Article Text

Abstract
Background Phase IIb clinical trial with isatuximab (Isa)-lenalidomide (Len)-dexamethasone (Dex) showed an improved progression-free survival (PFS) in patients with relapsed or refractory multiple myeloma (RRMM), but the efficacy varied by patient. Antibody-dependent cell-mediated cytotoxicity (ADCC) by natural killer (NK) cells plays a crucial role in arbitrating antitumor activities of therapeutic-antibodies. We tested if patient-specific genetic makeup known to set NK cell functional threshold influence response to Isa-Len-Dex therapy.
Methods We characterized 57 patients with RRMM receiving Isa-Len-Dex for polymorphisms of killer-cell immunoglobulin-like receptors (KIR), human leukocyte antigen (HLA) class I, and FCGR3A loci. In vitro ADCC assay, coincubating primary NK cells expressing specific KIR repertoire with multiple myeloma cell lines (MM cells) expressing selected HLA class I ligands, was used to confirm the identified genetic correlatives of clinical response.
Results Patients with KIR3DL2+ and its cognate-ligand HLA-A3/11+ had superior PFS than patients missing this combination (HR=0.43; p=0.02), while patients carrying KIR2DL1+ and HLA-C2C2+ compared with to patients missing this pair showed short PFS (HR=3.54; p=0.05). Patients with KIR3DL2+ and HLA-A3/11+ plus high-affinity FCGR3A-158V allele showed the most prolonged PFS (HR=0.35; p=0.007). Consistent with these clinical data, mechanistic experiments demonstrated that NK cells expressing KIR3DL2 trigger greater ADCC when MM cells express HLA-A3/11. Inversely, NK cells expressing KIR2DL1 do not kill if MM cells express the HLA-C2C2 ligand. NK cells expressing high-affinity FCGR3A-158VV-induced greater ADCC compared with those with low-affinity FCGR3A-158FF.
Conclusions Our results suggest that KIR3DL2+ and HLA-A3/11+ with FCGR3A-158V markers lead to enhanced Isa-dependent NK-mediated cytolysis against MM cells and results in improved PFS in patients with RRMM treated by Isa-Len-Dex. Moreover, the presence of KIR2DL1+ and HLA-C2C2+ identifies patients who may have a lower response to Isa-Len-Dex therapy linked to a reduced NK-mediated ADCC. These biomarkers could potentially identify, via precision medicine, patients more likely to respond to Isa-Len-Dex immunotherapy.
Trial registration number NCT01749969.
- genetic markers
- immunity
- innate
- immunotherapy
Data availability statement
Data are available upon reasonable request. The datasets generated and analyzed during the current study are available from the corresponding author on reasonable request.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Background
Multiple myeloma (MM) is a malignant plasma cell disorder characterized by the overproduction of immunoglobulin and/or light chain and can cause significant clinical symptoms, including hypercalcemia, renal insufficiency, anemia, and lytic bone disease.1 In the last 10 years, overall survival has improved due to the use of autologous stem cell transplantation and the development of novel therapeutics, including proteasome inhibitors (bortezomib, carfilzomib, and ixazomib), immunomodulatory drugs (thalidomide, lenalidomide (Len), and pomalidomide), and histone deacetylase inhibitors (panobinostat and vorinostat).2 Despite these therapeutic advancements, patients ultimately relapse or become refractory, and thus additional novel therapeutics are needed.3 Patients with relapsed or refractory multiple myeloma (RRMM) have a median event-free survival and overall survival of only 5 and 9 months, respectively, further supporting the need for new treatments.4
More recent immunotherapies targeting cell surface molecules with a monoclonal antibody (mAb) have shown significant clinical activity.5 Notably, the Food and Drug Administration approved three therapeutic mAbs, anti-SLAMF7 mAb elotuzumab, and anti-CD38 mAbs daratumumab and isatuximab (Isa) for MM. Several mechanisms have been postulated to account for the antitumor activities of these therapeutic antibodies,6 including complement-dependent cytotoxicity, antibody-dependent cellular cytotoxicity (ADCC), antibody-dependent cellular phagocytosis, and activation of apoptosis. Isa was recently approved for use in combination with pomalidomide and dexamethasone (Dex) (Pd) in RRMM based on improved PFS in a phase III trial (PFS: Isa-Pd 11.3 m vs Pd 6.5 m).7 Isa combines well with immunomodulatory drugs, and this was also noted in a phase I study of Isa combined with Len and Dex (Rd).8 Response rates in the Isa-Pd and Isa-Rd ranged between 60% and 70%, suggesting some resistance patterns and possibly some undiscovered biomarkers for response. Preclinical studies indicated that Isa elicits anti-MM activity through multiple mechanisms, including ADCC by natural killer (NK) cells.9–12
NK cells account for approximately 10%–15% of peripheral blood lymphocytes and are recognized as fast-acting cytotoxic innate lymphoid cells that trigger immunity to infection and cancer.13 NK cells use a very complex and sophisticated repertoire of activating and inhibitory receptors that are calibrated to ensure self-tolerance while eliminating diseased cells. The polymorphic human leukocyte antigen (HLA) class I-specific killer-cell immunoglobulin-like receptors (KIR) are crucial for human NK cell development and function.14 KIR receptors are encoded by a family of genes located in the leukocyte receptor complex on chromosome 19.15 Several haplotypes that differ by the number and type of KIR gene content have been described in populations. The most common haplotype that occurs nearly 50% in Caucasians is called A-haplotype, which contains a fixed set of nine genes (figure 1). The A-haplotype encodes inhibitory KIRs to all four HLA class I ligands, C2 (KIR2DL1), C1 (KIR2DL3), Bw4 (KIR3DL1), A3/A11 (KIR3DL2), and a single activating KIR2DS4. The remaining haplotypes are collectively referred to as group B-haplotypes, which have variable KIR gene content comprising several genes (ie, KIR2DL2, KIR2DL5, KIR2DS1, KIR2DS2, KIR2DS3, KIR2DS5, and KIR3DS1) that are not part of the A haplotype. The group-B haplotypes encode 1–6 activating KIRs and only two inhibitory KIRs that bind to HLA-C1 (KIR2DL2), HLA-A3/A11 (KIR3DL2). Little is known about the ligands for the activating KIRs. A stretch of 14 kb DNA enriched with L1 repeats upstream of KIR2DL4 divides the KIR haplotype into centromeric and telomeric halves (figure 1), and reciprocal recombination events between these two halves presumably diversify KIR gene content for haplotypes. The inheritance of paternal and maternal haplotypes comprising different KIR gene content generates extraordinary diversity between humans in KIR genotypes (such as AA, AB, and BB carriers).

