Article Text

Abstract
Expanding the US Food and Drug Administration–approved indications for immune checkpoint inhibitors in patients with cancer has resulted in therapeutic success and immune-related adverse events (irAEs). Neurologic irAEs (irAE-Ns) have an incidence of 1%–12% and a high fatality rate relative to other irAEs. Lack of standardized disease definitions and accurate phenotyping leads to syndrome misclassification and impedes development of evidence-based treatments and translational research. The objective of this study was to develop consensus guidance for an approach to irAE-Ns including disease definitions and severity grading. A working group of four neurologists drafted irAE-N consensus guidance and definitions, which were reviewed by the multidisciplinary Neuro irAE Disease Definition Panel including oncologists and irAE experts. A modified Delphi consensus process was used, with two rounds of anonymous ratings by panelists and two meetings to discuss areas of controversy. Panelists rated content for usability, appropriateness and accuracy on 9-point scales in electronic surveys and provided free text comments. Aggregated survey responses were incorporated into revised definitions. Consensus was based on numeric ratings using the RAND/University of California Los Angeles (UCLA) Appropriateness Method with prespecified definitions. 27 panelists from 15 academic medical centers voted on a total of 53 rating scales (6 general guidance, 24 central and 18 peripheral nervous system disease definition components, 3 severity criteria and 2 clinical trial adjudication statements); of these, 77% (41/53) received first round consensus. After revisions, all items received second round consensus. Consensus definitions were achieved for seven core disorders: irMeningitis, irEncephalitis, irDemyelinating disease, irVasculitis, irNeuropathy, irNeuromuscular junction disorders and irMyopathy. For each disorder, six descriptors of diagnostic components are used: disease subtype, diagnostic certainty, severity, autoantibody association, exacerbation of pre-existing disease or de novo presentation, and presence or absence of concurrent irAE(s). These disease definitions standardize irAE-N classification. Diagnostic certainty is not always directly linked to certainty to treat as an irAE-N (ie, one might treat events in the probable or possible category). Given consensus on accuracy and usability from a representative panel group, we anticipate that the definitions will be used broadly across clinical and research settings.
- immunotherapy
- autoimmunity
- guidelines as topic
- clinical trials as topic
- translational medical research
Data availability statement
All data relevant to the study are included in the article.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Immune checkpoint inhibitors (ICIs) have revolutionized management in a variety of tumor types and the number of patients being treated with these agents is rising dramatically.1 Since 2011, seven agents (ipilimumab, pembrolizumab, nivolumab, cemiplimab, atezolizumab, durvalumab, avelumab) targeting immune checkpoints have been approved by the US Food and Drug Administration for more than 60 indications.2–5 It is estimated that now over 233,000 patients with cancer are now eligible for treatment with an ICI annually in the USA alone.6 ICIs are being used not only as single agent therapy, or in combination with one another, but increasingly with chemotherapy, targeted therapy, or radiation.7 8
Side effects, termed immune-related adverse events (irAEs), limit the utilization and therapeutic potential of ICIs. The spectrum of irAEs and neurologic irAEs (irAE-Ns), sometimes called nirAE or nAE in the literature, has been reviewed.9–12 Incidence of irAE-Ns in patients treated with immunotherapy is estimated at approximately 1%–12%, with the peripheral nervous system (PNS) affected twice as commonly as the central nervous system (CNS).10 13–15 IrAE-Ns, along with irMyocarditis, have higher fatality rates than other irAEs.16 Oncologic societies developed initial consensus guidance statements regarding irAEs.17–20 However, a lack of standardized disease definitions causes syndrome misclassification, impedes further clinical and research progress,21 and can have significant downstream consequences in the management of patients.19 Additionally, the Common Terminology Criteria for Adverse Events (CTCAE) are a set of criteria that were developed to classify adverse events associated with chemotherapy and were not designed to accurately capture irAEs. CTCAE grade and irAE severity do not always correlate, underscoring the urgent need for refined CTCAE criteria, tailored for immunotherapy .22
Here we present consensus disease definitions for diagnosis and severity grading of irAE-Ns. Guidance statements were developed for an approach to irAE-Ns along with disease-specific definitions for CNS and PNS irAE-Ns.
Methods
A working group of neurologists (LBB, BKC, ACG, JH) drafted irAE-N guidance statements, disease definitions, and severity criteria, which were then reviewed by a panel of neurologists, oncologists, neuro-oncologists and irAE subspecialists (AAA through LZ). A modified Delphi consensus process was used, with two rounds of anonymous ratings by panelists and two virtual meetings to discuss controversial areas. Panelists rated the content for usability, appropriateness, and accuracy on 9-point scales and provided free text comments in an electronic survey. The working group aggregated survey responses and incorporated free text comments into revised definitions. Consensus based on numeric ratings was determined using the RAND/UCLA Appropriateness Method.23 Briefly, group medians were categorized into ranges (1–3 not usable, 4–6 uncertain, 7–9 usable). Agreement was defined as <1/3 of ratings outside the 3-point range containing the median. Consensus was reached when the median rating fell in the 7–9 range with agreement. Items that reached consensus in round 1 and did not undergo substantial revisions were not re-rated. The Delphi process was exempted by the Massachusetts General Brigham Institutional Review Board (Protocol #2020P003032).
Results
The Delphi panel consisted of 30 members who accepted invitations to participate, from American academic medical centers in the northeast (20), mid-west (2), south/southeast (4) and west (3), as well as one international oncologist. Of the 30 participants, 27 completed the round 1 survey and 24 completed the round 2 survey. The 27-member panel included neurologists (12), oncologists (8), neuro-oncologists (3) and irAE subspecialists (4). The panel first identified the following unmet needs for irAE-N disease definitions (% of panel members identifying the issue): (1) identifying subclinical or mild disease (85%); (2) recognizing the spectrum of presentations (96%); (3) differentiating irAE-Ns from alternative etiologies (89%); (4) grading irAE-N severity (100%); (5) classification of patients for cohort studies (85%); (6) classification of irAE-N phenotype for translational research (85%); and (7) adjudication of irAE-Ns in clinical trials (96%). Consensus guidance statements and disease definitions were developed with the goal of fulfilling unmet needs in these areas.
The first round of the Delphi included a total of 53 rating scales (6 general guidance statements, 18 PNS disease definition components, 24 CNS disease definition components, 3 severity criteria, and 2 clinical trial adjudication statements); of these, 41 (77%) received first-round consensus. Round 2 included ratings of 24 revised components and two new general guidance statements; 26 out of 26 (100%) received consensus in round 2. Medians and ranges from the Delphi process are presented with the consensus guidance statements and disease definition components in the tables.
