Article Text

Abstract
Background Human telomerase reverse transcriptase (hTERT) is frequently classified as a ‘universal’ tumor associated antigen due to its expression in a vast number of cancers. We evaluated plasmid DNA-encoded hTERT as an immunotherapy across nine cancer types.
Methods A phase 1 clinical trial was conducted in adult patients with no evidence of disease following definitive surgery and standard therapy, who were at high risk of relapse. Plasmid DNA encoding one of two hTERT variants (INO-1400 or INO-1401) with or without plasmid DNA encoding interleukin 12 (IL-12) (INO-9012) was delivered intramuscularly concurrent with the application of the CELLECTRA constant-current electroporation device 4 times across 12 weeks. Safety assessments and immune monitoring against native (germline, non-mutated, non-plasmid matched) hTERT antigen were performed. The largest cohort of patients enrolled had pancreatic cancer, allowing for additional targeted assessments for this tumor type.
Results Of the 93 enrolled patients who received at least one dose, 88 had at least one adverse event; the majority were grade 1 or 2, related to injection site. At 18 months, 54.8% (51/93) patients were disease-free, with median disease-free survival (DFS) not reached by end of study. For patients with pancreatic cancer, the median DFS was 9 months, with 41.4% of these patients remaining disease-free at 18 months. hTERT immunotherapy induced a de novo cellular immune response or enhanced pre-existing cellular responses to native hTERT in 96% (88/92) of patients with various cancer types. Treatment with INO-1400/INO-1401±INO-9012 drove hTERT-specific IFN-γ production, generated hTERT-specific CD4+ and CD8+ T cells expressing the activation marker CD38, and induced hTERT-specific activated CD8 +CTLs as defined by cells expressing perforin and granzymes. The addition of plasmid IL-12 adjuvant elicited higher magnitudes of cellular responses including IFN-γ production, activated CD4+ and CD8+ T cells, and activated CD8+CTLs. In a subset analysis of pancreatic cancer patients, the presence of immunotherapy-induced activated CD8+ T cells expressing PD-1, granzymes and perforin correlated with survival.
Conclusions Plasmid DNA-encoded hTERT/IL-12 DNA immunotherapy was well-tolerated, immune responses were noted across all tumor types, and a specific CD8+ phenotype increased by the immunotherapy was significantly correlated with survival in patients with pancreatic cancer.
- immunotherapy
- immunity
- cellular
Data availability statement
All data relevant to the study are included in the article or uploaded as online supplemental information.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
The telomerase complex is critical for maintaining telomere length at chromosome ends during the semiconservative DNA replication and is expressed mainly in embryonic cells.1–3 Reactivation of telomerase is a primary mechanism of cell immortalization leading to cancer.4–6 Human telomerase reverse transcriptase (hTERT), the catalytic subunit of the telomerase complex, is highly expressed in more than 85% of human tumors of diverse origin.6 7 In contrast, hTERT is not expressed in somatic cells, aside from low expression in a few cell types with high self-renewal capacity.8–11 Telomerase has additional functions beyond maintaining telomere ends that contribute to its oncogenic properties, which include promoting cell proliferation, resistance to apoptosis, epithelial to mesenchymal transition, transcriptional regulation and metabolic reprogramming.12 13 Furthermore, hTERT expression has been demonstrated in cells from all stages of cancer development, including cancer stem cells.5 6 14–16 Therefore, therapies that target hTERT have the potential to abrogate cancer stem cells as well as cells in all other stages of cancer development. Because hTERT is expressed in such a vast number of cancers and cancer cell stages, it has been classified as a ‘universal’ tumor-associated antigen.17 18
Several studies have shown that cancer cells process and present epitopes from hTERT on Major Histocompatibility Complex (MHC).19–24 In addition, CD8+ and CD4+T cells specific for hTERT can be detected in the blood of both healthy individuals and cancer patients.25–28 Telomerase-specific CD8 +T cells have been found in patients with cancers including prostate, breast, lung, gastric, and colorectal, as well as hepatocellular carcinoma, chronic myeloid leukemia and non-Hodgkin’s lymphoma.24–26 29–32 Telomerase-specific CD4 +T cells are also found in patients with cancers and associate with overall survival in non-small cell lung cancer, renal cell carcinoma, and anal squamous cell carcinoma.27 33 These studies demonstrate that although telomerase is a self-antigen, some hTERT-specific T lymphocytes are able to escape thymic deletion.