Killer-cell immunoglobulin-like receptors (KIR) haplotypes vary in gene content. Map of group A and B KIR haplotypes. Inhibitory KIR genes are depicted in white boxes with their specific human leukocyte antigen (HLA) class I ligands. Activating KIRs are shown in dark boxes. Hatched boxes represent pseudogenes or KIR genes with no apparent function. The centromeric and telomeric half are marked.
During NK cell development, the interaction of inhibitory KIR with cognate HLA class I ligand sets the functional threshold for NK cells, a process called “licensing” that allows the subsequent recognition and elimination of target cells with decreased expression of self HLA class I due to viral-infection or tumor-transformation.16–20 Given that KIRs on chromosome 19 and HLA loci on chromosome 6 are polymorphic and display substantial variations, the independent segregation of these unlinked gene families generates extraordinary diversity in the number and type of KIR+HLA combinations inherited in humans.21 These inter-individual differences in the KIR+HLA constellation constitute a significant source of variability in NK cell activity influencing clinical outcomes in diverse settings, including susceptibility to infections, tumor transformation, autoimmune diseases, pregnancy success, and success of allogeneic transplantation.22
The CD16 (FcRIIIa), a low-affinity activating NK cell-surface receptor for the Fc portion of IgG antibodies, regulates NK cell ADCC.23 The CD16 receptor is encoded by the FCGR3A gene, which carries a functional single nucleotide dimorphism that substitutes phenylalanine (F) for valine (V) at residue 158 and influences binding affinity to IgG. Despite equivalent levels of CD16 expression, 158V homozygotes have a high affinity for IgG compared with 158F homozygotes.23 Cancer immunotherapy studies have reported that the high-affinity genotype of FCGR3A is beneficial for the improved clinical response rate to rituximab, trastuzumab, and cetuximab.24–26 Preliminary studies revealed no significant associations between tumor response and FCGR3A dimorphism in patients with RRMM treated with Isa-Pd.27
In preclinical studies, the ADCC was recognized as the dominant effector mechanism in the therapeutic effects of the Isa in patients with MM.12 However, studies supporting a role for low and high-affinity FCGR3A genotypes in the clinical response to Isa in patients with MM are limited.28 Moreover, the role of KIR+HLA mediated licensing and inhibition in human ADCC is poorly defined. Herein, we tested the hypothesis that individualized constellation of genes that control the NK cell effector function influence the efficacy of Isa mAb therapy through Isa-dependent NK cytolysis. The data from this study show, for the first time, a correlation between genes contributing to NK cell ADCC and the clinical efficacy of Isa therapy. The outcome of this study may lead to new treatment strategies in predicting which patients would be best suited for antibody immunotherapy.
Methods
Patients
We analyzed 57 patients with RRMM enrolled in a phase IIb clinical trial (registered at www.clinicaltrials.gov) evaluating treatment with Isa-Len-Dex.8 These patients had a confirmed diagnosis of MM, had progressed or were refractory to the immediate prior therapy, and had received more than two lines of anti-MM treatment, including many of whom were Len exposed and refractory.8 This phase Ib dose-escalation study assessed the safety, efficacy, and pharmacokinetics of Isa given in two schedules (3, 5, or 10 mg/kg every other week (Q2W) or 10 or 20 mg/kg weekly (QW) for 4 weeks and then Q2W thereafter (QW/Q2W)), in combination with Len 25 mg (days 1–21) and Dex 40 mg (QW). Patients received 28-day treatment cycles. Twenty-four patients were treated in the dose-escalation phase (3 mg/kg Q2W, n=4; 5 mg/kg Q2W, n=3; 10 mg/kg Q2W, n=6; 10 mg/kg QW/Q2W, n=3; 20 mg/kg QW/Q2W, n=8). Three patients were replaced for dose-limiting toxicity evaluation due to grade 3 infusion-associated reactions that occurred during the first Isa infusion and led to treatment discontinuation (1 patient at 3 mg/kg Q2W and 2 patients at 20 mg/kg QW/Q2W). We included all 57 patients with RRMM that received at least one dose of Isa-Len-Dex therapy despite dosage level 3, 5, 10, or 20 mg/kg and treatment schedule. Baseline patient demographics, disposition, and disease characteristics were summarized previously.8
Antibodies and reagents
Isa mAb was provided by Sanofi. Anti-HLA-ABC-FITC (clone: REA230), anti-CD38-FITC-PE (clone: IB6), anti-CD3-FITC (clone: BW264/56), and anti-CD56-PE (clone: AF12-7H3) were purchased from Miltenyi Biotec Inc (Auburn, California, USA). EasySep Direct human NK cell isolation kit was purchased from STEMCELL Inc (Vancouver, British Columbia, Canada). Carboxyfluorescein succinimidyl ester (CFSE), fixable viability dye (FVD), fetal bovine serum (FBS), interleukin 6 (IL-6), IL-2, and Penicillin-Streptomycin antibiotics (10,000 U/mL) were purchased from Thermo Fisher Scientific (Waltham, Massachusetts, USA).