Consensus statements
Approach to irAE-N diagnosis and general guidance statements
The workflow outlined in figure 1 first broadly classifies irAE-Ns into one of four central or three PNS core syndromes. Each irAE-N diagnosis consists of the following components: (1) core syndrome and syndrome subtype, (2) level of diagnostic certainty, (3) severity grading (reflecting the maximum severity of the irAE-N), (4) exacerbation of prior condition or de novo presentation, (5) antibody association, and (6) concurrent neurologic or non-neurologic irAEs (figure 1). These descriptions are intended to be dynamic: as the diagnostic workup evolves and a patient undergoes further evaluation, including by a neurologist when appropriate, the diagnosis may be updated or made more specific. For example, a central irAE-N may initially be classified by the type of CNS disorder (meningitis, encephalitis, vasculitis, or demyelinating disease) and then the specific subtype (ie, limbic encephalitis or optic neuritis) (median consensus score 8, range 3–9).

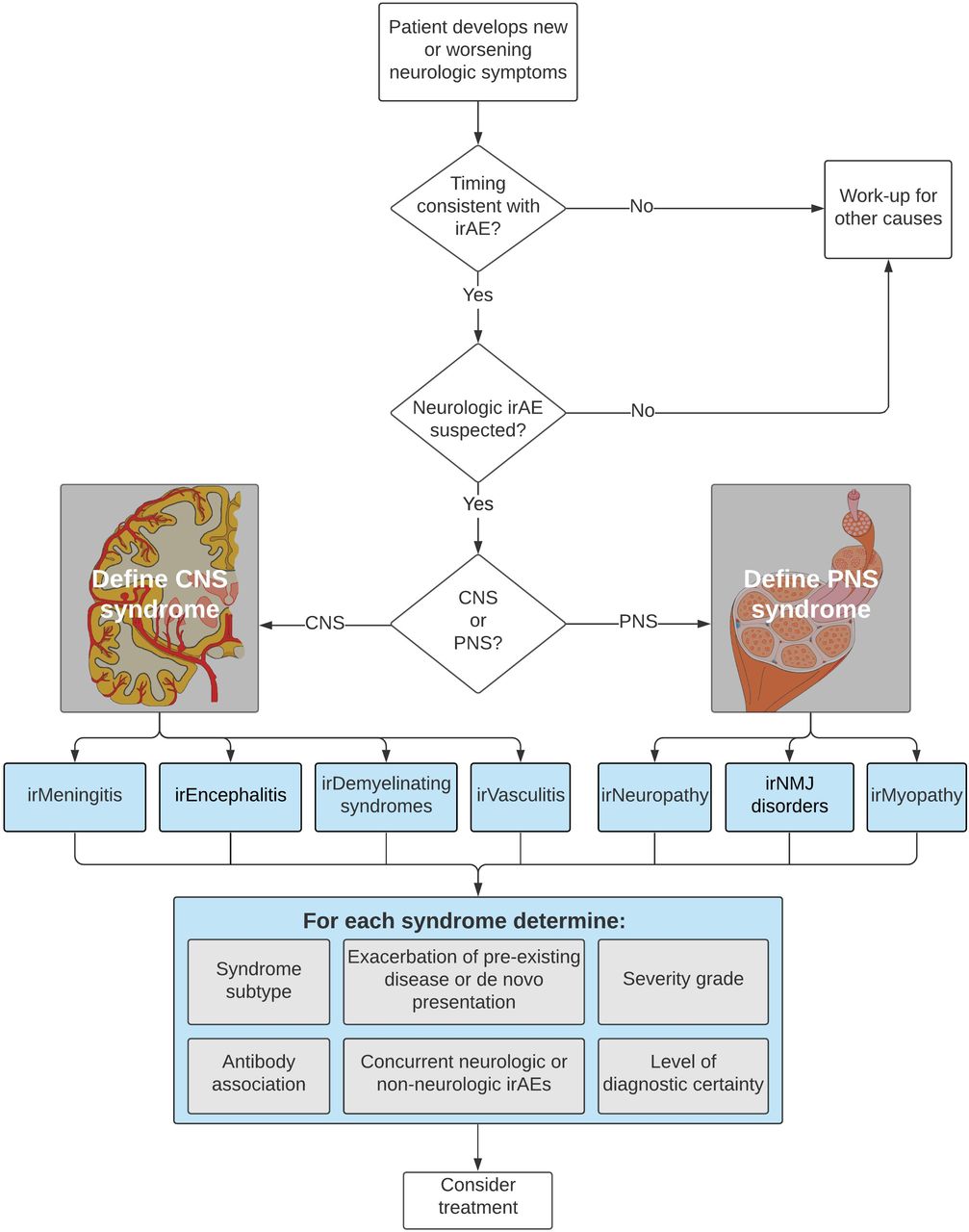{kind=link}
Approach to Classification of Neurologic Immune-related Advserse Events. CNS, central nervous system; irAEs, immune-related adverse events; PNS, peripheral nervous system.
Attribution of neurologic syndrome to checkpoint inhibitor therapy
Several features increase the likelihood that neurologic signs or symptoms starting after ICI administration represent an irAE-N. Diagnosis of irAE-Ns requires that other potential etiologies have been reasonably excluded by a workup tailored to each patient.
Timing
Most irAE-Ns occur early in treatment, usually within 6 months of starting or switching ICIs, although irAE-Ns may occur anytime while on treatment or after completion.24 To be considered an irAE-N, symptoms must begin within 12 months of the last ICI infusion. In general, new neurologic autoimmunity presenting more than 12 months after the last ICI infusion is unlikely to be an irAE. IrAE-Ns occurring between 6 and 12 months after completing an ICI are possible but less common. Diagnosis and evidence of relatedness in these later onset irAEs may require a higher ‘burden of proof’ in the individual patient. This area warrants additional research (median 8, range 2–9).
