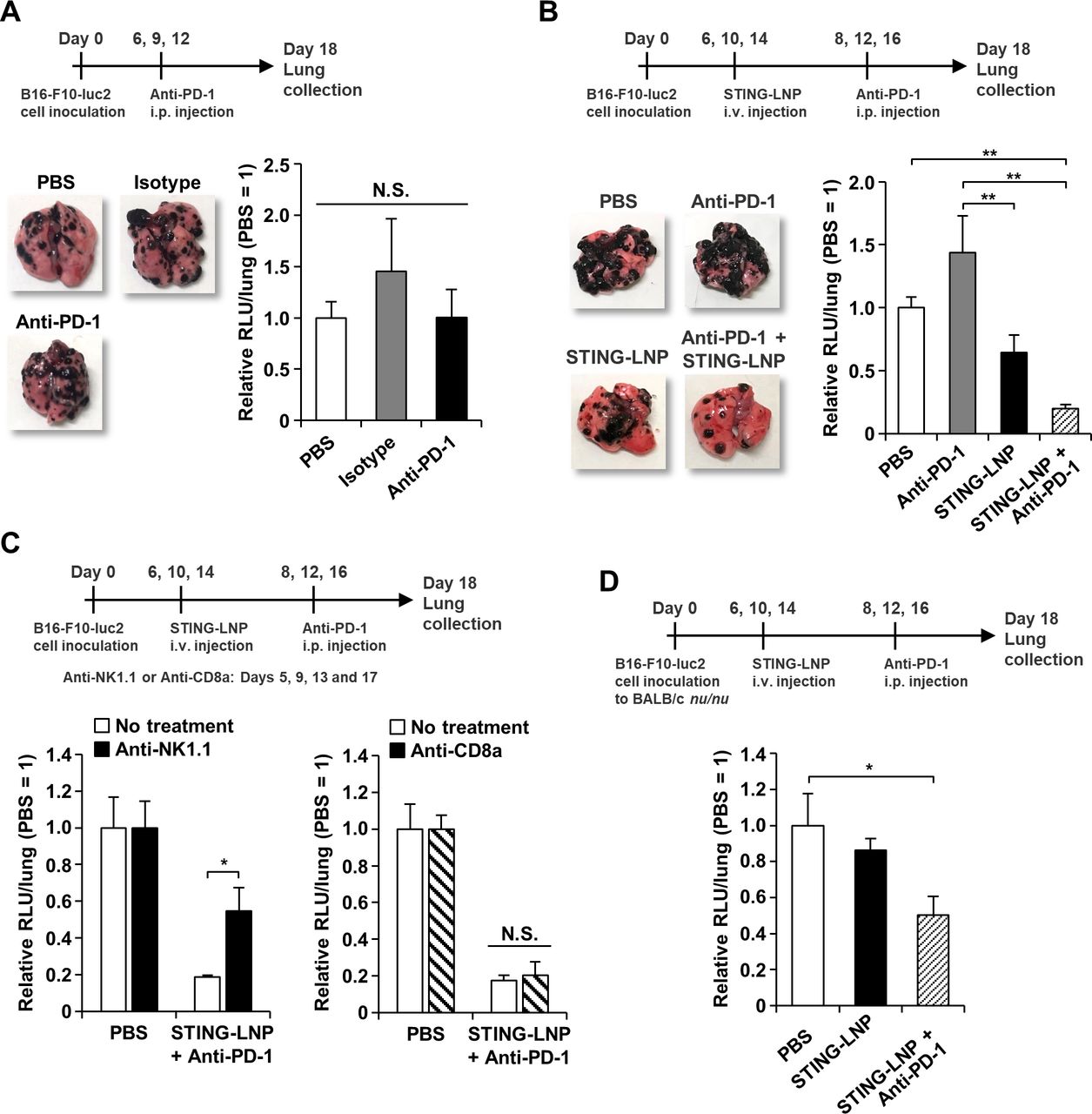
B16-F10 cells and 4T1 cells that were subcutaneously inoculated in mice are resistant to anti-PD-1 monotherapy,31 32 but information on anti-PD-1 resistance in a mouse model of lung metastasis of B16-F10 is not available. Anti-PD-1 monotherapy was performed following a typical protocol that showed efficacy (200 µg/mouse, 3 doses).33 The anti-PD-1 monotherapy showed no antitumor effect, indicating that the B16-F10 lung metastasis mouse was resistant to anti-PD-1 therapy (figure 2A).
We next investigated the effect of using a combination of the STING-LNP and anti-PD-1. The dose of anti-PD-1 was decreased to 50 µg because we found that the dose could be reduced to this level and still be effective. Anti-PD-1 monotherapy was showed no antitumor effect and the STING-LNP monotherapy showed only a moderate antitumor effect (figure 2B). Interestingly, the combination therapy by STING-LNP and anti-PD-1 significantly inhibited tumor growth and the antitumor effect was synergistic (p=0.00789) (figure 2B). The antitumor effect caused by the STING-LNP was abolished when the dose of c-di-GMP was decreased to 1 µg (online supplemental figure S2). Comparable effects were observed for doses of 3 and 6 µg of c-di-GMP. In contrast, in the combination therapy, an antitumor effect was observed, even at a dose of 1 µg of c-di-GMP, although this effect was not statistically significant (online supplemental figure S2). In addition, a slight, dose-dependent increase in the antitumor effect was observed. The result indicates that the resistance to anti-PD-1 can be reduced by the use of STING-LNPs.
We also assessed the contribution of NK cells and T cells to the antitumor effect by the combined therapy. In STING-LNP monotherapy against the same B16-F10 lung metastasis mouse model, the antitumor effect is mainly dependent on NK cells.8 When treated with anti-NK1.1, the antitumor effect by the combined therapy was significantly impaired (figure 2C). In contrast, the anti-CD8a treatment did not affect the antitumor effect (figure 2C). When we evaluated the antitumor effect in nude mice (BALB/c nu/nu), the combination therapy showed a significant antitumor effect (figure 2D). The results indicate that NK cells, but not CTL and CD4+ T cells, can be main effector cells for achieving antitumor activity by the combined therapy.