MM cell lines
We obtained genomic DNA samples from >40 human myeloma cell lines from Keats lab (https://www.keatslab.org/myeloma-cell-lines). HLA typing was performed using a Luminex-based oligonucleotide probe hybridization method according to the manufacturer’s instructions (One Lambda, Canoga Park, California, USA). Based on the HLA class I types, 12 MM cell lines with specific HLA class I ligands (online supplemental table 1), such as those having homozygous HLA-C (C1C1 or C2C2) and with or without HLA-A3/11 or Bw4 ligands were identified for purchasing from DMSZ (https://www.dsmz.de/catalogues/catalogue-human-and-animal-cell-lines.html) or JCRB cell banks (http://cellbank.nibiohn.go.jp/english/). The MM cell lines were cultured in Roswell Park Memorial Institute 1640 (RPMI-1640) medium (Invitrogen, Carlsbad, California, USA) supplemented with 10% heat-inactivated FBS (Sigma, St. Louis, MO), IL-6, and antibiotics (100 units/mL penicillin, 100 mg/mL streptomycin) at 37°C in a humidified atmosphere with 5% CO2. These MM cell lines were routinely tested using MycoAlert PLUS Mycoplasma Detection Kit from Lonza (Walkersville, MD) and STR profiling to ensure they were free from mycoplasma.
Supplemental material
HLA and KIR genotyping
Genomic DNA from human PBMC and MM cell lines was extracted using the Qiagen DNA extraction kit (Qiagen, Valencia, California, USA). The HLA class I and KIR genotyping were performed using Luminex-based oligonucleotide probe hybridization methods according to the manufacturer’s instructions (One Lambda, Canoga Park, California, USA). Prediction of KIR haplogroups and centromeric and telomeric gene clusters were performed as described previously.29 Briefly, those having only the genes of group A haplotype KIRs (3DL3-2DL3-2DL1-2DP1-3DP1-2DL4-3DL1-2DS4-3DL2) were assigned as AA genotype carriers. The BB genotype refers to be lacking any of the four A haplotype-specific KIR genes (2DL1, 2DL3, 3DL1, and 2DS4). The AB genotype is heterozygous, carrying all A haplotype-specific KIR genes plus other KIR genes.
FCGR3A-158 genotyping
The single-nucleotide polymorphisms of FCGR3A-F158V were analyzed by a single-step multiplex allele-specific PCR and melting curve-based assay using modified primers sets.30 The primer sequences are: V allele-specific reverse primer: 5′-AGACACATTTTTACTCCCAAC-3′; F allele-specific reverse primer: 5′-GCGGGCAGGGCGGCGGGGGCGGGGCCGGTGATGTTCACAGTCTCTGATCACACATTTTTACTCCCAAA-3′; FCGR3A-specific common forward primer: 5′-TCCAAAAGCCACACTCAAAGTC-3′). The sequence and specificity of the primers were verified by direct sequencing of PCR amplicons from nine subjects (online supplemental figure 1). Only 50 patients were typed for FCGR3A-V158F dimorphism, and the quantity of DNA from the remaining 7 patients was not sufficient to perform this genotyping.
NK cell isolation
Peripheral blood samples were obtained from 11 selective donors with specific KIR+HLA compositions (online supplemental table 2). The NK cells were isolated by an immunomagnetic negative-selection method using the EasySep direct procedure according to the manufacturer’s instructions. The purity of NK cells following isolation was evaluated by flow cytometry, and those with >95% were used. These freshly isolated NK cells were stimulated overnight in IL-2 (100 U/mL) added RPMI-1640 medium supplemented with 10% heat-inactivated FBS.
Supplemental material
Flow cytometric ADCC analysis
To evaluate the impact of KIR receptors and cognate HLA class I ligand interactions on Isa-dependent NK cytolysis of MM cell line targets, a recently developed flow cytometry method using CFSE labeling and FVD staining was employed.31 Briefly, MM cell line targets were prelabeled with CFSE for 8 min and then divided into 10,000 cells/well in a 96-well culture plate. Target killing was initiated by the addition of following six different coincubation treatments overnight at 37°C: (1) no treatment as normalization control; (2) Len (10 µM); (3) Dex (10 µM); (4) Len and Dex (10 µM each); (5) Isa (10 µg/mL); (6) IL-2 activated NK cells (2.5:1 as the effector:target ratio); (7) IL-2 activated NK cells+Isa. The cells were harvested and stained with FVD for 20 min for the detection of dead cells. The cells were then washed with phosphate-buffered saline containing 2% FBS and then subjected to flow cytometry analysis using Fortessa x-20 (BD Biosciences, San Jose, California, USA). Data were collected, analyzed using Flowjo software. MM cell death rate was calculated by numbers of normalized CFSE+FVD+ cells/numbers of normalized CFSE+ cells. Len or Dex treatment for 72 hours with dosages ranging from 0 to 100 µM resulted in 0%–20% of death with all 10 MM cell lines except Dex-treated KAS61, which showed up to 40% cell death (online supplemental figures 2 and 3). Len and Dex (10 µM each) combined showed an additive effect on MM cell line death during 24 hours (online supplemental figure 3). To avoid the Len/Dex-mediated cytotoxicity background, we used untreated MM cell lines in all subsequent experiments to precisely examine the impact of KIR+HLA interactions on Isa-dependent NK cytolysis.