To date, a number of clinical trials have been performed using immunotherapies to target hTERT, including vaccination.9 24 The trials competed thus far have shown that targeting hTERT with vaccines is safe and can induce immunological responses in 50%–100% of vaccinated patients.18 34 35 Several trials have also demonstrated a clear correlation between vaccine-induced immune responses and clinical response.36–39 Vaccines tested in clinical trials thus far include mainly peptide vaccines and dendritic cell vaccines.9 24 DNA vaccines targeting hTERT are less common but have also been tested.40 41 In a phase I clinical trial using INVAC-1, a modified full-length hTERT vaccine, hTERT-specific CD8+ and CD4+T cell responses were detected in 25% and 63% of patients, respectively, and disease stabilization was observed in 58% of patients.41 However, the length of stable disease was only 2.7 months, which is shorter than previous studies using hTERT peptide or dendritic cell vaccines.36 Taken together, the results of clinical trials demonstrate that while hTERT-based vaccination can induce immune responses in patients, these immune responses to date have not generally been enough to control tumor growth or disease progression in advanced cancer patients.24
Improvements in DNA vaccines can be accomplished using modifications to increase processing and immunogenicity, which may lead to increased vaccine efficacy, and be engineered to help evade tolerance.42 The INO-1400 and INO-1401 plasmids encode a modified version of full-length hTERT that are RNA and codon optimized and both contain a highly efficient leader sequence. The INO-1400 plasmid includes two point mutations in the hTERT sequence to aid in breaking tolerance43 44 and remains over 99% homologous to the native hTERT sequence, while the INO-1401 plasmid encodes a further modified hTERT sequence with approximately 95% similarity to the native hTERT protein, which may further impact host immunity uniquely. The INO-1400/INO-1401 plasmids encode for full length hTERT, which is processed endogenously, resulting in presentation of peptides across multiple HLA alleles. Importantly, this hTERT DNA vaccine was shown to induce strong immunological responses in both mice and non-human primates.44
Here, we explore the safety and immunogenicity of plasmids encoding modified hTERT (INO-1400/INO-1401) alone or in combination with interleukin 12 (IL-12) plasmid (INO-9012) in patients with various solid tumors (breast, lung, pancreatic, head and neck, ovarian, colorectal, gastric, esophageal or hepatocellular) who are at a high risk of relapse post definitive surgery and standard adjuvant therapy.
Materials and methods
Study population
Patients, at least 18 years old, with one of nine specific solid tumors: breast carcinoma (stage II with axillary node-positive disease, stage III or stage IV following metastatectomy), non-small cell lung cancer (stage IB, II, IIIA; stage IIIB or stage IV following metastatectomy), pancreatic ductal adenocarcinoma (stage I, II, III; stage IV following metastatectomy), head and neck squamous cell carcinoma (AJCC 7 stage III; stage IV following metastatectomy), ovarian carcinoma (stage III; stage IV following metastatectomy), colorectal adenocarcinoma (stage III; stage IV following metastatectomy), gastric or esophageal carcinoma (Stage IIB, III), or hepatocellular carcinoma (any stage, ineligible for and not post-liver transplantation; Child-Pugh class A required) who were at high risk of relapse, had been treated with curative intent, and had no evidence of disease (NED) following front-line therapy were enrolled. Therapy was completed no fewer than 4 weeks, and no later than 25 weeks, before first dose of study drug(s). Patients had an Eastern Cooperative Oncology Group performance status of 0–1, adequate organ function and no higher than National Cancer Institute Common Terminology Criteria for Adverse Events (CTCAE; version 4.03) grade 1 or 2 laboratory parameters at screening. Patients were excluded if they received treatment with any hTERT targeted or IL-12-based therapy in the past or any immune modulators within the past 3 years, had metastasis in the brain or central nervous system, had any malignant condition requiring active treatment, any clinically significant autoimmune disease or were chronically immunosuppressed. Written informed consent was obtained from each patient prior to performing any assessments.
Immunotherapy and delivery using CELLECTRA device
INO-1400 and INO-1401 are synthetic DNA plasmids encoding for a modified human telomerase protein. INO-9012 consists of a DNA plasmid encoding for synthetic human IL-12 (p35 and p40 subunits). All constructs were designed using proprietary technology (Inovio Pharmaceuticals, Inc.). The CELLECTRA 2000 adaptive constant current electroporation device (EP; Inovio Pharmaceuticals) delivers three 52 ms controlled electric pulses, spaced in 1 s intervals, through a sterile, disposable array to the injection site. INO-1400/1401 with or without INO-9012 was formulated in sterile water for injection and delivered intramuscularly (IM) in a 1 mL volume followed immediately by EP.