STING-LNP changes the immune status in B16-F10 lung metastasis from immunologically cold tumor to hot tumor
To analyze the immune status in the lung with B16-F10 metastasis, we measured gene expression at the mRNA level. The mRNA levels of CD3, CD4, NK1.1, PD-1 and IFN-γ were significantly increased by the treatment of STING-LNP (figure 3A). This suggests that, in a lung with B16-F10 metastasis, the number of NK cells increased as the result of the STING-LNP treatment, resulting in the main production of IFN-γ. Furthermore, the increase in CD3 and CD4 expression indicates that CD4+ T cells had infiltrated into the lung with B16-F10 metastasis. We also investigated the gene expression after the STING-LNP and anti-PD-1 combination therapy. As a result, NK1.1 and IFN-γ levels were increased in the STING-LNP monotherapy and the combination therapy compared with the PBS group (online supplemental figure S3). However, there was no difference between the STING-LNP monotherapy and the combination therapy. In addition, no changes in other gene expressions were observed. Thus, this fact suggests that the combination of anti-PD-1 may induce no additional activation of immune cells in a lung with a B16-F10 melanoma.

Analysis of lungs with B16-F10 metastasis after the STING-LNP treatment. (A) Gene expression at mRNA level in the lung with B16-F10 metastasis. Mice were intravenously injected with B16-F10-luc2 cells. The mice were intravenously injected with the STING-LNP (6 µg of c-di-GMP) on days 2, 4 and 8. On day 9, the lungs were collected, and the mRNA levels measured by RT-qPCR. The values for the PBS-treated mice were set to 1. Data are the mean+SEM (n=3, **p<0.01). (B) Analysis of NK cell population. (C) The NK1.1 expression in NK cells. (D) Analysis of PD-1+ NK cell population. B16-F10-luc2 cells were intravenously injected to mice. The mice were intravenously injected with the STING-LNP (6 µg of c-di-GMP) on day 2. On day 3, the lungs were collected, and the lymphocyte fraction obtained. The lymphocytes that were stained with anti-CD3, anti-NK1.1 and anti-PD-1 were analyzed by flow cytometry. NK cells were identified as CD3-NK1.1+ cells. Data are the mean+SEM (n=6–7, *p<0.05, **p<0.01). N.S., no significant difference. (E) Killing of B16-F10 cells by NK cells. NK cells from mice treated with PBS or the STING-LNP were mixed with B16-F10-luc2 cells at effector/target (E/T) ratio of 1:1, 5:1 and 10:1. Data are the mean+SEM (n=3, **p<0.01 vs PBS). (F) Analysis of gene expression at mRNA level in the B16-F10 colonies. The mice were intravenously injected with the STING-LNP (6 µg of c-di-GMP). The values for the PBS-treated mice were set to 1. Data are the mean+SEM (n=3, *p<0.05, **p<0.01). c-di-GMP, cyclic di-GMP; NK, natural killer; PD-1, programmed cell death 1; STING-LNP, lipid nanoparticle containing a stimulator of an interferon gene; PBS, phosphate-buffered saline.
Because the antitumor effect mediated by STING-LNPs is largely attributed to the activation of NK cells,8 NK cells in the lung with B16-F10 metastasis were analyzed by flow cytometry. Mice with B16-F10 lung metastasis were intravenously injected with STING-LNPs on day 2 and the lungs were collected on day 3 because the alteration of mRNA levels associated with NK cells was comparable between the 3-times injection and the 1-time injection (online supplemental figure S4). The percentage of NK cells (identified as CD3-NK1.1+ cells) was comparable between the PBS treatment and STING-LNP treatment (online supplemental figure S5 and figure 3B). In contrast, the expression of NK1.1 was significantly increased compared with the PBS treatment (figure 3C). In addition, the percentage of PD-1+ NK cells was also increased (figure 3D). The NK cells from mice that had been treated with the STING-LNP showed a significant enhancement in killing against B16-F10 cells (figure 3E). These findings suggest that NK cells in the lung with B16-10 metastasis were activated and expressed PD-1.
The STING-LNP treatment caused no change in the population of CD4+ T cells and CD8+ T cells (online supplemental figure S6). Although the population of CD69+CD4+ T cells was comparable between the PBS group and the STING-LNP group, the STING-LNP treatment caused a significant increase in the population of CD69+CD8+ T cells (online supplemental figure S6). The exposure of type I IFNs enhances the expression of CD69 in CD8+ T cells, whereas type I IFNs have only minor effects on CD4+ T cells.34 This suggests that the CD69+CD8+ T cells were not B16-F10-specific CTL because the contribution of CD8+ T cells should have a minor effect on antitumor activity (figure 2C,D). The collective facts suggest that T cells were activated by the STING-LNP treatment, but they did not function as effector cells for antitumor activity.
The exposure of type I IFNs and IFN-γ results in the upregulation of the expression of PD-L1 on cancer cells.35–37 Thus, we hypothesized that type I IFNs are induced by the treatment with the STING-LNPs and IFN-γ produced by activated NK cells upregulated the expression of PD-L1 in B16-F10-luc2 cells, resulting in immune suppression via PD-1/PD-L1. In in vitro cell cultures, the IFN-β or IFN-γ treatments resulted in a drastic upregulation in the expression of PD-L1 in B16-F10-luc2 cells (online supplemental figure S7). We also intravenously injected mice with B16-F10 lung metastasis with the STING-LNP on days 12, 14 and 16, and collected tumor colonies on day 18 (figure 3F). In order to accurately collect only tumor colonies, the STING-LNP was administered after the tumor colonies had grown. The analysis of mRNA expression showed a significant increase in NK1.1, PD-1, IFN-γ, PD-L1, NKG2D and FasL (figure 3F). The mRNA levels of CD11b and CD69 in mice that had been treated with the STING-LNP were not statistically significant compared with that of PBS but tended to be high. The gene expression levels of NK1.1, PD-L1, and IFN-γ increased in a dose-dependent manner, whereas no dose-dependent increase in PD-1 expression was observed (online supplemental figure S8). In addition, the intravenous administration of recombinant IFN-γ showed no antitumor effect, no NK cell activation and no increase in PD-L1 expression, indicating that the local production of IFN-γ is necessary (online supplemental figure S9). These results indicate that the expression of PD-L1 in the B16-F10 tumor was upregulated by IFN-γ exposure and that activated NK cells had infiltrated into the tumor colony.