Pre-existing neurologic disorders
A careful history, baseline neurologic examination, and ancillary data help confirm or exclude pre-ICI neurologic disease. Most patients with history of mild symptoms or stable, well-established neurologic disorders do not require an additional evaluation by a neurologist when starting an ICI. However, patients with known immune-mediated neurologic disorders or patients with systemic autoimmune conditions with neurologic involvement may benefit from evaluation prior to initiating ICI therapy or soon afterward. Such evaluation is especially important for neuro-immunologic conditions of a relapsing or fluctuating nature, such as myasthenia gravis (MG) or multiple sclerosis, where ascertainment of clinical and/or radiologic disease activity provides a baseline. Additional considerations in this patient group may include risk/benefit discussions, modification of baseline immunomodulation prior to starting an ICI, and assistance with interpretation of changes in neurologic status after starting ICI treatment (median 9, range 5–9).
Consideration of concurrent irAEs and overlap syndromes
Patients frequently have irAEs affecting multiple organ systems or multifocal involvement of the nervous system. A concurrent, non-neurologic irAE may increase the likelihood that neurologic symptoms represent an irAE-N. A neurologic irAE also prompts consideration of other irAEs when known patterns of overlapping disease exist (ie, a diagnosis of myopathy or MG prompts evaluation for myocarditis, or diagnosis of meningitis prompts consideration of encephalitis). Sometimes it may be artificial to separate overlapping syndromes, particularly when they cause similar clinical manifestations and/or have similar management. When multiple syndromes occur, neurologic irAEs are listed separately with as much specificity regarding diagnosis, level of certainty, and severity as possible for each irAE-N, understanding that it may be difficult to ascertain which irAE-N accounts for maximum severity (median 9, range 7–9).
Improvement with holding drug and/or initiating corticosteroids
While improvement on holding ICI therapy and/or initiating corticosteroids or other immunomodulatory therapy is non-specific, it is expected in most patients with irAE-Ns and further supports the diagnosis of an irAE-N. Some irAE-Ns, however, may be treatment refractory/resistant or result in chronic disease or irreversible neurologic deficits. Lack of improvement with treatment typically prompts broadening of the workup for alternate etiologies but does not exclude the possibility of an irAE-N that is treatment-resistant or irreversible (median 9, range 6–9).
Non-specific symptoms or undefined events
Symptoms such as headache, confusion, fatigue and, in some cases, tremor are too non-specific to assign an irAE-N syndrome and may be related to other irAEs (eg, headache as a manifestation of an immune-related endocrinopathy) or due to other etiologies in patients with cancer, including cancer progression. Caution is advised when attributing these symptoms to an ICI. These symptoms, however, may trigger additional workup for irAEs along with other etiologies including toxic/metabolic disorders. Symptoms that are persistent and/or severe, even if unexplained, may lead to treatment delays or discontinuation of ICI therapy, but in isolation should not be classified as a neurologic irAE. If evaluation of symptoms leads to diagnosis of a neurologic irAE (eg, presentation with headache leads to a diagnosis of irMeningitis), an appropriate core syndrome and syndrome subtype is then assigned. If there are presentations that do not fit into a category outlined, or causal association with ICI is unknown (eg, posterior reversible encephalopathy syndrome), a general description of the syndrome is recommended (median 8, range 5–9).
Autoantibodies
Some irAEs are associated with well-characterized neural-specific antibodies. These antibodies may be known prior to ICI administration or detected during evaluation of an irAE-N. When naming an irAE-N in a patient with a known antibody, we recommend including the antibody as part of the diagnosis (eg, ‘definite immune-related myasthenia gravis with acetylcholine receptor antibodies’ or ‘probable immune-related myelitis with CRMP5 antibody’). Patients may additionally have low titer abnormal antibodies after ICI therapy. As many of these antibodies may be unrelated and are non-specific (eg, N-type VGCC antibody, GAD antibody), a patient’s syndrome should be referenced back to known antibody-associated syndromes before establishing a diagnosis. Recommendations for antibody evaluation and their assessment of association in irAE-N is an evolving area with extensive published literature.25 26 References to antibodies in the diagnostic evaluation and criteria have intentionally been kept general and to the discretion of the clinician evaluating the patient, except for key selected antibody recommendations which are syndrome specific (eg, acetylcholine receptor (AChR) antibodies, Hu or aquaporin-4 antibodies) (median 8, range 2–9).
Paraneoplastic syndromes
Patients may have paraneoplastic syndromes exacerbated or triggered by ICI therapy.27 28 The distinction between a process driven by an underlying cancer and an irAE can have significant treatment implications, although many aspects of diagnosis and management overlap. Similar to the approach to autoantibodies, disease definitions have been constructed to include criteria that ensure a relationship. A patient may therefore initially have a ‘possible’ or ‘probable’ diagnosis before it becomes ‘definite’ and these patients typically benefit from multidisciplinary collaboration (median 9, range 5–9).
IrAE-N consensus disease definitions
For each irAE-N syndrome, we achieved consensus on syndrome subtypes, possible presenting symptoms and examination findings, diagnostic workup, and diagnostic criteria. Some tests included in the workup are not part of the diagnostic criteria but can provide clinically useful information in the management of these patients (eg, bedside pulmonary function testing in a patient with MG).
IrAE-Ns affecting the CNS
For purposes of this discussion, the CNS is defined to include the meninges and the parenchyma of the brain, spinal cord and optic nerves. Meningitis is a syndrome of headache, fever and neck stiffness; focal neurological deficits or seizures should be taken as evidence of parenchymal CNS involvement.18 29–33 The parenchyma can be affected by inflammation of the gray and/or white matter (encephalitis), demyelination or vasculitis (which can lead to CNS ischemia).18 27 31–43An attempt should be made to attribute neurological symptoms that localize to the CNS to one of these major categories, recognizing that this may, in some cases, be difficult for non-specific symptoms (as discussed above).
The evaluation of CNS disease relies primarily on a combination of head and spinal cord imaging and cerebrospinal fluid (CSF) analysis. Head imaging (CT or MRI) should be performed prior to lumbar puncture (LP) to exclude mass lesions that may cause herniation and because LP can cause pachymeningeal enhancement.
MRI has superior sensitivity to detect many abnormalities compared with CT. MRI of the brain with contrast is recommended to evaluate for leptomeningeal and/or pachymeningeal enhancement, and for evidence of CNS metastasis, encephalitis, vasculitis, and demyelination. Head CT evaluates for cerebral edema, strokes and hemorrhages but is unlikely to demonstrate leptomeningeal or pachymeningeal inflammation. If MRI brain is not possible, then head CT is preferably performed with contrast. A head CT rather than an MRI may also be warranted in more acute presentations given the rapidity with which it can be obtained.