Study design
This phase 1, multicenter, open-label, dose-escalation study was performed in adults with NED following definitive surgery and standard adjuvant therapy, who are at high risk of relapse. Following informed consent, each patient was enrolled into one of 10 treatment arms (online supplemental table 1) for a total of nine patients per treatment arm: Arm 1: 2 mg INO-1400, Arm 2: 8 mg INO-1400, Arm 3: 2 mg INO-1400 +0.5 mg INO-9012, Arm 4: 2 mg INO-1400 +2 mg INO-9012, Arm 5: 8 mg INO-1400 +0.5 mg INO-9012, Arm 6: 8 mg INO-1400 +mg INO-9012, Arm 7: 2 mg INO-1401, Arm 8: 8 mg INO-1401, Arm 9: 8 mg INO-1401 +0.5 mg INO-9012, Arm 10: 8 mg INO-1401 +2 mg INO-9012. Patients received the first of four doses of either INO-1400 or INO-1401 with or without INO-9012 on day 0, followed by three additional doses each given 4 weeks apart. Blood collection for immunological analyses occurred at weeks 0, 2, 6, 10 and 14, and every 12 weeks thereafter. Radiological disease assessment was requested at screening and every 12 weeks.
Supplemental material
A modified ‘3+3’ design was used to assess safety and tolerability in up to the first 6 patients in each arm regardless of tumor type. Patients were then enrolled to the next treatment if there was 0/3 or 1/6 dose limiting toxicities (DLTs) in the arm. Arms were opened sequentially once each patient had been assessed for a minimum of 6 weeks followed by a safety review of all available data. Enrollment to Arm 7, the first dosing arm of INO-1401, was not contingent on an additional safety review of INO-1400. DLTs were defined as related CTCAE ≥grade 3 injection site erythema, swelling or induration after study treatment; pain or tenderness at the injection site that required overnight hospitalization despite proper use of non-narcotic analgesics; ≥grade 3 headache in patients who did not have a history of recurrent headaches; or ≥grade 3 laboratory abnormalities.
The primary objective was to evaluate the safety and tolerability of INO-1400 or INO-1401 alone or in combination with INO-9012, delivered IM, followed by EP. The secondary objectives were to determine the humoral and cellular immune responses and the exploratory objectives were to evaluate disease-free survival (DFS) and overall survival (OS).
The study was registered on ClinicalTrials.gov with the identifier NCT02960594.
Safety assessments
Local and systemic adverse events (AEs), vital signs, targeted physical assessments, and the development of laboratory abnormalities were monitored from the date of informed consent through the last follow-up visit. In particular, injection site reactions, including local skin erythema, induration, pain and tenderness were assessed via a participant reminder card on the day of each treatment and for three consecutive days post-treatment. Patients were queried at each visit for new AEs or disease and use of concomitant medications. All events were graded in accordance with CTCAE V.4.03 and all injection site reactions were measured in accordance with the U.S. Food and Drug Administration (FDA) Guidance for Industry- Toxicity Grading Scale for Health Adult and Adolescent Volunteers Enrolled in Preventive Vaccine Clinical Trials (September 2007) and coded with MedDRA V.21.0.
Further enrolment and treatment were to be stopped if one third or more patients experienced a DLT; unexpected grade 4 toxicity; potentially life-threatening AE or death assessed as related to study treatment; three or more patients experienced the same related grade 3 or 4 AE; or any report of grade 3 anaphylaxis.
Women of childbearing potential completed a β-HCG test at screening and within 3 days prior to each dose. Laboratory parameters including hematology, coagulation, serum chemistry (including liver function) and creatine phosphokinase were monitored throughout the study and assessed locally at the center.
Sample collection
Whole blood was collected in ACD-A tubes and peripheral blood mononuclear cells (PBMCs) were isolated, followed by cryopreservation. For analysis, PBMCs were thawed, recovered overnight in cell culture medium, spun, washed and resuspended the following day for cellular immune analyses in batches.
Interferon gamma ELISpot
A standard ELISpot assay was performed (MabTech, OH). Briefly, PBMCs were stimulated with native hTERT peptides or media and DMSO (negative control) at the same concentration (2 µg/mL) for 18–24 hours. The peptides were 15–amino acids in length, overlapping by eight amino acids, and encompass the entire native hTERT sequence. The peptides were split up into four pools such that each pool includes approximately a quarter of the peptides in a sequential format. The hTERT-specific values graphed are the sum of the background subtracted four hTERT peptide pool stimulated wells at baseline (pre) and the study week with the highest value (peak).