STING-LNP induces systemic NK cell activation
We next examined systemic NK cell activation after the treatment of STING-LNP. The number of NK cells in the spleen was significantly reduced (online supplemental figure S10A), but the expression of NK1.1 was significantly increased (online supplemental figure S10B). The decrease in NK cells after activation was consistent with previous reports.8 38 The number of CD69+ NK cells was drastically increased (online supplemental figure S10C), and the expression of CD69 was also significantly increased (online supplemental figure S10D). Although the number of NKG2D+ NK cells was significantly decreased (online supplemental figure S10E), the expression of NKG2D itself was significantly increased (online supplemental figure S10F). In the case of PD-1, the number was significantly increased (online supplemental figure S10G), but the level of expression was comparable (online supplemental figure S10H). These collective findings indicate that systemic activation of NK cells was induced, and a part of the activated NK cells expressed PD-1.
Liver macrophages mainly internalize STING-LNP
To reveal where the STING-LNP actually functioned, we examined the biodistribution and the cellular uptake in tissue after 1 hour. The percentages of STING-LNP accumulation in the liver, spleen, lung and kidney were 74%, 6%, 3% and 1%, respectively (figure 4A). That is, the STING-LNPs had mainly accumulated in the liver.
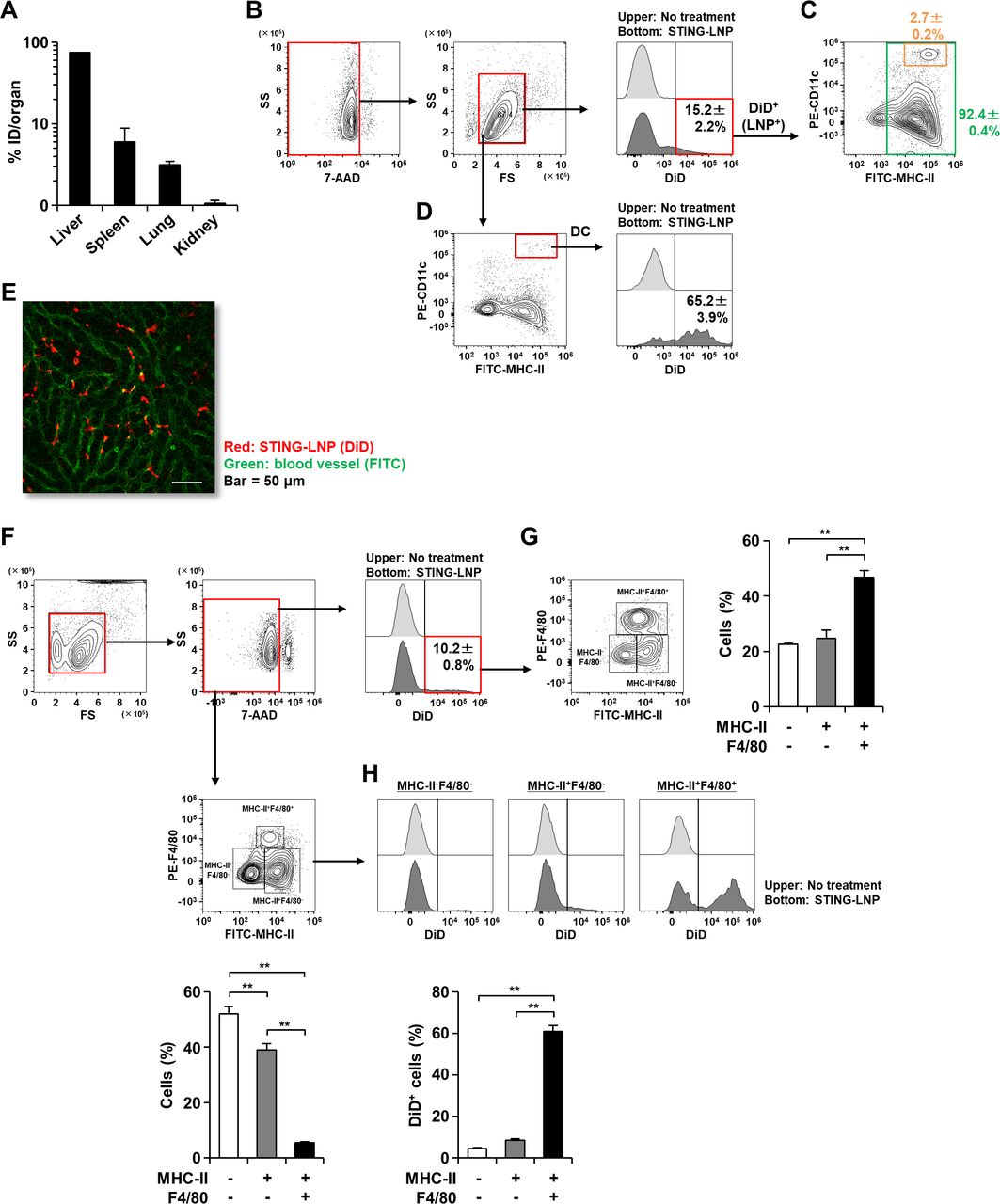
Analysis of STING-LNP distribution. Mice were intravenously injected with the DiD-labeled STING-LNP and tissues were collected after 1 hour. (A) Biodistribution of STING-LNP. The FIs of each tissue were measured. The values represent the accumulation rate to each organ with respect to the injected dose (ID). Data are the mean+SEM (n=3). (B) Gating strategy and percentage of DiD+ cells in all splenocytes. (C) Analysis of DiD+ cells on MHC-II/CD11c plot. (D) Percentage of DiD+ cells in DCs. The splenocytes were stained with anti-I-Ab and anti-CD11c and were then analyzed by flow cytometry. DCs were identified as MHC-II+CD11c+ cells. Data are the mean±SEM (n=3). (E) CLSM image of liver. Red and green represent STING-LNP (DiD) and blood vessel (FITC), respectively. Bar=50 µm. (F) Gating strategy and percentage of DiD+ cells in liver lymphocytes. (G) Analysis of DiD+ cells on MHC-II/F4/80 plot. (H) Percentage of DiD+ cells in MHC-II-F4/80- cells, MHC-II+F4/80- cells and MHC-II+F4/80+ cells. The lymphocyte fraction was collected from the liver. After staining with anti-I-Ab and anti-F4/80, the lymphocytes were analyzed by flow cytometry. Data are the mean+SEM (n=3, **p<0.01). FI, fluorescence intensity; DC, dendritic cell; MHC-I, major histocompatibility complex class I; CLSM, confocal laser scanning microscopy; STING-LNP, lipid nanoparticle containing a stimulator of an interferon gene.