Supplemental material
Statistical methods
In the phase IIb study, progression-free survival (PFS) was defined as the time from the first dose of study medication to either disease progression or death, whichever came first. PFS was censored at the date of the last valid assessment before the cut-off date in the absence of either disease progression or death before the analysis cut-off date. The PFS estimates were calculated using the Kaplan-Meier method.32 All cytolysis experiments were conducted in triplicates and repeated at least twice. The mean percentage ±SD of each experimental condition was calculated from 3 to 6 replicate wells. A paired t-test was used to compare cell killing by different conditions. The coefficient of drug interaction (CDI) was used to calculate combination synergism, as previously described.33 The CDI is calculated as following: CDI = AB/(A×B). AB is the ratio of the combination groups to the control; A or B is the ratio of the single-drug group to the control. The CDI value <1, = 1, or >1 indicates that the combination is either synergistic, additive, or antagonistic, respectively. The statistical analysis was performed using GraphPad Prism V.6 (GraphPad Software, La Jolla, California, USA). Differences were considered to be significant at a p value below 0.05.
Results
Contrasting effects of KIR3DL2+HLA-A3/11+ and KIR2DL1+HLA-C2C2+ combinations on PFS
To examine whether KIR and its cognate HLA class I ligand combinations known to set NK cell functional threshold influence response to Isa-Len-Dex therapy, we determined the correlation between specific KIR+HLA combinations and median PFS time (MST). Nearly one-fourth of the patient cohort (24.6%; 14/57) carried the KIR3DL2+HLA-A3/11+ combination, and these patients had significantly prolonged PFS compared with the rest of the cohort lacking this combination (MST=19.35 vs 6.11 months, HR=0.43 (95% CI 0.22 to 0.88), p=0.02) (figure 2A). In contrast, patients with KIR2DL1+HLA-C2C2+ had significantly reduced PFS (MST=5.73 vs 13.33 months, HR=3.54 (95% CI 0.08 to 0.98), p=0.05) compared with the patients missing this combination (figure 2B). It is important to note that none of the 14 patients who carried the KIR3DL2+HLA-A3/11+ combination had a KIR2DL1+HLA-C2C2+ combination.
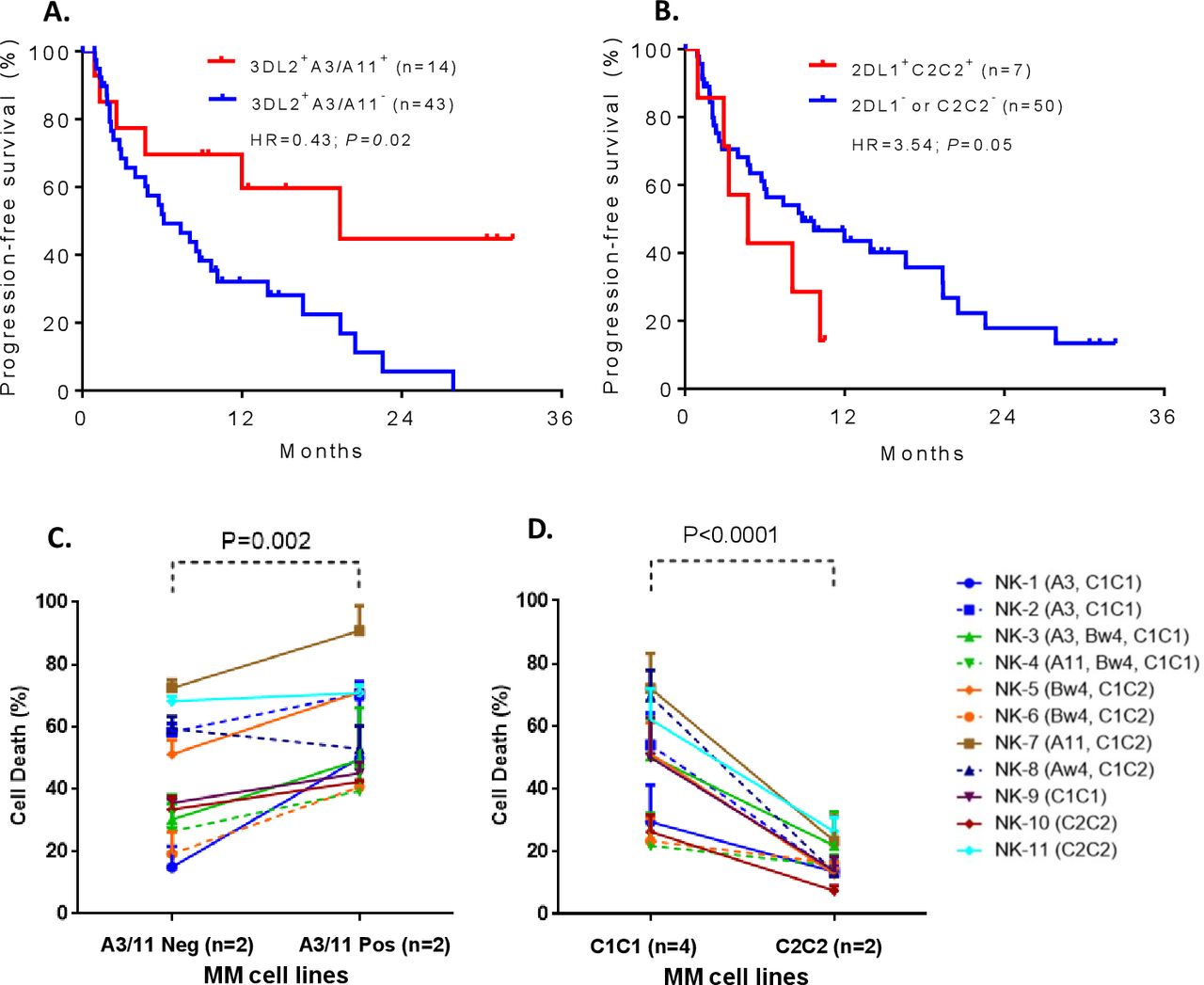
The Kaplan-Meier curves showing the impact of specific killer-cell immunoglobulin-like receptors (KIR)+human leukocyte antigen (HLA) combinations on progression-free survival (PFS) of relapsed or refractory multiple myeloma (RRMM) patients with RRMM treated by isatuximab+lenalidomide+dexamethasone (A, B). Patients carrying KIR3DL2+HLA-A3/11+ had significantly prolonged PFS compared with the patients missing those combinations (A). In contrast, patients with KIR2DL1+HLA-C2C2+ had significantly reduced PFS compared with patients missing this combination (B). Mechanistic studies confirm the role of these KIR+HLA interactions in modulating isatuximab-dependent natural killer (NK) cytolysis of MM cells (C, D). Multiple myeloma (MM) cells expressing HLA-A3/11 (ANBL6 expresses HLA-A3, and KMS18 expresses HLA-A11) showed a significantly improved isatuximab-dependent NK cytolysis compared with those that were negative for HLA-A3/11 (SKMM2 and