In all cases, it is important to rule out infectious disease as well as CNS involvement by the primary malignancy. In certain situations, serologic tests can identify antibodies associated with autoimmune encephalitis or demyelinating disease. Antinuclear antibody and extractable nuclear antigen positivity may suggest an autoimmune tendency but are of unclear pathogenic significance in isolation. Serum evaluation for specific infectious agents should be dictated by local prevalence, seasonal incidence, travel history and other patient risk factors such as intravenous drug use or high-risk sexual exposure. If concern about undiagnosed infectious disease persists, additional testing including metagenomic next-generation sequencing can be considered, typically with guidance from an infectious disease expert. Given its invasive nature, brain biopsy is not commonly pursued but may very rarely be needed to rule out alternative etiologies.
irMeningitis
Subtypes, evaluation and definitions of irMeningitis syndromes are presented in table 1. The rationale for common, possible and uncommon disease-specific testing is outlined.
irMeningitis
Common
In addition to the diagnostic tests described above, LP with CSF studies evaluates for inflammation (expect pleocytosis, elevated protein) and excludes other causes of meningeal disease (expect negative cytology and flow cytometry, negative Gram stain and culture, negative infectious studies). Specific CSF studies are recommended for universally common infectious agents such as herpes simplex virus (HSV) and cryptococcal disease. Further CSF and/or serum infectious studies may be determined by local epidemiology, seasonal incidence, travel history and patient-specific risk factors. Blood cultures evaluate for septic meningitis.
Possible
As previously discussed, MRI is generally preferred to head CT except in very urgent circumstances.
Uncommon
Meningeal biopsy is needed only in exceptional circumstances (eg, chronic pachymeningitis or continued concern of leptomeningeal carcinomatosis despite unrevealing CSF studies). Sometimes, CSF cytokine profiles are not widely available but can sometimes differentiate between infectious and aseptic meningitis, and etiologies such as CNS lymphoma.
irEncephalitis
Subtypes, evaluation and definitions of irEncephalitis syndromes are presented in table 2. The rationale for common, possible and uncommon disease-specific testing is outlined.
irEncephalitis
Common
In addition to brain MRI, spinal cord MRI with dedicated short tau inversion recovery (STIR) and pre-contrast and post-contrast T1 sequences may be obtained to evaluate for inflammatory, demyelinating, ischemic, or metastatic lesions. LP with CSF studies evaluates for evidence of inflammation (expect lymphocytic pleocytosis, elevated protein, CSF-restricted oligoclonal bands) and excludes other causes of encephalitis (expect negative cytology and flow cytometry, negative Gram stain and culture, negative viral and other infectious studies). Of note, HSV and varicella-zoster virus (VZV) encephalitis should either be ruled out prior to initiation of steroids or other immune suppression or be concurrently treated with antivirals while test results are pending. They can typically be excluded with commonly available tests (HSV and VZV PCR), although under certain circumstances advanced testing such as determination of a VZV CSF to serum antibody index may be appropriate.
Electroencephalogram (EEG) may reveal (subclinical) seizures or status epilepticus as a complication of encephalitis or as a cause of persistently depressed sensorium, although they are not specific to irEncephalitis. Screening metabolic tests are appropriate to assess for alternative etiologies or exacerbating factors. Serum and/or CSF autoimmune antibody evaluations assess for specific malignancy-associated neurologic syndromes.
Possible
For specific encephalomyelitic syndromes, additional antibody evaluation is considered, for example, autoimmune encephalitis panel, GQ1b antibodies (rhombencephalitis, Bickerstaff encephalitis), celiac antibody panel (ataxia). TPO and thyroglobulin antibodies are of limited utility, especially given their seroprevalence in healthy individuals, although they may be thought to reveal an autoimmune tendency. Brain biopsy may be obtained in very rare circumstances following multispecialty collaboration to rule out infectious and/or neoplastic causes when not possible by other methods. Brain PET (positron emission tomography) may be helpful in differentiating the cause of parenchymal lesions.
Uncommon
If concern about undiagnosed infectious disease persists, additional tests such as metagenomic next-generation sequencing can be considered.
irDemyelinating syndromes
Subtypes, evaluation and definitions of irDemyelinating syndromes are presented in table 3. The rationale for common, possible and uncommon disease-specific testing is outlined.
irDemyelinating
Common
Diagnostic evaluation for irDemyelinating syndromes commonly involves MRI with contrast of the brain, orbit, and cervical and thoracic spinal cord to the level of the conus medullaris to evaluate for evidence of parenchymal involvement. LP with CSF studies excludes other diagnoses and evaluates for evidence of CSF-restricted antibody production with oligoclonal bands. Serologic testing includes antibodies to aquaporin 4 and myelin oligodendrocyte glycoprotein, which are thought to be pathogenic in CNS demyelinating disease.
Possible
Ophthalmic or neuro-ophthalmic evaluation may be indicated. In addition, optical coherence tomography can provide evidence of prior optic neuropathy, but typically abnormalities follow clinical changes by several weeks. CSF autoimmune antibody evaluation (ie, for aquaporin 4 antibody) is unlikely to increase the sensitivity beyond serum testing alone. Evoked potential testing may provide supportive evidence of demyelination of visual, auditory or somatosensory nerve fiber pathways. A negative CSF PCR test for JC virus may exclude progressive multifocal leukoencephalopathy.
Uncommon
Biopsy may provide definitive evidence of CNS demyelination but is typically not necessary, as per the irEncephalitis discussion.
irVasculitis
Subtypes, evaluation and definitions of irVasculitis syndromes are presented in table 4. 44 45 The rationale for common, possible and uncommon disease-specific testing is outlined.
irVasculitis
Common
Diagnostic evaluation for irVasculitis commonly includes MRI brain to evaluate for infarcts and other parenchymal changes; where available, post-contrast vessel wall studies can also evaluate for concentric vessel wall enhancement that suggests a vasculitic process.46 Intracranial MR (MRA) or CT angiogram (CTA) evaluates for vascular abnormalities including narrowing and beaded vessels. MRA neck or CTA neck evaluates for vascular abnormalities more proximally including vasculitic changes and carotid atherosclerosis. Lumbar puncture with CSF studies, including VZV testing and syphilis testing (if serum testing positive), evaluates for evidence of CNS inflammation as well as other causes of vasculitis. These can typically be excluded with commonly available tests (VZV PCR in CSF, serum testing for syphilis), although under certain circumstances advanced testing such as determination of a VZV CSF to serum antibody index may be appropriate. A workup for embolic stroke including carotid Doppler, electrocardiogram (EKG), heart rhythm monitoring and echocardiogram may be performed if an infarct is found. Serum markers including C reactive protein (CRP), erythrocyte sedimentation rate (ESR), anti-neutrophil cytoplasmic antibody (ANCA) and anti-nuclear antibody (ANA) and other markers associated with systemic vasculitis provide further support of vasculitis.