We subsequently analyzed the cellular uptake of the STING-LNPs in the spleen because the spleen is a lymphatic organ. The percentage of cells with internalized STING-LNPs (DiD+ cells) in the total splenocytes was 15.2%±2.2% (figure 4B). Subsequently, the DiD+ cell population was analyzed on a CD11c/MHC-II plot. Most of the DiD+ cells were MHC-II+ cells, and 2.7%±0.2% of the cells were DCs (identified as MHC-II+CD11c+ cells) (figure 4C). In contrast, of the total DC population, 65.2%±3.9% of the cells with internalized STING-LNPs (figure 4D). These results indicate that the STING-LNP was essentially taken up by antigen presenting cells (APCs) and the majority of DCs contained the STING-LNPs.
We also analyzed the cellular uptake of STING-LNPs in the liver, the main organ for STING-LNP accumulation. We first visually assessed the distribution of STING-LNPs in the liver. Most of the red dots (STING-LNPs) were observed in the blood vessels (green dots) and not in the liver parenchyma (figure 4E). Moreover, the red dots were not colocalized with the green dots, but existed alone (figure 4E). This observation suggests that the STING-LNP can be taken up by blood cells in the liver. To examine this further, we collected the lymphocyte fraction from the liver at 1 hour after the STING-LNP treatment and carried out a flow cytometry analysis. The percentage of cells with internalized STING-LNPs (DiD+ cells) in the total liver lymphocytes was 10.2%±0.8% (figure 4F). The DiD+ cell population was subsequently analyzed on a F4/80/MHC-II plot. The MHC-II+F4/80+ cells (macrophages) showed a significantly higher cellular uptake of the STING-LNP than the MHC-II-F4/80- cells and the MHC-II+F4/80- cells (figure 4G). In addition, although the percentages of DiD+ cells in the total MHC-II-F4/80- cells and MHC-II+F4/80- cells were less than 10%, 60% of the total MHC-II+F4/80+ cells were DiD+ cells (figure 4H). These results suggest that the STING-LNPs were largely internalized by macrophages in the liver.
Liver macrophages with internalized STING-LNP initiate innate immune responses
We examined which cells are involved in the innate immunity caused by the STING-LNP. Contrary to our expectations, significant increases in the gene expression of interleukin-6 (IL-6), tumor necrosis factor-α (TNF-α) and IFN-β were observed only in the liver (figure 5A). The mRNA levels in the lung and spleen remained unchanged (figure 5A). Figure 5B shows the results for IFN-β production in the serum in the same mice. We subsequently plotted the values for each mouse using the y-axis as the mRNA expression of IFN-β in the liver and the x-axis as the IFN-β concentration in the serum (figure 5C). The coefficient of correlation was 0.977, showing a strong correlation. This suggests that the innate responses mediated by the STING-LNP can be initiated in the liver.

Analysis of major cells initiating innate immunity by STING-LNPs. (A) Gene expression at mRNA level. (B) IFN-β concentration in the serum. (C) Plot of relative mRNA level/IFN-β concentration. Mice were intravenously injected with B16-F10-luc2 cells. The mice were intravenously injected with the STING-LNPs (6 µg of c-di-GMP) on day 2. Tissue and blood samples were collected after 1.5 hours. The mRNA levels in each tissue and the IFN-β concentration in the serum were measured by RT-qPCR and ELISA, respectively. The values for the PBS-treated mice were set to 1 in the analysis of mRNA expression. Data are the mean+SEM (n=3, *p<0.05, **p<0.01). The relative mRNA levels of each mouse were plotted against the IFN-β concentrations for each mouse. The plot of PBS group was used the average values. (D) IFN-β concentration in the serum under macrophage depletion. (E) Gene expression at the mRNA level under macrophage depletion. Mice were intravenously injected with clodronate liposomes (Clodro). After 2 days, the mice were intravenously injected with the STING-LNPs (6 µg of c-di-GMP). After 2 hours, the blood and liver were collected. The values for the PBS-treated mice were set to 1 in the analysis of mRNA expression. Data are the mean+SEM (n=4, *p<0.05, **p<0.01). N.D., not detected. (F) The antitumor effect caused by the depletion of macrophages. Mice were then intravenously injected with the clodronate liposomes (Clodro) and the STING-LNP (6 µg of c-di-GMP). The mice were also intraperitoneally injected with 50 µg of anti-PD-1. The images represent collected lungs. The value of RLU per whole lung for the mice group that had not been treated with the clodronate liposomes (no treatment) was set to 1. Data are the mean+SEM (n=3–4, *p<0.05, **p<0.01). IFN, interferon; PD-1, programmed cell death 1; RLU, relative light unit; STING-LNP, lipid nanoparticle containing a stimulator of an interferon gene; PBS, phosphate-buffered saline.
We subsequently investigated the effect of macrophages on the innate responses in the liver by the STING-LNP. The level of IFN-β in the serum and the mRNA expression in the liver were measured under conditions in which macrophages were depleted by using clodronate liposomes. The efficiency of depletion of macrophages by clodronate liposomes in the liver, spleen and lung were 87%, 58% and 30%, respectively (online supplemental figure S11). The IFN-β level in the serum was significantly impaired under the depletion of macrophages (figure 5D). The value dropped to 1/16 compared with the value without the depletion of macrophages. In addition, the IL-6, TNF-α and IFN-β expressions were also largely impaired under conditions of depleted macrophages (figure 5E). The facts indicate that the liver macrophages had a major role in the induction of innate immune responses by the STING-LNPs.