JK6L) (C). All these four MM cell lines (ANBL6, KMS18, SKMM2, and JK6L) coexpress HLA-C1C1 and HLA-Bw4I80 ligands. The MM cell lines expressing HLA-C2C2 (Karpas620 and ARD) were resistant to isatuximab-dependent NK cytolysis compared with the MM cell lines (AMO1, KAS61, KMS20, and XG2) that are negative for HLA-C2C2 (D). These six MM cell lines (Karpas620, ARD, AMO1, KAS61, KMS20, and XG2) co-express HLA-Bw4T80 ligand. The cognate HLA class I ligands of the effector NK cells are given in parenthesis. Data provided in (C) and (D) represent the mean±SD of 11 independent experiments.
The KIR3DL2 is a framework gene and exists ubiquitously in all individuals, and thus the prolonged PFS is arguably ensured only by HLA-A3/11. To confirm the prolonged PFS is mediated by KIR3DL2+HLA-A3/11+ combination and not by HLA-A3/11 alone, we assessed the presence of other core HLA-A antigens that do not bind KIR3DL2 (eg, HLA-A1, A2, A24, A26, A29, and A68) and found no correlation between these core HLA-A antigens and PFS (online supplemental figure 4).
Supplemental material
Varying KIR and HLA interactions diversify Isa-dependent NK cytolysis of MM cells
We performed comprehensive mechanistic studies employing in vitro cell-killing experiments to confirm the role of ADCC in inter-individual variations of efficacy to Isa treatment for MM. We used primary NK cells with distinct KIR receptor repertoire (online supplemental table 2) and MM cell lines expressing minimal HLA class I ligands (online supplemental table 1). The NK cells alone can kill MM cell lines, but the degree of killing differs substantially among effector (NK cells) and target (MM cell lines) combinations (online supplemental figure 5A,B). Such variation in NK cytolysis is presumably due to the “missing ligand” phenomenon, where the absence of HLA class I ligand from MM cell targets to which the effector NK cells were initially licensed by their cognate HLA class I ligands. Supplementing Isa in NK+MM reactions enhanced the NK cytolysis into several folds via Isa-dependent NK cytolysis mechanism (online supplemental figure 5A, C and D).
Supplemental material
Opposing effects of KIR3DL2+HLA-A3/11+ and KIR2DL1+HLA-C2C2+ interactions on Isa-dependent NK cytolysis
We performed in-depth mechanistic studies using in vitro cell-killing experiments to confirm the opposing effects of KIR3DL2+HLA-A3/11+ and KIR2DL1+HLA-C2C2+ combinations on PFS are arbitrated by Isa-dependent NK cytolysis. The MM cell lines that are positive or negative for HLA-A3/11 or HLA-C2C2 ligands were subjected to killing by NK cells of 11 donors with different KIR+HLA constellations in the presence of Isa mAb. Consistent with clinical survival results, MM cells expressing HLA-A3/11 (ANBL6 expresses HLA-A3, and KMS18 expresses HLA-A11) showed a significantly improved Isa-dependent NK cytolysis compared with those MM cells (SKMM2 and JK6L) that were negative for HLA-A3/11 (p<0.002, figure 2C). The MM cells expressing HLA-C2C2 (Karpas620 and ARD) were resistant to Isa-dependent NK cytolysis compared with those MM cells (AMO1, KAS61, KMS20, and XG2) that were negative for HLA-C2C2 (p<0.0001, figure 2D). These effects were independent of the varied KIR receptor repertoire of NK cells used in these ADCC assays.