Lytic granule loading
1×106 PBMCs were stimulated 5 days with peptides corresponding to hTERT pooled at a concentration of 2 µg/mL, while an irrelevant peptide was used as a negative control (OVA) and concanavalin A was used as a positive control (Sigma-Aldrich). No co-stimulatory antibodies or cytokines were added to cell cultures at any point. After 5 days, cells wee stained for CD3, CD4, CD14, CD16, CD137, granulysin, CD19, CD38, CD8, granzyme B (BD Biosciences), granzyme A (ThermoFisher), Programmed Cell Death-1 (PD1), perforin, and CD69 (BioLegend). Staining for extracellular markers (CD4, CD8, CD137, CD69, CD38, PD-1) occurred first, followed by permeabilization to stain for the remaining markers. The frequency of hTERT-specific responses per output are calculated by subtracting the background frequency in the negative control wells. Acquired data were analyzed using the FlowJo software version X.0.7 or later (Tree Star).
Statistical methods
The study was designed to be analyzed descriptively and did not include any formal statistical hypothesis. Any patient who received at least one treatment dose and had data available post first dose was included in the primary and safety analyses. Patients were allocated to the modified intention-to-treat (mITT) and per-protocol (PP) populations for secondary cellular and humoral immunogenicity and exploratory Kaplan-Meier analyses of DFS efficacy analyses. The mITT population included all patients who received at least one dose of study treatment. For the primary objective, the percentage of patients with AEs and DLTs was summarized along with corresponding exact 95% Clopper-Pearson CIs for each treatment arm and overall. Continuous outcomes such as laboratory observations and vital signs were summarized using the mean/median, SD, range and 95% CI. Binary outcomes such as number of relapsed patients were summarized using proportion and exact Clopper-Pearson 95% CI. Time to event outcomes were analyzed using the Kaplan-Meier method on all treatment cohorts combined as well as separately by indication. The Kaplan-Meier statistics included the number of relapses and deaths, number of censored patients, Kaplan-Meier estimates at 3, 6, 12 and 18 months from the time of first dose (day 0), and the median time to DFS from time of first dose (day 0) with 95% CI based on the log-log transformation.
Wilcoxon-Mann-Whitney rank sum tests (between cohorts) or Wilcoxon signed-rank tests (within cohorts) were used to assess the significance of immunogenicity analyses due to the non-normality of these types of data. Because these analyses were intended to be hypothesis-generating for future studies, p values were not corrected for multiplicity and therefore do not account for type I errors.
All statistical analyses were conducted in SAS V.9.4 and Prism V.8.4.3.
Results
Clinical characteristics
A total of 93 patients with one of nine solid tumor types were enrolled between February 6 2015 and January 26 2018 (table 1). The largest cohort of patients had pancreatic cancer (34 of 93; 37%), followed by colorectal cancer (18 of 93; 19%) and non-small cell lung cancer (14 of 93; 15%) (online supplemental table 2). The median age was 58 years (range 28 to 76 years). Sixty-six percent of patients were female, 90% were white, and 93% were non-Hispanic/Latino. All 93 patients received at least one dose PP.
Patientdemographics (mITT population)
Safety
Of 93 patients who received at least one dose, 88 (94.6%) had at least one AE with 77.4% of these being grade 1 or 2. Sixteen patients (17.2%) reported at least one grade 3 event, and one patient had a grade 4 event of sepsis (online supplemental table 3); no grade 5 events were reported. Of the AEs reported across all arms, injection site reactions (grades 1 and 2) were reported most often, with injection site pain (76 patients; 81.7%), injection site swelling (18; 19.4%), injection site erythema (18; 19.4%), and injection site bruising (11; 11.8%) reported. Fatigue was reported in 19 (20.4%) patients, abdominal pain in 10 (10.8%), diarrhea in 10 (10.8%), and pain in extremity in 10 (10.8%). There were 19 SAEs reported in 11 (11.8%) patients, only two grade 3 events (breast cellulitis and abdominal pain) were considered treatment related. There was one treatment-related AE (rash, maculopapular) that was considered dose-limiting and led to study drug being permanently discontinued in this patient. (online supplemental table 3) lists grade 3 and 4 AEs by System Organ Class and Preferred Term reported, of any relationship.
Survival and disease-free analysis
Online supplemental figure 1 shows Kaplan-Meier analysis of DFS across all treatment arms in the mITT population (N=93). At 18 months, 54.8% of all patients were disease-free, with median DFS not reached by end of the study. For patients with pancreatic cancer, the median DFS was 9 months with 95% CI (4.5 to not available), with 41.4% of these patients disease-free at 18 months (figure 1). Online supplemental figure 2 shows time from diagnosis to first dose (day 0) of study treatment, and time on study from day 0 by tumor type.