We also evaluated the effect of macrophage depletion on the antitumor effect (figure 5F). The use of clodronate liposomes for macrophage depletion showed no effect on the activity of NK cells.39 The clodronate liposome treatment significantly inhibited the antitumor effect achieved by the STING-LNP monotherapy, suggesting that liver macrophages contributed to the antitumor effect exerted by the STING-LNP (figure 5F). In contrast, interestingly, the antitumor effect observed for the combination therapy of the STING-LNP and anti-PD-1 was significantly enhanced by the clodronate liposome treatment (figure 5F). This indicates that the macrophages inhibit the antitumor effect caused by the anti-PD-1.
The effect of type I IFN on antitumor activity
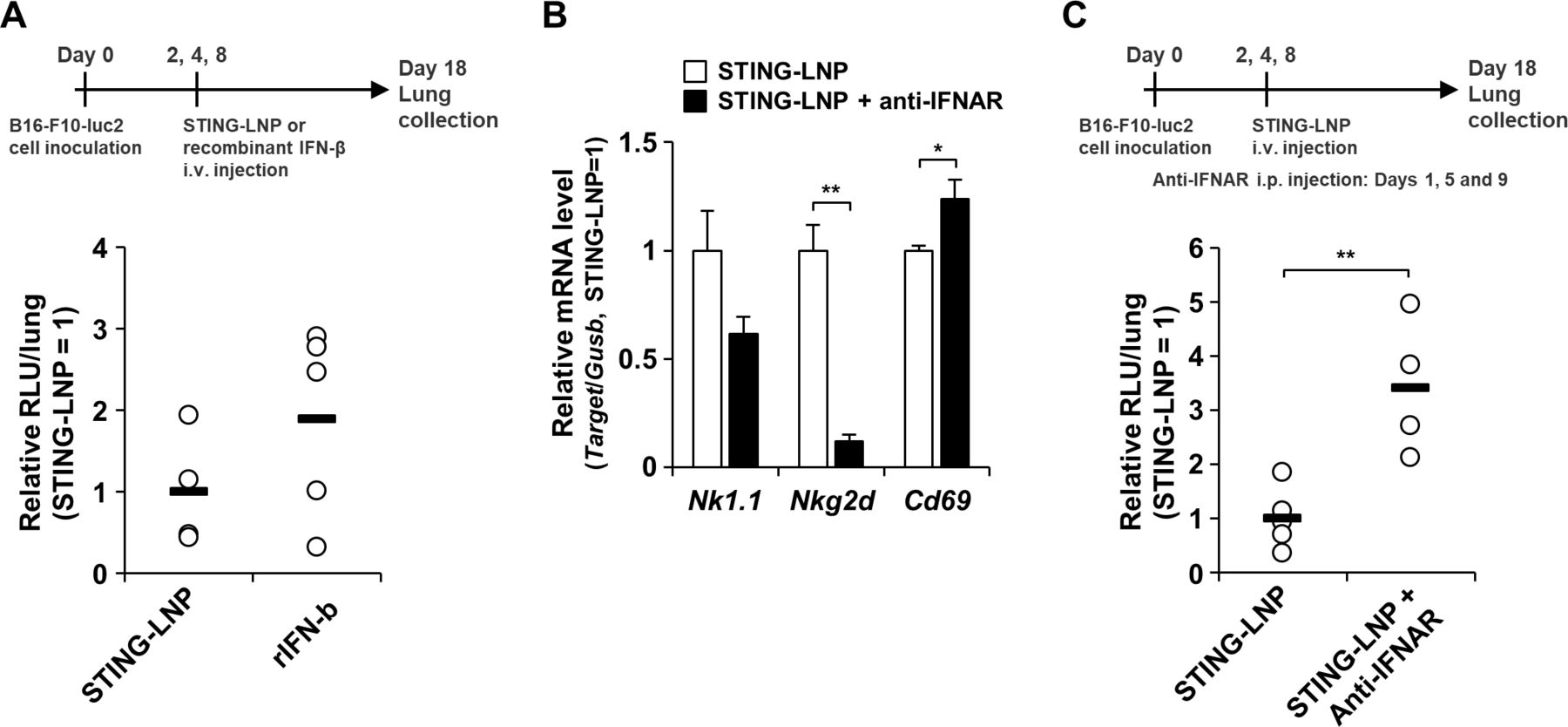
Finally, we investigated the effect of type I IFN on antitumor activities. Although there was no statistical difference in the antitumor effect between the two treatments, the antitumor effect for the STING-LNP tended to be higher than that for the recombinant IFN-β (figure 6A). We also examined the mRNA level associated with NK cells in the lung after the STING-LNP treatment under conditions of an IFNAR blockade. Under the IFNAR blockade, NK1.1 expression tended to be reduced and NKG2D expression was significantly reduced (figure 6B), indicating that type I IFN signaling contributed to the NK cell activation by the STING-LNP in the lung with the B16-F10 metastatic melanoma. On the other hand, the CD69 expression was significantly elevated under conditions of the IFNAR blockade (figure 6B). We next assessed the antitumor effect of the STING-LNP under conditions of the IFNAR blockade. The IFNAR blockade significantly inhibited the antitumor effect by the STING-LNP (figure 6C). These results indicate that the signaling pathway for type I IFNs contributes to the antitumor activity caused by the STING-LNP.