Patients with KIR2DL3+HLA-C1C1+ or KIR3DL1+HLA-Bw4I80+ combinations showed a tendency toward an improved PFS
Although the difference was not statistically significant, patients with KIR2DL3+HLA-C1C1+ combination showed a tendency toward an improved PFS compared with patients missing this combination (MST=9.69 vs 6.11 months, HR=0.81 (95% CI 0.42 to 1.57), p=0.55) (online supplemental figure 6A). No difference was observed in PFS with patients carrying KIR3DL1+HLA-Bw4+ combination compared with patients missing this combination (online supplemental figure 6B). The strength of KIR3DL1-mediated inhibition differs from HLA-Bw4 subtypes.34 35 The patients with strongly inhibiting ligand HLA-Bw4I80 and KIR3DL1 combinations showed a trend towards improved PFS compared with patients carrying weakly inhibiting KIR3DL1+HLA-Bw4T80+ combination (MST=11.99 vs 5.98 months, HR=1.81 (95% CI 0.71 to 4.60), p=0.4); however, this difference was not significant (online supplemental figure 6C). No difference was observed in PFS in patients having KIR3DL1+HLA-Aw4 (ie, HLA-A23, A24, A25, and A32) (online supplemental figure 6B) or in patients having or missing KIR2DL2+HLA-C1C1+ combinations (online supplemental figure 6D). Similarly, the activating KIR genes do not contribute to the PFS (online supplemental figure 7).
Supplemental material
Patients with high-affinity FCGR3A-158V allele showed an improved PFS
To determine if dimorphic residue 158 of FcRIIIa/CD16 influences the therapeutic outcome of Isa mAb by modulating Isa-dependent NK cytolysis, we compared the PFS between patients having high-affinity or low-affinity FCGR3A genotype. The distribution of FCGR3A-158 dimorphism in the patient cohort was 30% F/F, 28% V/F, and 7% V/V. Although not statistically significant, patients with high-affinity FCGR3A-158VV/VF genotype showed a trend of improved PFS compared with those carrying low-binding FF genotype (MST=13.93 vs 9.35 months, HR=0.55 (95% CI 0.24 to 1.27), p=0.37) (figure 3A). Next, we evaluated the functional implications of FCGR3A-158 dimorphism in Isa-dependent NK cell-mediated cytotoxicity. The presence of Isa in the in vitro killing assay increased NK cell cytolysis of MM cell line targets (figure 3B). The Isa-mediated NK cytolysis increased significantly with the presence of FCGR3A-158VV by over a factor of 3 compared with the NK cells that are homozygous for phenylalanine (p=0.0008) (figure 3B).
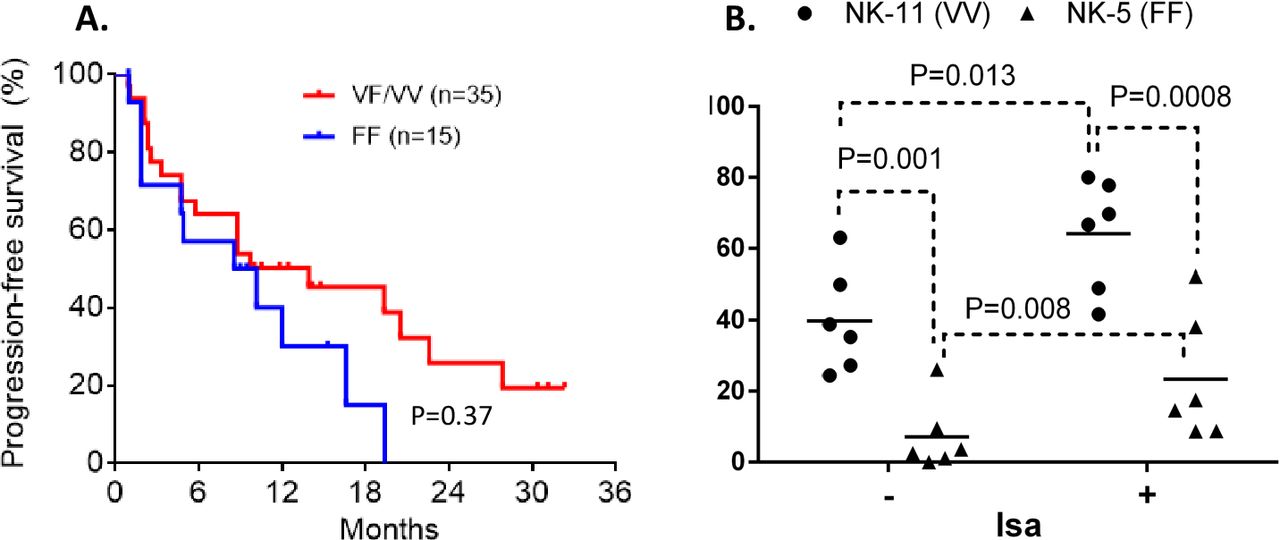
Impact of FCGR3A genotypes on progression-free survival (PFS) and isatuximab-dependent natural killer (NK) cell-mediated cytotoxicity. The Kaplan-Meier curves showing PFS with patients having a high-affinity FCGR3A-158VV/VF genotype compared with the patients having low-binding FCGR3A-158FF genotype (A). Six multiple myeloma (MM) cell lines expressing a similar set of human leukocyte antigen class I ligands (C1C1+Bw4+) were treated with and without isatuximab (10 µg/mL), and then exposed to primary NK cells carrying either homozygous FCGR3A-158VV or FF genotype. Effector NK cells with FCGR3A-158VV mediate greater isatuximab-dependent antibody-dependent cell-mediated cytotoxicity compared with the NK cells expressing FCGR3A-158FF (B).