Possible
Formal rheumatology and/or dermatology evaluation may provide additional consideration to systemic manifestations of vasculitis. Brain biopsy can provide definitive proof of CNS vasculitis and should include sampling of both brain and meninges. Extracranial biopsy, including temporal artery biopsy or skin biopsy, may provide evidence of a systematic vasculitis and obviate the need for brain biopsy. Retinal fluorescein angiography can provide evidence of a small vessel vasculitis. Conventional (digital subtraction) angiogram may demonstrate vessel abnormalities if MRA or CTA do not provide sufficient clarity. Angiography of other vascular beds such as the renal or splanchnic circulation can assist with establishing the presence of a systemic vasculitis. Antiphospholipid antibody panel and hypercoagulability evaluation can evaluate for alternative causes of stroke.
Uncommon
Body PET imaging may provide evidence of systemic vasculitis.
IrAE-Ns affecting the PNS
For purposes of this discussion, the PNS is defined to include nerves (including cranial nerves; axons and cell bodies), the neuromuscular junction, and muscle. Peripheral neuropathy is a rare but likely under-reported complication of ICI therapy with an incidence rate of approximately 1%.24 Cranial neuropathies with and without acute polyradiculoneuropathy syndromes are the two most common neuropathy phenotypes.47 Reporting of neuropathy in large databases and meta-analyses has focused on Guillain-Barré syndrome. Painful length-dependent sensory and motor axonal neuropathies, polyradiculopathies and sensory neuronopathies following ICIs are likely under-recognized.18 48
Patients may present with new onset MG or exacerbation of previous MG after ICI therapy. Compared with idiopathic MG, there is a high rate of concurrent myopathy and myocarditis, which may increase disease severity and mortality.49–51 Additionally, myopathy after ICIs can mimic MG with an ocular, bulbar, axial and respiratory pattern of weakness.52 Therefore, some cases of irMyopathy may be misdiagnosed as MG. Immune-related Lambert-Eaton syndrome (LEMS) has been rarely reported and it remains unclear whether it is a paraneoplastic disorder or an irAE.49 As such, it is not specifically included as a separate category in this classification system.
ICI-related muscle disease has been referred to as both myopathy and myositis. This group selected the broader term irMyopathy, which encompasses both inflammatory myositis and immune-mediated necrotizing myopathy. IrMyopathy most commonly presents with ptosis, diplopia, dysphagia, dyspnea, neck weakness, and/or hip flexor weakness.10 53 54 It may also present with myalgias and gait dysfunction. irMyopathy can be focal, sometimes involving only one muscle.10 Presentations limited to ocular muscles (orbital myositis) have been reported.53 Although ptosis and diplopia are uncommon in other forms of myositis, irMyopathy may have a specific predilection for these muscles, or these symptoms may result from superimposed neuromuscular junction (NMJ) dysfunction.53 Given the substantial overlap, differentiating symptoms resulting from myopathy from NMJ dysfunction can be challenging.
The evaluation of PNS disease relies primarily on a combination of electrodiagnostic studies (EDX), serologic tests, and imaging. EDX include nerve conduction studies and needle electromyography and are preferably performed by neurologists with familiarity with irAE-N to increase utility. Nerve conduction studies are used to confirm and characterize large fiber neuropathy. Repetitive nerve stimulation may be included with nerve conduction studies to test for NMJ dysfunction. Needle electromyography (EMG) can detect the presence of neuropathic or myopathic disorders and provide information about chronicity. Because PNS syndrome overlap is common, screening for NMJ dysfunction (with EDX) and myopathy (with EDX and serum creatine kinase (CK)) is recommended for patients presenting with motor predominant symptoms thought to be peripheral. Additionally, patients presenting with immune-related NMJ disorders or myopathy should have screening for irMyocarditis with serum troponin and EKG given the potential severity of this condition. Serologic and radiographic workup is disease specific and discussed in the following sections.
Immune-related neuropathy (irNeuropathy)
Subtypes, evaluation and definitions of irNeuropathy syndromes are presented in table 5. The rationale for common, possible and uncommon disease-specific testing is outlined.
irNeuropathy
Common
Serum testing evaluates for alternative causes of neuropathy and signs of autoimmunity/inflammation. Serum testing may be abnormal but not explain the clinical phenotype (ie, B12 deficiency would not explain a demyelinating neuropathy; a mildly elevated hemoglobin A1c (HbA1c) would not explain an immune-mediated polyradiculopathy). Definite irNeuropathy can be achieved in such cases as long as concordance between clinical phenotype and testing is considered when applying the diagnostic criteria. EDX confirm or exclude large fiber neuropathy and characterize the type, severity, and chronicity of neuropathy if present (ie, sensory and/or motor, axonal and/or demyelinating, length-dependent or non-length-dependent). Demyelinating features or a non-length-dependent and/or asymmetric pattern of neuropathy can help distinguish irNeuropathies from chemotherapy-related neuropathies, which are more typically symmetric and axonal. EDX also evaluates for other etiologies (ie, focal entrapment neuropathy, radiculopathy, myopathy). Coexisting myopathy and NMJ disorders must be excluded in patients with prominent motor involvement with EDX and CK. Spinal imaging is often obtained to exclude metastatic disease, focal structural radiculopathy, and spinal stenosis; it may demonstrate spinal nerve root enhancement/enlargement in inflammatory radiculopathies. MRI brain with contrast is obtained when cranial nerve involvement is suspected, which may show smooth enhancement of cranial nerves.
Possible
If the diagnosis or the etiology is uncertain, LP with CSF analysis may provide further support of an immune-mediated process in certain neuropathy phenotypes (such as Guillain-Barré syndrome) and exclude infection or malignancy. CSF shows cytoalbuminologic dissociation in immune-mediated neuropathies; however, lymphocytic pleocytosis (white blood cell count <50) is common in this patient group and cytoalbuminologic dissociation may be absent in the first week after symptom onset. Selected additional laboratory tests may be performed based on patient factors and the neuropathy phenotype (ie, mononeuritis multiplex would warrant a vasculitis workup) but are not required for all patients. Bedside pulmonary function tests and/or swallowing evaluation is obtained in patients with a Guillain-Barré phenotype to screen for or monitor respiratory dysfunction or dysphagia but are not included in the diagnostic criteria.