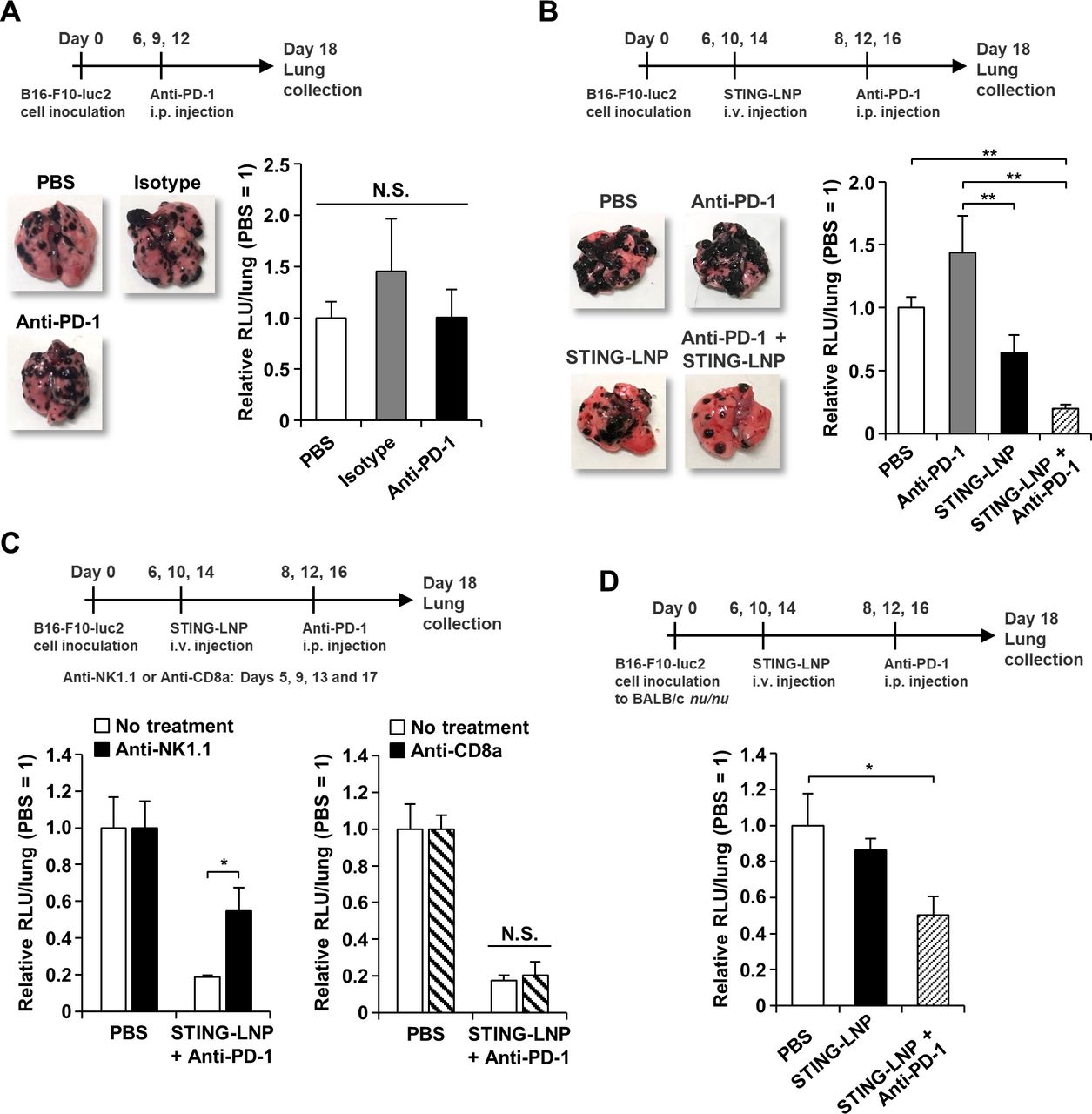
Combination therapy against anti-PD-1-resistant B16-F10 lung metastasis. (A) Antitumor effect of anti-PD-1 monotherapy against B16-F10 lung metastasis. The mice were intraperitoneally injected with 200 µg of anti-PD-1. The images represent collected lungs. The value of RLU per whole lung for the PBS-treated mice was set to 1. Data are the mean+SEM (n=4). N.S., no significant difference. (B) Antitumor effect of combination therapy against B16-F10 lung metastasis. The mice were intravenously injected with the STING-LNP (6 µg of c-di-GMP) and were intraperitoneally injected with 50 µg of anti-PD-1. The images represent collected lungs. The value of RLU per whole lung for the PBS-treated mice was set to 1. Data are the mean+SEM (n=8–10, **p<0.01). (C) The effect of NK cell or CTL depletion on the antitumor activity by the combination therapy. Mice with B16-F10 lung metastasis were intraperitoneally injected with anti-NK1.1 (200 μg) or anti-CD8a (200 µg). The value of RLU per whole lung for the PBS-treated mice was set to 1. Data are the mean+SEM (n=3–7, *p<0.05). (D) Antitumor effect of combination therapy against B16-F10 lung metastasis in BALB/c nu/nu. The mice were intravenously injected with the STING-LNP (6 µg of c-di-GMP) and were intraperitoneally injected with 50 µg of anti-PD-1. The value of RLU per whole lung for the PBS-treated mice was set to 1. Data are the mean+SEM (n=5–6, *p<0.05). CTL, cytotoxic T lymphocyte; PD-1, programmed cell death 1; RLU, relative light unit; STING-LNP, lipid nanoparticle containing a stimulator of an interferon gene; PBS, phosphate-buffered saline.
Discussion
The findings presented in this study demonstrate that the STING-LNPs activated NK cells and converted the immune status from immunologically cold to immunologically hot, resulting in reducing the anti-PD-1 resistance and a synergistic antitumor effect in a B16-F10 lung metastasis model. NK cells, but not CD8+ T cells and CD4+ T cells, can be dominant effector cells in the synergistic antitumor effect. In addition, the innate immune responses were initiated by liver macrophages that contained STING-LNPs. We summarize the mechanism regarding the synergistic antitumor tumor immunity by NK cell activation in figure 7 and is discussed below.

Summary for reducing anti-PD-1 resistant by STING-LNP. (1) The intravenously injected STING-LNPs accumulate in the liver and are taken up by liver macrophages, leading to the activation of the STING pathway. (2) The liver macrophages secret type I IFNs to the blood circulation. (3) The type I IFNs activate systemic NK cells such as in the spleen and the lung. A portion of NK cells express PD-1. (4) The activated NK cells produce IFN-γ in the B16-F10 tumor. (5) The exposure of type I IFNs and IFN-γ promotes the expression of PD-L1 on the cancer cells, leading to the establishment of immunosuppression via PD-1/PD-L1 axis between PD-1+ NK cells and cancer cells. (6) An anti-PD-1 treatment blocks the immunosuppression, resulting in the reactivation of NK cells and the development of cytotoxicity against cancer cells. c-di-GMP, cyclic di-GMP; IFN, interferon; NK, natural killer; PD-1, programmed cell death 1; STING-LNP, lipid nanoparticle containing a stimulator of an interferon gene.