We also investigated the effects of other KIR genotypes and the combined effects of multiple favorable KIR+HLA combinations (online supplemental table 3). Patients carrying high-affinity FCGR3A-158V allele and KIR3DL2+HLA-A3/11+ combination showed the highest MST (MST >32 vs 6.11 months, HR=0.35 (95% CI 0.16 to 0.74), p=0.007) (figure 4A). Similarly, patients carrying KIR3DL2+HLA-A3/11+ and KIR2DL3+HLA-C1C1+ combinations showed improved PFS (MST=19.35 vs 7.39 months, HR=0.45 (95% CI 0.21 to 0.96), p=0.04) (figure 4B). Of interest, 15.8% of patients carrying centromeric BB motif or centromeric AA motif with HLA-C2C2+ ligand showed worse PFS (online supplemental table 3).
Supplemental material

{kind=link}
{kind=link}
{kind=link}
{kind=link}
The Kaplan-Meier curves showing the impact of specific killer-cell immunoglobulin like receptors (KIR)+human leukocyte antigen (HLA) and FCGR3A gene polymorphisms on progression-free survival (PFS) of patients with relapsed or refractory multiple myeloma treated by isatuximab+lenalidomide+dexamethasone. Patients carrying KIR3DL2+HLA-A3/11+ plus FCRG3A-158V (A) or KIR2DL3+HLA-C1C1+ (B) had significantly prolonged PFS compared with the patients missing those combinations.
Discussion
Targeted mAb-based immunotherapies have revolutionized the treatment of cancer.36 In this phase Ib trial, the combination of Isa with Len and Dex resulted in encouraging clinical activity with an overall response rate of 56% and a clinical benefit rate of 71% in heavily pretreated patients with RRMM.8 The median PFS was 8.5 months. However, there was notable individual heterogeneity in the efficacy of Isa therapy, which we hypothesized was mediated by host genes that control NK cell response. Consistent with our hypothesis, we observed a strong correlation between the effectiveness of Isa mAb therapy and the specific constellation of KIR-HLA-FCGR3A genetic variables that control NK cell responsiveness.
The inhibitory KIR-mediated NK cell interactions with self-HLA class I molecules beget NK cell maturation (ie, licensing or education) and the subsequent ability to survey, recognize, and destroy virally infected or transformed cells that have downregulated HLA class I expression16–20—a recognition strategy described as “missing-self” hypothesis.37 38 The patient survival data and pertinent in vitro experiments presented in this study challenge this paradigm. Although all these inhibitory KIR+HLA interactions are presumably delivering inhibitory signals within the NK cells, the biological consequences of the signals produced by specific KIR+HLA interactions on ADCC appear to be paradoxical.
The most prolonged PFS was observed with patients carrying inhibitory KIR3DL2+HLA-A3/11+ combination. Consistent with the improved clinical outcome, the in vitro ADCC assays confirm a vigorous NK cytolysis of MM cell lines expressing HLA-A3/11 ligand. Indeed, the specific residues involving in KIR3DL2+HLA-A3/11+ interaction have been studied poorly.39 40 Peptide bound by HLA-A3/11 influences its binding to KIR3DL2,41 and KIR3DL2+HLA-A3/11+ recognition does not appear to educate NK cells.42 Although the mechanism of anti-MM activity rendered by inhibitory KIR3DL2+HLA-A3/11+ interaction is unclear, our results indicate that NK cell recognition through KIR3DL2+HLA-A3/11+ interaction may trigger more robust NK cytolysis of cancerous plasma cells via the ADCC pathway.
The inhibitory signals triggered by the high-affinity KIR2DL1+HLA-C2+ interaction were shown to be relatively more potent compared with those triggered by KIR2DL2/3+HLA-C1+ interactions.43 Presumably, the stronger inhibitory signals elicited by the KIR2DL1+HLA-C2+ might lead to vigorous suppression of NK cell effector function, including ADCC, and consequently compromise the efficiency of Isa-mediated anti-MM response in patients. Using in vitro mechanistic experiments, we confirmed such a dominant KIR2DL1+HLA-C2+ mediated inhibition of NK cytolysis of MM cell lines. These observations are congruent with prior findings showing an association between KIR2DL1+HLA-C2+ and increased susceptibility to childhood acute lymphoblastic leukemia44 and myeloid leukemia.45 Overexpression of KIR2DL1 on the entire NK cell population was shown to lead to severe immunodeficiency.46
Patients with acute myelogenous leukemia who underwent HLA-matched hematopoietic cell transplantation revealed that a strong inhibiting KIR3DL1+HLA-Bw4I80+ combination predicts higher relapse and mortality, whereas weak combinations were protective.47 In contrast to KIR2DL1+HLA-C2+ interaction, the strongly inhibiting KIR3DL1+HLA-Bw4I80+ interaction in the current study showed a propensity of improved PFS compared with the weakly inhibiting KIR3DL1+HLA-Bw4T80+ combination. The notion of potent inhibition-arbitrated NK cell hyporesponsiveness does not apply to all clinical settings and requires further investigation to understand the mechanisms.