Uncommon
Immune-related neuromuscular junction disorders (irNMJ disorders)
Subtypes, evaluation and definitions of irNMJ syndromes are presented in table 6. The rationale for common, possible and uncommon disease-specific testing is outlined.
irNeuromuscular junction disorder
Common
Evaluation for irNMJ disorders includes diagnostic antibody testing for MG and evaluation for concurrent myopathy, myocarditis, and thyroid dysfunction. A positive acetylcholine receptor (AChR) binding or MuSK antibody is consistent with a diagnosis of MG. AChR modulating antibodies in isolation are not sensitive or specific. AChR binding with modulating antibodies has the highest specificity.55 Rate of ACHR ab positivity in irMG has not been definitively established. Anti-straited muscle antibodies are frequently present in irMyopathy and/or irMG but are not diagnostic for irMG.53 Chest imaging is performed to exclude thymoma once an irNMJ disorder is confirmed. Patients are screened for myopathy with CK and EDX and for myocarditis with a troponin and EKG.
The role of EDX in patients with AChR antibodies and suspected irMG has not been well studied and was debated by the panel. In idiopathic MG, AChR binding or MuSK antibody positivity in the appropriate clinical scenario is diagnostic and EDX is unnecessary. However, patients may have AChR antibodies after ICI therapy without evidence for a disorder of neuromuscular transmission. Demonstrating abnormal neuromuscular transmission on EDX either through abnormal repetitive nerve stimulation or single fiber EMG is needed for definite irMG diagnosis.
Possible
Testing for the paraneoplastic Lambert-Eaton syndrome with P/Q VGCC antibodies is performed if LEMS is suspected clinically or EDX show a characteristic pattern of facilitation and decrement. MuSK antibody testing is performed in this context if electrodiagnostic studies show evidence for a disorder of neuromuscular transmission and AChR antibodies are absent. MuSK antibody testing assesses primarily for pre-existing disease since it has not been reported de novo after ICI therapy. Single fiber EMG can provide evidence of a disorder of neuromuscular transmission but may rarely show mild abnormalities in myopathy; it is typically performed at specialized centers if MG is suspected but routine EDX and antibody testing are normal/negative. The ice pack test, performed by applying ice to a ptotic lid for 2 min, can show improvement in ptosis at the bedside and further support a diagnosis of MG.56 An EKG, serum biomarkers, transthoracic echocardiogram and cardiac MRI may be performed for further evaluation of concurrent myocarditis given syndrome overlap.56 57
Uncommon
Edrophonium testing may not be available and is usually not needed in this patient group.
Immune-related myopathy (irMyopathy)
Subtypes, evaluation and definitions of irMyopathy syndromes are presented in table 7. The rationale for common, possible and uncommon disease-specific testing is outlined.
irMyopathy
Common
Diagnostic evaluation for irMyopathy includes CK to assess for muscle breakdown. Aspartate aminotransferase (AST) and alanine aminotransferase (ALT) are typically elevated along with CK. Gammaglutamyl transferase (GGT) evaluates for liver-specific injury and is normal when AST/ALT elevations are due to muscle disease. EDX evaluate for muscle membrane irritability (indicated by fibrillation potentials and positive sharp waves), which can be seen in inflammatory or necrotizing myopathy, and for myopathic motor unit potentials (characterized by short duration, with or without low amplitude and/or early recruitment). EMG should include needle examination of clinically weak muscles and thoracic paraspinals. Of note, abnormalities in motor unit potentials may be patchy or subtle, and recruitment is often normal, particularly in the acute setting. EDX are often performed with repetitive nerve stimulation of proximal nerve-muscle combinations to test for an overlapping neuromuscular junction disorder. Troponin-I, EKG, and echocardiogram may be used for initial screening for concurrent myocarditis.57 While troponin-T may be53 58 elevated with myositis, troponin-I is more specific for cardiac injury.57
Possible
Muscle biopsy of affected muscle, ideally 4/5 MRC grade strength, allows direct assessment of muscle inflammation, but may not be needed in many cases. T-cell infiltrate identified by biopsy is supportive of irMyopathy, although other histopathological patterns have been described.53 58 MRI showing muscle edema on STIR images and/or contrast enhancement can suggest inflammation and may help select site for muscle biopsy; however, these changes are not specific for myositis and can also be seen in denervated muscle, so imaging findings are not sufficient for diagnosis. Myositis-specific antibodies (including Jo-1, PL-7, PL-12, EJ, OJ, Mi-2, SRP, TIF-1 gamma) and anti-HMGCR antibodies may be measured to further characterize myositis subtypes. ESR, CRP, and ANA are non-specific markers of inflammation, which support a diagnosis of inflammatory myositis and may be tracked longitudinally. Aldolase is another marker of muscle breakdown, although may not provide additional information to the CK level. AChR antibodies can evaluate for a superimposed NMJ disorder, particularly in patients with ocular or bulbar symptoms. Beside pulmonary function tests may be appropriate if there is concern for respiratory muscle weakness, dysphagia or head drop. Swallowing evaluation can be considered for patients with dysphagia to ensure safe oral intake. Finally, an elevated troponin and abnormal cardiac MRI can suggest a concurrent myocarditis and consultation with a cardiology service would be advised.59
Uncommon
Genetic testing may be performed when there is clinical suspicion for a hereditary myopathy. Although more commonly used to evaluate malignancy, fluorodeoxyglucose (FDG)-PET can also detect skeletal muscle inflammation.
Severity criteria
The panel adapted the CTCAE for irAE-Ns with illustrative examples (online supplemental table 1).
Supplemental material
Clinical trial adjudication
Use of consensus definitions and severity criteria would standardize reporting, allowing for data pooling and cross-trial comparisons for irAEs-N. To facilitate usability, it may be beneficial to have a centralized data safety monitoring capacity to classify irAE-Ns according to consensus criteria in clinical trials based on primary source documentation. Source documents for irAE-N adjudication are outlined (online supplemental table 2). For overlap syndromes, classification recommendations are followed for each individual toxicity. Priority, relevance and feasibility of individual items depends on setting and toxicity (median 8, range 6–9).