It should be noted that the starting point of immune responses should be the liver, whereas the responses for eliminating tumor cells would occur in the lung. After the STING-LNP treatment, no IL-6, TNF-α and no IFN-β production were detected in the spleen and the lung, whereas a large increase in the production of their cytokines was observed in the liver (figure 5A–C). This result provides evidence to show that the STING-LNP that accumulated in the lung with B16-F10 did not cause an innate immune response. The depletion of liver macrophages largely reduced both cytokine production and antitumor activity (figure 5D–F). If the STING-LNP were to directly function in the lung, then cytokine production and antitumor activity would be expected to be maintained, even in cases of treatment with clodronate liposomes. This is because the clodronate liposome treatment had no influence on the population of lung macrophages (online supplemental figure S11). In addition, the type I IFN signaling pathway largely contributed to the observed NK cell activation and antitumor activity (figure 6B,C). These facts indicate that the STING-LNP that had accumulated in the lung did not initiate an immune response. We therefore concluded that the innate immune response in the liver led to the activation of lung NK cells, resulting in the induction of antitumor activity.
Our findings demonstrate the importance of liver as a target organ for effective induction of innate immunity. When nanoparticles are intravenously injected, up to 90% accumulate in the liver and are then cleared. Liver macrophages, Kupffer cells, are largely responsible for this clearance.40 41 Generally, the liver and Kupffer cells are major barriers to the delivery of a drug to a target. In contrast, the spleen is considered to be the main organ for controlling immune responses, when nanoparticles are intravenously injected.28 29 42–44 If therefore follows that the liver and Kupffer cells were not targeted by nanoparticles to activate cancer immunity. Meanwhile, the liver has an important role as a lymphoid organ in terms of the detection of pathogens entering our body.45–47 The main role of liver macrophages is in the detection, capture and clearance of pathogens and consist of Kupffer cells and monocyte-derived liver macrophages.48 The main player is Kupffer cells that represent around 30% of the non-parenchymal cells in the liver and 90% of all tissue macrophages.47 Kupffer cells reside in the liver sinusoids, namely, the blood stream, placing them in an ideal position for sensing antigens and pathogens. Although there is no report regarding the induction of NK cell activation via the STING pathway in the Kupffer cells, it has been reported that the activation of the STING pathway in the Kupffer cells contributes to the antitumor immunity against hepatocellular carcinomas and the control of cytomegalovirus infections in the liver, in which type I IFNs play an important role.49 50 Type I IFNs can strongly prime the cytotoxicity of NK cells. Therefore, it appears that the STING-LNPs activated the STING pathway in Kupffer cells and the activated Kupffer cells produced type I IFNs, thus leading to systemic NK cell activation. In addition, the findings provide the first demonstration to show that Kupffer cells are a potential target for adjuvant delivery.
Interestingly, the macrophages appeared to have a negative role in the STING-LNP and anti-PD-1 combination therapy. Macrophages capture anti-PD-1 via Fc/Fcγ receptors, leading to a reduction in antitumor activity caused by anti-PD-1.51 In addition, anti-PD-1 is systemically distributed after intraperitoneal injection.52 Thus, the clodronate liposome treatment would inhibit the internalization of anti-PD-1 by macrophages, thus resulting in an enhancement in the antitumor effect. It is possible that the activation of macrophages by the STING-LNP may also enhance the engulfment of anti-PD-1.
The findings reported herein suggest that, among the activated NK cells, PD-1+ NK cells are critical effector cells for killing B16-F10 cells in the lung. The contribution of CD4+ T cells and CD8+ T cells would be expected to be minor (figure 2C,D and online supplemental figure S6), whereas NK cells would be expected to be largely involved in the antitumor effect (figures 2C, 6B,C, online supplemental figures S3 and S4). The importance of PD-1+ NK cells in the antitumor effect was deduced from the dose–response data for antitumor effects (online supplemental figure S2) and mRNA levels in the tumor colony (online supplemental figure S8). The STING-LNP monotherapy resulted in an increase in the expression of NK1.1, PD-L1 and IFN-γ and this increase was dose-dependent, resulting in dose-dependent NK cell activation and the expression of PD-L1 in B16-F10 cells (online supplemental figure S8). In contrast, the PD-1 expression was not dose-dependent, and a constant PD-1 expression was observed (online supplemental figure S8). In this situation, the function of PD-1+ NK cells appeared to be inhibited by the PD-L1 produced in B16-F10 cells. Furthermore, the NK activation at a c-di-GMP dose of 1 µg was insufficient for inducing an antitumor effect because the antitumor effect disappeared when the 1 µg dose was administered (online supplemental figure S2A). In contrast, the combination therapy showed an antitumor effect even at a c-di-GMP dose of 1 µg (online supplemental figure S2B). The antitumor effect can be attributed to the lack of inhibition of PD-1+ NK cells. Thus, these results indicate that the activated NK cells eliminate cancer cells, but that PD-1+ NK cells can eliminate them more strongly even if the population of PD-1+ NK cells in the lung with B16-F10 was small (figure 3D). Recent studies have emerged to show that PD-1 is expressed on human NK cells in several cancers and that PD-1 expression inhibits the function of NK cells against tumors via a PD-1/PD-L1 axis.53–55 In addition, the PD-1+ NK cells are the most active cells for killing tumor cells, and the blockade of PD-1/PD-L1 axis drastically improved the antitumor effect, even though only a part of the NK cells expressed PD-1.56 That is, the PD-1/PD-L1 axis can have a quite important role in the antitumor activity mediated by NK cells. However, it is true that the number of PD-1+ NK cells was lower than we expected. If the PD-1+ NK cells made a large contribution to the antitumor activity, it is likely that the number of cells could be easily increased. Therefore, if PD-1+ NK cells could be activated more efficiently, the antitumor activity would be expected to be correspondingly enhanced.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Effect of type I IFN on the antitumor activity by STING-LNP. (A) Comparison of the antitumor effect between the STING-LNP and the recombinant IFN-β. The mice were intravenously injected with the STING-LNP (6 µg of c-di-GMP) or recombinant IFN-β (rIFN-β) (14.6 ng). The RLU value per whole lung for the STING-LNP-treated mice was set to 1. Data are the mean+SEM (n=4–5). (B) Mice with B16-F10 lung metastasis were intraperitonially injected with 200 µg of anti-IFNAR on day 1 and intravenously injected with the STING-LNP (6 µg of c-di-GMP) on day 2 and mRNA levels were measured on day 3. The values for the STING-LNP without the anti-IFNAR group were set to 1 in the analysis for mRNA expression. Data are the mean+SEM (n=3, *p<0.05, **p<0.01). (C) Mice with B16-F10 lung metastasis were intraperitonially injected with 200 µg of anti-IFNAR and intravenously injected with the STING-LNP (6 µg of c-di-GMP). The RLU value per whole lung for the STING-LNP without anti-IFNAR group was set to 1. Data are the mean+SEM (n=4–5, **p<0.01). IFN, interferon; RLU, relative light unit; STING-LNP, lipid nanoparticle containing a stimulator of an interferon gene.