Kaplan-Meier analysis of disease-free survival in pancreatic carcinoma Patients (mITT population, N=34). mITT, modified intention-to-treat.
INO-1400/1401 induces hTERT-specific interferon gamma production from T cells
IFN-γ ELISpot was used to assess levels of cellular reactivity to the endogenous form of hTERT before, during and following completion of immunotherapy in all treated patients with available sample (n=92). The peak response (defined as the highest magnitude observed above the day of treatment initiation) was identified for each individual and graphed together with the predose time point value for each treatment arm. Magnitudes of hTERT-specific IFN-γ secreting cells significantly increased in 9 out of 10 arms (online supplemental figure 3A). Immune responses against hTERT antigen were similar when comparing the increases over baseline for INO-1400 and INO-1401 immunotherapies (online supplemental figure 3B) and cohorts were therefore combined for further analysis. The impact of hTERT dose as well as the inclusion and dose of IL-12 on the immune response following immunotherapy was also explored. Patients were also grouped by tumor type to assess the response to immunotherapy across various cancers.
Patients given a low (2 mg) or high (8 mg) dose of hTERT immunotherapy had similar significant increases in the number of hTERT-specific IFN-γ secreting cells from pre-dose to peak post-dose time points, p=0.003 and p=0.015 for low and high-dose groups, respectively (figure 2A, top row). The median spot-forming unit (SFU, cells per million PBMCs) values post treatment were 20 and 14.5 with a maximum response of 48.9 and 88.9, respectively. Low-dose and high-dose cohorts that received IL-12 also had significant increases in the number of hTERT-specific IFN-γ secreting cells (p<0.001) from predose to peak postdose time points (figure 2A, bottom row). Moreover, the inclusion of IL-12 significantly improved immunotherapy induced responses, as defined by the change in hTERT-specific SFU from predose to the peak postdose response time point in the low-dose group (p=0.028, figure 2B), and increased the median change in SFU 2.5-fold with a maximum magnitude of 617.8 SFU. Magnitudes in the high-dose cohort that received IL-12 had higher SFU values than the high-dose cohort without IL-12 (peak response of 338.9 vs 67.8, respectively), as well as a 2.8-fold higher median change from baseline that did not reach significance.

IFN-γ ELISpot responses broken out by hTERT dose, IL-12 dose, and tumor type. (A, C, E) Open symbols represent individual patients, the box extends from the 25 th to the 75 th percentile, line inside the box is a the median, and the whiskers extend from the minimum to maximum values. Wilcoxon signs rank test was used to assess significance between the magnitude of IFN-g in patients before (PRE) and after (POST) immunotherapy. The number of patients in each group is displayed below the graph, N. (B, D) The increase over baseline is shown for each group. Wilcoxon ranked sum test was used to assess significance of the increase over baseline between treatment groups. The number of patients, means, medians and ranges of the delta magnitudes for each treatment group are shown. IFN-γ ELISpot responses broken out by hTERT dose, IL-12 dose, and cancer type. hTERT, human telomerase reverse transcriptase; IFN-γ, interferon-γ; IL-12, interleukin 12.
The effect of the dose of the cytokine adjuvant, IL-12, was also explored, irrespective of antigen dose (figure 2C). Patients who received no IL-12, a low dose of IL-12 (0.5 mg), or a high dose of IL-12 (2.0 mg) had significant increases in the number of hTERT-specific IFN-γ secreting cells (p<0.001 in each case) from predose to peak postdose time points (figure 2C). Both doses of IL-12 examined resulted in numerically higher hTERT-specific SFUs above predose compared with the arms without IL-12 (figure 2D), but the delta magnitudes were not significantly different from one another (figure 2D).
The number of patients with samples for immunology testing were as follows pancreatic (n=34), colorectal (n=18), lung (n=13), ovarian (n=9), breast (n=10), and ‘other’ (n=8). The ‘other’ subgroup consisted of patients with gastric (n=1), head and neck (n=3), esophageal (n=2), and hepatocellular (n=2) cancer types. When grouped together by cancer types, significant increases in IFN-γ secreting cells following immunotherapy were observed in pancreatic (p<0.001), lung (p<0.001), and ovarian (p=0.012) cancer patients (figure 2E). Patients with colorectal and breast cancer had numerical increases as well but not statistically significant (p=0.062 and p=0.088, respectively).