Patients expressing high-affinity FCGRs showed prolonged PFS in this study, possibly due to increased Isa-binding and Isa-mediated NK cytolysis of cancerous plasma cells. Consistent with the direct in vivo effect of ADCC, our in vitro experimental data also convincingly show that MM cell lines are more susceptible to Isa-mediated ADCC when NK cells express high-affinity FCGR3A-158VV. In agreement with these data, the enhanced effect of the 158V polymorphism on ADCC was previously demonstrated using rituximab and NK cells from 52 healthy donors.48
With the rapid expansion of therapeutic options available for patients with MM, biomarkers predictive of clinical responses are becoming increasingly relevant.49 Our results may reveal the genetic and molecular basis of heterogeneous NK cell responses mediated by ADCC in patients with MM receiving Isa-Len-Dex immunotherapy. Hierarchical clustering based on the correlation between host NK cell immunogenetics distinguishes patients that benefit from Isa-Len-Dex combination therapy versus those who appear resistant. At the extremes of this hierarchy, over 17.5% of the patients carrying the best responding genotype KIR3DL2+HLA-A3/11+ plus FCGR3A-158V, while 15.8% have the worst responding mutually exclusive genotypes cen-BB or cen-AA+HLA-C2C2. These quantitative data-derived models may identify patients with a higher risk of resistance to Isa-Len-Dex combination therapy and offer a potential precision medicine approach for optimizing cancer immunotherapies. Additional strategies, including combinations with reagents that can enhance NK cell expansion and responsiveness, such as IL-2, are needed for all patients, especially those who may be genetically predisposed to resistance.50 Blocking the selective inhibitory KIR+HLA interactions using anti-inhibitory KIR mAbs may remove the overwhelming inhibition and, thereby potentially improve the anti-MM activity in patients with resistant KIR2DL1+HLA-C2+genotypes.51
Both KIR and HLA systems are polygenic and polymorphic and exhibit abundant diversity between human populations. The A and B haplotypes are equally distributed in Caucasians. Conversely, the A haplotype is over-represented in Northeast Asians (Chinese, Japanese, and Koreans), while the B haplotype occurs most frequently in the natives of India, Australia, and the USA.52 The A and B haplotypes differ primarily by activating KIRs—B haplotypes can have 1–5 activating KIRs while A haplotypes have only a single activating KIR. The inhibitory KIRs do not differ significantly between A and B haplotypes. Therefore, most individuals in any given population carry all inhibitory KIR genes. However, the HLA class I ligand distribution is diverse in populations. For example, Northeast Asians have a high incidence of HLA-A11 and HLA-C1, while Africans have a higher frequency of HLA-C2 compared with Northeast Asians (54% vs 24%). The HLA class I ligand diversity can affect inhibitory KIR+HLA class I ligand constellations, which in turn can influence ADCC-mediated Isa therapy in distinct populations. Therefore, the finding observed from this study can not be directly applicable to other populations. For example, the African population enriched with HLA-C2 ligands may frequently display inadequate ADCC-mediated response to Isa therapy.
To our knowledge, these data are the first to indicate that genotypic KIR+HLA and FCGR3A profiles of NK cells may affect their ability to induce ADCC and correlate with clinical outcomes in patients with RRMM treated with Isa therapy. Given the small size of our study, further investigations are needed in larger and potentially randomized trials with a fixed Isa dose and treatment schedule to test our hypothesis. Besides, the small number of patients studied here prevented evaluation of the impact of multiple KIR+HLA combinations. Therefore, further systematic analyses of associations should focus on determining the predictive value of multiple KIR+HLA and FCGR3A combinations in patients receiving Isa immunotherapies.
Data availability statement
Data are available upon reasonable request. The datasets generated and analyzed during the current study are available from the corresponding author on reasonable request.
Ethics statements
Ethics approval
The study includes 57 patients with RRMM enrolled in a phase Ib clinical trial (registered at www.clinicaltrials.gov as #NCT01749969) evaluating treatment with Isa-Len-Dex. The study protocol was approved by the ethics committee at every institution and conducted per recommendations of Good Clinical Practice and the Declaration of Helsinki. All patients provided written informed consent before participation in the study.
Acknowledgments
We thank all participants in this study.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors Conception and design: HS, TGM, JMV, RR. Provision of study materials or patients: TGM. Collection and assembly of data: HS, TGM, JM, DK, JK, SM, MC, JLW, JMV, RR. Data analysis and interpretation: HS, TGM, RR. Manuscript writing and editing: all authors. Final approval of manuscript: all authors. Accountable for all aspects of the work: all authors.
Funding This research was funded by Sanofi (to RR and TGM) and in part through the National Cancer Institute, National Institutes of Health Cancer Center Support (P30CA082103 to TGM).
Competing interests HS: no relationship to disclose. TGM: research funding: Sanofi. JM: no relationship to disclose. DK: no relationship to disclose. JK: no relationship to disclose. SM: employee of Sanofi. MC: employee of Sanofi. JLW: consulting or advisory role: Onyx/Amgen, Takeda, and Celgene. JMV: no relationship to disclose. RR: research funding: Sanofi; consulting or advisory role: CareDx.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.