Our results indicate that the signaling pathway for type I IFNs strongly contributed to the NK cell activation in the lung in the case of the B16-F10 melanoma (figure 6B) and the antitumor activity caused by the STING-LNP (figure 6C). On the other hand, it appears that the recombinant IFN-β treatment failed to induce the same antitumor activity as the STING-LNP (figure 6A). Liang et al also reported that the the intratumoral injection of recombinant IFN-β induces an antitumor effect, while an intravenous injection did not.57 These facts indicate that recombinant IFN-β only is not sufficient to induce a strong antitumor effect similar to that for the STING-LNP. Since the STING-LNP can induce the production of IFN-α in addition to IFN-β, the signaling pathway for type I IFNs may cause a strong stimulation compared with only the recombinant IFN-β. The difference in stability and biodistribution between the STING-LNP and the recombinant IFN-β may have an influence on the stimulation of the signaling pathway. Collectively, these findings suggest that the stimulation of the signaling pathway of type I IFNs by the STING-LNP has advantages in terms of activating NK cells and inducing antitumor activity.
Our results suggest that the activated NK cells produced IFN-γ in the B16-F10 tumor tissue, leading to an increase in the expression of PD-L1 in the B16-F10 cells (figure 3, online supplemental figure S9). In addition, type I IFNs produced by the liver macrophages appear to cause an increase in the expression of PD-L1 (figure 3F). The engagement of immunosuppression via the PD-1/PD-L1 axis between PD-1+ NK cells and B16-F10 cells can be then established, resulting in the appearance of an antitumor effect by anti-PD-1 (figure 2B). It is well known that the exposure of IFN-γ enhances the expression of PD-L1 on cancer cells35 36 and that IFN-γ is produced by CTL.58 Until recently, it was assumed that NK cells were not involved, but that IFN-γ secreted by NK cells increased the expression of PD-L1 in cancer cells.59 The blockade of the PD-1/PD-L1 axis enhances the antitumor activity of NK cells.56 59 Although our results are consistent with these conclusions, an important point to emphasize is the fact that we first found a series of these responses when a combination of STING-LNP and anti-PD-1 was used. That is, the mechanism by which IFN-γ is produced by activated NK cells induces the expression of PD-L1 on cancer cells and establishes immunosuppression via the PD-1/PD-L1 axis should be a promising strategy for improving PD-1 resistance.
The high level of type I IFNs (figure 1B) can be effective for producing a strong antitumor immunity, while it appears to hamper a comparable use in humans. We also reported that the levels of IL-6 and TNF-α in serum were high.8 The results of a biochemical examination of a blood sample indicates that the STING-LNP treatment resulted in the development of liver toxicity (online supplemental figure S12). Since the appearance of the mice after 2 days was the same as that of the control group, we conclude that this toxicity is likely transient. However, this nevertheless is an important issue that will need to be addressed prior to studies regarding clinical applications. Changing STING agonists can be a potent strategy. Because the structure of the STING agonist influences the complex structure of STING and agonists.60 Furthermore, cytokine profiles of type I IFNs, IL-6 and TNF-α can differ depending on the agonist structure.61 This strategy can reduce the inflammation that causes liver toxicity while also maintaining the antitumor activity.
In conclusion, we first demonstrated that a STING-LNP treatment reduced the PD-1 resistance in B16-F10 lung metastasis. The mechanism responsible for this provided new insights into this problem: the production of type I IFNs by STING-LNP was found to be initiated in the liver and liver macrophages were the main cell population that produce the type I IFNs; the mechanism by which IFN-γ produced by activated NK cells induced PD-L1 expression on cancer cells and establishes immunosuppression via PD-1/PD-L1 axis can improve PD-1 resistance. Collectively, the STING-LNP represents a promising candidate for combination therapy with anti-PD-1 against tumors without MHC/HLA class I loss, neoantigen loss and incomplete antigen presentation such that NK cells are effector cells.
Data availability statement
Data are available on reasonable request.
Ethics statements
Ethics approval
All animal experiments were approved by the Ethics of Pharmaceutical Science Animal Committee of Hokkaido University (approval number: 16-0014). All experiments were carried out in accordance with National University Corporation Hokkaido University Regulations on Animal Experimentation.
Acknowledgments
The Japanese Government (MEXT), Platform Project for Supporting in Drug Discovery and Life Science Research (Platform for Drug Discovery, Informatics, and Structural Life Science) from the Japan Agency for Medical Research and Development (AMED) and Hokkaido University, Global Facility Center (GFC), Pharma Science Open Unit (PSOU), funded by MEXT under 'Support Program for Implementation of New Equipment Sharing System'. We also appreciate Dr Milton S. Feather for his helpful advice in inspecting the English in this manuscript.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
TN and TS contributed equally.
Contributors TN and HH designed the research, conducted the data analysis and wrote the manuscript. TS mainly performed the experiments. NT carried out the STING-LNP preparation and a part of experiment of antitumor effect. RE and SS performed the gene expression analysis and antitumor effect that appears in the revised version of this paper. YS performed the synthesis of YSK12-C4. MH and YH provided c-di-GMP.
Funding JSPS KAKENHI (Grant Numbers 16K15102, 17H03974, 18K19888 and 20H03373) (TN), the Special Education and Research Expenses from the Ministry of Education, Culture, Sports, Science and Technology (HH) and Ono Pharmaceutical (TN and HH).
Competing interests TN and HH received research funding from Ono Pharmaceutical.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.