To determine whether patients with pre-existing hTERT-specific IFN-γ secreting cells responded more favorably to immunotherapy, patients were divided into two groups based on the presence or absence of hTERT-specific IFN-γ secreting cells at baseline. The increase over baseline following immunotherapy was not statistically different between the two groups, although the mean was numerically higher in the group that had pre-existing responses (online supplemental figure 3C).
INO-1400/1401 induces hTERT-specific activated CD4+ and CD8+T cells
To characterize the function of the immune response elicited by the immunotherapy, hTERT-specific responses were assayed on CD4+ and CD8+T cells using flow cytometry based on percent expression of the activation marker CD38. The change in expression of CD38 on CD4 +T cells was similar post treatment regardless of dose (figure 3A). Specifically, 29.6% of patients receiving the 2.0 mg dose of immunotherapy exhibited an increase in hTERT-specific CD4 +T cells after treatment compared with 20% of patients receiving the 8.0 mg dose. For both hTERT doses, overall magnitudes of CD4 +CD38+T cells increased after treatment with a difference in the means of 0.06% and 0.1% in the 2 mg and 8 mg dose groups, respectively (figure 3A, left panel). When assessing CD8 +T cells expressing CD38, the frequency of patients exhibiting hTERT-specific responses was again similar with 25.9% in the 2.0 mg group and 20.6% in the 8.0 mg group displaying increases in frequencies after treatment. However, the overall magnitude of response was higher in the 2.0 mg group showing an increase in the difference of the means of 0.37% after treatment, while the 8.0 mg group showed a decrease in the difference of the means after treatment of 0.19% (figure 3A, right panel).

Activated CD4+ (left column) or CD8+ (right column) T cells broken out by hTERT dose, IL-12 dose and cancer type. (A–C) Open symbols represent individual patients, the mean is represented with ‘+’, the box extends from the 25 th to the 75 th percentile, line inside the box is a the median, and the whiskers extend from the minimum to maximum values. Wilcoxon signs RANK test was used to assess significance between the magnitude of IFN-g in patients before (PRE) and after (POST) immunotherapy. The number and percent of patients in each group that had an increase, decrease or no change from baseline is displayed below each graph. hTERT, human telomerase reverse transcriptase; IFN-g, interferon-γ; IL-12, interleukin 12.
We additionally profiled immune responses to INO-1400/1401 as a function of IL-12 plasmid dose, which was tested at 0.5 mg (low) and 2.0 mg (high) (figure 3B). Of the patients who did not receive any IL-12 adjuvant, 15.4% had an increase in the frequency of hTERT-specific CD4 +T cells following treatment. In the patients who received the IL-12 adjuvant, 25.0% of the low-dose and 35.0% of the high-dose recipients had an increase in the frequency of activated CD4 +T cell frequencies after treatment. There was in increase in the difference of the means postdose versus predose of 0.12% in the low-dose and 0.20% in the high-dose IL-12 treatment groups. Similar trends were observed in the CD8 +T cell compartment, for which a numerically higher number of patients had increases in activated hTERT-specific cells in the IL-12 treated groups. Specifically, 33.3% of the low-dose IL-12% and 31.6% of the high-dose IL-12 recipients had increases in the frequency of these cells compared with 11.1% of patients who did not receive IL-12. The largest mean difference of 0.37% was observed in the high-dose IL-12 group, followed by 0.03% in the low-dose IL-12 group. The postdose mean value in the no IL-12 group decreased by 0.15% compared with the predose mean value.
Last, we looked at the ability of INO-1400/1401 to induce CD4+ and CD8+T cell activation based on disease condition. To that end, we considered patients based on the six tumor types, as noted above in (figure 3C). Colorectal, lung and pancreatic cancer diagnoses had the highest patient numbers for analysis with 15, 11 and 19, respectively. Observation of the CD4 +T cell compartment revealed that patients with breast, colorectal, ovarian and pancreatic tumors exhibited hTERT-specific activation as evidenced by CD38 upregulation (figure 3C left panel). In particular, the patients with colorectal and pancreatic cancer saw the highest frequency of increase over predose values with 26.7% and 36.8% of patients responding, respectively. In the CD8 +T cell compartment, similar trends were noted, that is, breast, colorectal and pancreatic patients exhibited hTERT-specific activation as evidenced by CD38 upregulation (figure 3C right panel). Similar to CD4 +T cell activation, 33.3% of patients with colorectal cancer had an increase in activated CD8 +T cells postimmunotherapy, although the magnitude of increase over predose values was low (difference in means of 0.07%) In addition, 31.6% of patients with pancreatic cancer exhibited an increase over predose frequencies, with a difference in means of 0.67%.