Discussion
This multidisciplinary, multi-institutional group has developed consensus disease definitions and severity criteria for irAE-Ns after ICI therapy. After reaching agreement on the critical unmet need for standardized disease definitions, the 27 panelists completed a two-round anonymous, modified Delphi voting process with two virtual meetings to obtain consensus on guidance statements. A comprehensive literature review, existing guidance from major oncologic organizations, the CTCAE and a methodologically rigorous process to gain input from experts across multiple disciplines were used. By process completion, all statements and disease definitions reached consensus. The high response rate, multidisciplinary panel, detailed nature of the experts’ comments and high retention rate of 80% through both voting rounds were notable study strengths. Therefore, these consensus irAE-N definitions will likely be representative of, and applicable to, neurologists, oncologists and other subspecialists involved in the clinical care or research of patients with irAEs. It is well established that a broad spectrum of irAEs-Ns exists, which indicates there may be unique or novel pathophysiologic underpinnings. Using these consensus definitions to accurately phenotype irAEs-Ns in both clinical trials and in biobanks will allow for an in-depth analysis with potential to detect diagnostic and predictive biomarkers that separate these heterogenous presentations. These are the first detailed definitions of irAEs-Ns, which forms the critical foundation for both clinical and translational research in this area.
The Delphi process revealed areas of controversy and challenges in irAE-N, which were discussed and debated at the virtual meetings, but not entirely resolved. Most fundamentally, these issues were related to (1) balancing phenotypic accuracy with usability by a wide range of clinicians; and (2) integration of disease definitions, diagnostic certainty and treatment recommendations.
Regarding the balance of accuracy and usability, a key remaining question is how much of the disease definition framework can be used in real-world and clinical trial applications by oncologists and how much will require neurology or neuro-oncology input, when available. Neurology consultation has been recommended previously for any irAE-N grade 2 or higher.18 The panel anticipated oncologists would begin the classification into CNS or PNS disorders, and possibly into one of the seven core syndromes, in addition to helping exclude other non-neurologic etiologies; the neurologist would further refine the diagnosis into a core syndrome and specific subclassification and assist with treatment and symptom management.18 Ordering of specialized diagnostic testing is often performed in consultation with neurology. Collaboration between specialists and referral systems to allow for expedited neurologic evaluation and testing will be critical. Thoughtful approaches to documentation and information sharing will be needed to ensure clear communication across different care settings and specialists. In certain settings where neurology consultation may not be readily available, future guidance regarding a set of symptoms that would necessitate referral to a neurologist and/or more urgent evaluation may increase usability.
One challenge that was identified at the second virtual meeting, after both rounds of consensus ratings, was how to classify myelitis cases that did not appear demyelinating. A proposal was made to alter the core disorders such that they were defined anatomically (ie, switch irDemyelinating to irMyelitis to focus on the spinal cord). A change at that stage, however, would not have permitted further review on the already agreed definitions and diagnostic criteria from the Delphi process. For clinical use, we therefore created an ‘Other myelitis’ subtype under irDemyelinating recognizing that, even though such cases may not strictly be demyelinating, the irDemyelinating diagnostic criteria include spinal cord imaging criteria that these cases will likely fulfill. For cases of encephalomyelitis that do not appear demyelinating, they may fulfill the irEncephalitis diagnostic criteria for the encephalitis component and could be included there.
Regarding integration of disease definitions and treatment guidelines, similar to other disease definitions in neurology,60 the panel emphasized that diagnostic certainty is not always directly linked to treatment decisions (ie, the panel would generally treat probable and sometimes possible irAE-Ns). To maximize accuracy of the definitions and to prevent misclassification of ‘definite’ patients, definitions were constructed so that most patients would likely reach a ‘probable’ category and that this would be sufficient in most cases to treat as an irAE-N. The panel also emphasized that improvement with corticosteroids is not specific for irAE-Ns and should not be used as a primary criterion to establish etiology as an irAE. Additionally, patients must be treated with appropriate doses of immunomodulatory or immunosuppressive therapies before being labeled treatment resistant or refractory. Evidence-based definitions for toxicity response and resistance to treatment will be needed.
These disease definitions provide a systematic approach to the spectrum of irAE-N types, which may be applicable to other toxicities in the future. This system provides flexibility for increasing level of diagnostic specificity to accompany patients through their workup through relevant settings including oncology and neurology clinics, clinical trials and translational research. The framework captures the spectrum of irAE-N from mild to fatal and accounts for diagnostic uncertainty. In circumstances such as combination immunotherapy and chemotherapy, where attribution of new neurologic symptoms to ICIs may be challenging, the statements provide guidance. These definitions are a key step in moving toward future evidence-based management recommendations for irAE-Ns.
Data availability statement
All data relevant to the study are included in the article.
Ethics statements
Patient consent for publication
Acknowledgments
Medical writing assistance was provided by Carina Storrs, PhD, independent science writer. Assistance in figures preparation was provided by Johnathan Rine, MSPH, MBA, of Project Data Sphere. We acknowledge Shaily Arora, Acting Associate Director for Safety at the Food and Drug Administration, and Nicky Wallis, Executive Director of Safety Science at BeiGene, for advising the disease definitions and irAE-N registry projects.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Twitter @DrJNaidoo
BKC and JH contributed equally.
Collaborators In addition to the named authors, medical writing assistance was provided by Carina Storrs, PhD, independent science writer. Assistance in figures preparation was provided by Johnathan Rine, MSPH, MBA, of Project Data Sphere. We acknowledge Shaily Arora, Acting Associate Director for Safety at the Food and Drug Administration, and Nicky Wallis, Executive Director of Safety Science at BeiGene, for advising the disease definitions and irAE-N registry projects.
Contributors ACG and LBB contributed equally to this work as co-first authors with the addition that ACG and KLR conceptualized and designed the study and Dr Burton designed, analyzed and interpreted the Delphi surveys and data. BKC and JH contributed equally. All authors provided (1) substantial contributions to the conception or design of the work, or the acquisition, analysis or interpretation of data, (2) drafting the work or revising it critically for important intellectual content, (3) final approval of the version published, (4) agreement to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. AAA, ABW, PKB, TAC, SLC, JC, JD, MD, CTD, DD, JG, JTG, DBJ, VCJ, RK, NK, NRL, JL, AM, MM-L, MJM, JN, TGN, DR, KMR, BDS, RS, NW, KW, and LZ contributed equally as part of the Neuro irAE disease definition panel.
Funding A portion of this work was supported by Project Data Sphere and the Global Oncology Big Data Alliance.