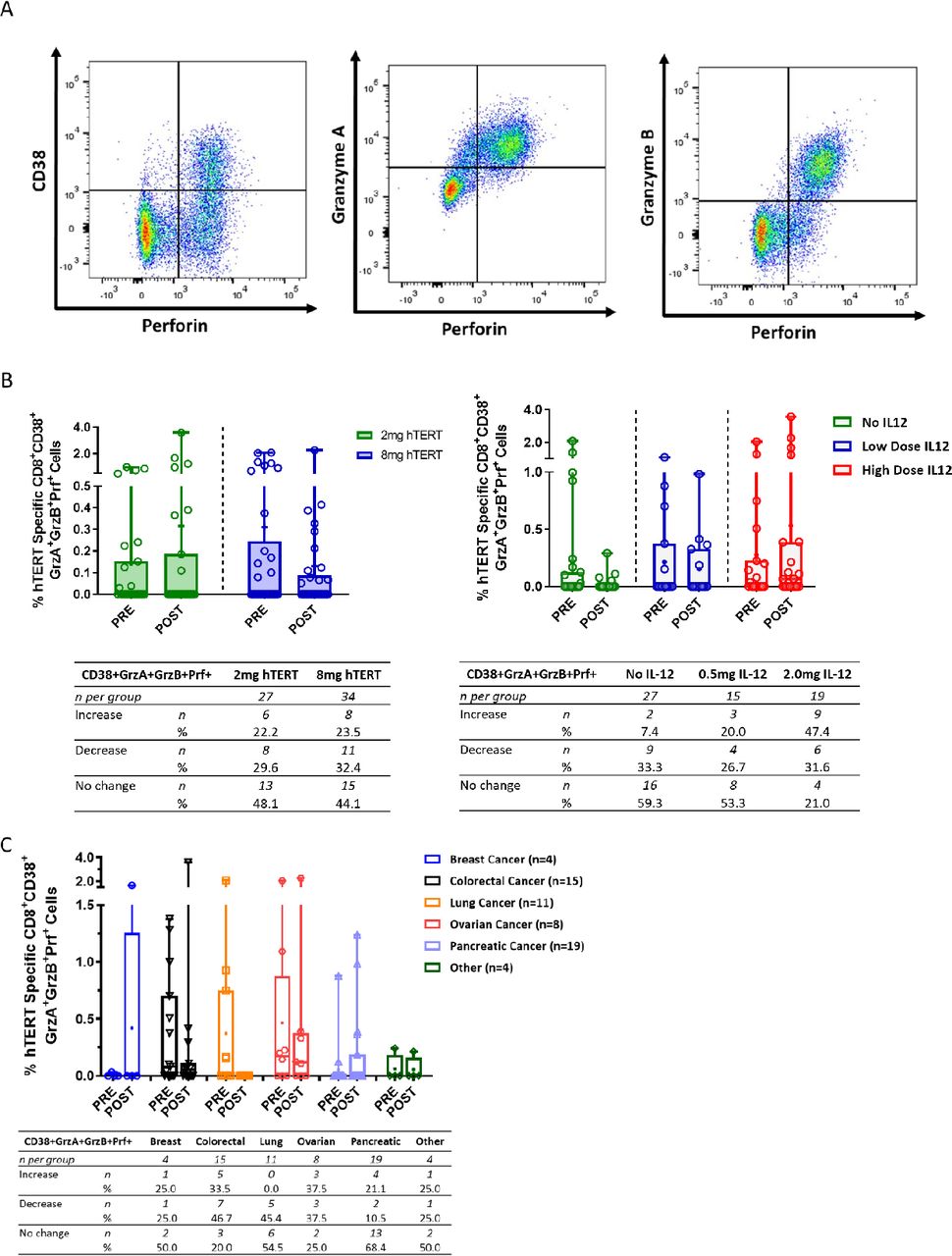
CD8+ T cells activated by INO-1400/1401 exhibit lytic potential
To further characterize the function of the immune response elicited by the immunotherapy, hTERT-specific responses were assayed using a lytic granule loading (LGL) assay that examines the activation status and lytic potential of CD8 +T cells. Example flow plots of live CD3 +CD8+T cells expressing the activation marker CD38 and lytic proteins perforin, granzyme A and granzyme B are shown in figure 4A. The low-dose hTERT immunotherapy arms had numerical increases after treatment in the means of hTERT-specific activated CD38 +CD8+T cells with lytic potential, coexpressing granzyme A, granzyme B, and perforin (0.17%, figure 4B, left panel). In contrast the high-dose hTERT arms had a trend toward higher frequencies that were not boosted following immunotherapy and resulted in numerical decreases in the difference of the means (0.18). However, a similar number of patients had an increase in activated CD8 +T cells with lytic potential- 22.2% (6/27) and 23.5% (8/34) in the low-dose and high-dose study arms, respectively (figure 4B, left panel).