Competing interests ACG has served on an advisory board and/or consulted with Alexion, Ra Pharma/UCB and Momenta pharmaceuticals/Janssen. She has received royalties from Oakstone Publishing and research support from the Myasthenia Gravis Foundation of America, the MG Rare Disease Network and Project Data Sphere® for an irAE-N registry. She has received clinical trial support to the institution from Ra Pharma and Momenta. LBB receives salary support from Biogen, royalties from Oakstone Publishing, and research support from Project Data Sphere for an irAE-N registry. BKC reports research support from Project Data Sphere for an irAE-N registry. JH receives research support from Project Data Sphere for an irAE-N registry and from GE Healthcare. THS is an employee at the not-for-profit corporation Project DS, with a trade name of Project Data Sphere. AAA served on medical advisory boards for Alexion, Sarepta, CSL Behring, Strongbridge Pharma, Argenx, Ra Pharmaceuticals, and as a neurology consultant for Johnson & Johnson COVID-19 vaccine trials. ABW served on a medical advisory board or as a consultant with Iovance, Novartis, Shanghai Jo’Ann Medical Technology. PKB has consulted for Angiochem, Genentech-Roche, Lilly, Tesaro, Voyager Therapeutics, ElevateBio, Pfizer (Array), Pfizer, SK Life Sciences and Dantari, received grant/research support (to Massachusetts General Hospital) from Merck, BMS and Lilly and honoraria from Merck, Pfizer, Genentech-Roche and Lilly. TAC has received royalties from Wolters Kluwer. SLC serves as the Editor of the Neurology®Podcast and Neurology Minute™. She has received research and/or clinical fellowship support from the Western Institute for Veterans Research, the Siegel Rare Neuroimmune Association, the Immune Deficiency Foundation, Alexion, the Barbara Gural Steinmetz Foundation, and the Sumaira Foundation for NMO. She has served on an advisory board and/or consulted with Alexion, Genentech, VielaBio, Guidepoint, Clarion Healthcare Consulting, ExpertConnect (majority fees paid to University of Utah). JC has received consulting fees from Sanofi-Genzyme and BMS. JD is a consultant for Blue Earth Diagnostics, Magnolia, Gamaka Bio, and Unum Therapeutics. JD has received research support from Beacon Biosignals, Boehringer Ingelheim, Brystol-Myers Squibb, Eli Lilly, Medimmune, Acerta Pharma, Orbus Therapeutics, and Novartis Pharmaceuticals. JD has received royalties from Wolters Kluwer for serving as an author for UpToDate. MD has research funding from Novartis and Eli Lilly, has served as a consultant for Roche-Genentech, Tillotts Pharma, ORIC Pharmaceuticals, Partner Therapeutics, and Moderna, and is a member of the Scientific Advisory Board for Neoleukin Therapeutics. CTD has served on advisory boards for Argenx and Dysimmune Diseases Foundation. He has received royalties from Oakstone Publishing. He has received grant support from NINDS/NeuroNext. DD has served on an advisory board and/or consulted for UCB, Astellas and Immunovant pharmaceuticals. All compensation for the consulting activities is paid to Mayo Clinic. He has patents pending for KLHL11 and LUZP4 as markers of neurological autoimmunity and germ cell tumors. JG has received research to UCSF support from Genentech/Roche for a clinical trial, performed consulting for Biogen, and received personal compensation for medical-legal consulting. JTG has served as a consultant in past 12 months for Immunovant, Alexion, Momenta, Ra Pharma, Grifols, Jacobus, Cabaletta, Regeneron, Argenx, Signant, UCB, Toleranzia and Piedmont Pharmaceuticals. He receives industry grant support from UCB pharma for a fellowship training grant. Full disclosure statement available at: https://dcri.org/about-us/conflict-of-interest/. DBJ has served on advisory boards for Array Biopharma, BMS, Catalyst Biopharma, Iovance, Jansen, Merck, Novartis, and Oncosec, and has received research funding from BMS and Incyte. VCJ is a site investigator in myasthenia gravis research trials sponsored by PCORI and by Alexion Pharmaceuticals. RK receives research funding from Alexion Pharmaceuticals. NK reports no relevant disclosures. NRL is a consultant and has received honoraria from Bayer, Seattle Genetics, Sanofi, Fortress Biotech, Silverback Therapeutics and SYNOX Therapeutics. JL reports no disclosures. AM reports no disclosures. MM-L reports no relevant disclosures. MJM has served on an advisory board and/or consulted with AstraZeneca Pharmaceuticals, Nektar Therapeutics, Catalyst Pharmaceuticals and Immunai. JN has received research funding from Merck and AstraZeneca; served as a consultant/advisory board member with Merck, AstraZeneca, BMS, Pfizer, Takeda, Roche/Genentech, Daiichi/Sankyo and has received honoraria from Merck, AstraZeneca, BMS, Pfizer, Takeda, Roche/Genentech. TGN reports acting as a consultant for Parexel, Bristol Myers-Squibb, H3 Biomedicine, AbbVie, and Intrinsic Imaging unrelated to the current research. TGN reports grant funding from AstraZeneca unrelated to current research. DR has been an advisor or consultant with AbbVie; Advantagene; Agenus; Agios; Amgen; Bayer; Boston Biomedical; Boehringer Ingelheim; Bristol-Myers Squibb; Celldex; Deciphera; DelMar; EMD Serono; Genenta; Genentech/Roche; Imvax; Inovio; Kintara; Kiyatec; Medicenna Biopharma; Merck; Merck KGaA; Monteris; Neuvogen; Novartis; Novocure; Oncorus; Oxigene; Regeneron; Stemline; Sumitono Dainippon Pharma; Taiho Oncology. Research support to his institution has been provided by Acerta Phamaceuticals; Agenus; Celldex; EMD Serono; Incyte; Inovio; Omniox; Tragara. KMR has served on an advisory board and/or consulted with Merck, BMS, and Eisai. BDS has served on an advisory board and/or consulted with Janssen, Kite/Gilead, and Celgene/BMS. RS has served on an advisory board and/or consulted with AstraZeneca, Bristol-Myers Squib, Eisai, Iovance, Merck, Novartis, Oncosec, Pfizer, and Replimune. He has received research funding from Merck. NW has served on an advisory board with Seattle Genetics, received royalties from Wolters Kluwer and research support from Merck. KW reports no disclosures. LZ has been a consultant for Merck. WCL is an employee of the not-for-profit corporation Project DS, with a trade name of Project Data Sphere. KLR has received personal compensation from Teladoc research support from Project Data Sphere for a irAE-N registry.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.