Activated CD8 +T cells with lytic potential broken out by hTERT dose, IL-12 dose and cancer type. (A) Representative dot plot showing CD8 +T cells expressing the activation marker CD38 and lytic granules—perforin, granzyme A and granzyme B. (B, C) Open symbols represent individual patients, the mean is represented with ‘+’, the box extends from 25 th to 75 th percentile, line inside the box is a the median, and the whiskers extend from the minimum to maximum values. Wilcoxon signs rank test was used to assess significance between the magnitude of IFN-g in patients before (PRE) and after (POST) immunotherapy. The number and per cent of patients in each group that had an increase, decrease or no change from baseline is displayed below each graph. hTERT, human telomerase reverse transcriptase; IFN, interferon; IL-12, interleukin.
The effect of the addition of IL-12 to the immunotherapy regimen was also examined in the LGL assay, for which the high dose of IL-12 trended toward a numerically higher mean of activated cells coexpressing lytic proteins (difference in means of 0.44%), which was not observed in the low-dose IL-12 and no IL-12 arms (figure 4B, right panel). Similarly, a trend toward the number of patients with an increase in the frequency activated CD8 +T cells with lytic following immunotherapy was observed with increased amounts of IL-12, that is, 7.4% (2/27) of patients in the no IL-12 arm, 20% (3/15) in the low-dose IL-12 arm, and 47.4% (9/19) in the high-dose IL-12 arm (figure 4B, right panel).
When grouped together by specific tumor type, a range of responses was observed depending on the tumor type (figure 4C). Similar mean increases in activated T cells with lytic potential were observed in the breast and pancreatic cancer subgroups, 25.0% and 21.1%, respectively (figure 4C). 33.5% of colorectal and 37.5% of ovarian patients had an increase in activated CD8 +T cells with lytic potential; however, the mean frequency of these cells decreased post treatment. Interestingly, 20% or more of patients in all subgroups except for those with lung cancer, had an increase in activated hTERT-specific CD8 +T cells with lytic potential after immunotherapy. Patients with lung cancer had a high frequency of these cells at study entry and they were not boosted following treatment.
INO-1400/1401 induced hTERT-specific CTLs are associated with DFS in patients with pancreatic cancer
Given the relatively high number of patients on study with pancreatic cancer and an immune profile suggesting the induction of both hTERT-specific CD4 T cells and CTLs (figures 3C, 4C, respectively), we analyzed immune responses induced by INO-1400/1401 relative to overall survival. To perform these analyses, a subset of pancreatic cancer patients (n=23) were followed prospectively during the conduct of the study and following its completion (figure 5A). Overall, 74% (n=17) of patients were alive at the last date of long-term follow-up (mean 428 days, range 1–1062 days). Of the 17 surviving patients, (10/17, 59%) exhibited NED at last contact (online supplemental table 4). The amount of time between the initial diagnosis of local disease and study day 0, as well as the time between study day 0 and last contact, is shown in figure 5A. The majority of confirmed deaths (5/6, 83%) were recorded prior to 3.5 years from diagnosis. The longest noted survival for the final deceased patient was 5.1 years (1876 days from diagnosisin patient 51045) while the longest tracked survival for a patient not yet deceased is 7.8 years (2844 days from diagnosis in patient 51034).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Association of an immunotherapy induced cytolytic T cell response with survival in a subset of pancreatic cancer patients. (A) Time (in days) from diagnosis to study day 0, from study day 0 and date of last contact, and survival status for a subset of pancreatic cancer patients. (B) A representative dot plot of CD8+T cells expressing PD1 and CD69 activation markers. (C) Activated CD8+T cells expressing granzyme B and perforin in pancreatic patients who were alive or deceased at the last date of contact. open symbols represent individual patients with a line connecting the preimmunotherapy and postimmunotherapy magnitudes. The bar extends to the mean. The number and percent of patients in each group that had an increase, decrease or no change from baseline is displayed below the graph. (D) The change in the frequency of these cytolytic CD8+T cells (post-pre) in patients based on survival status at the last date of contact. open symbols represent individual patients, the mean is represented with ‘+’, the box extends from 25th to 75 th percentile, line inside the box is a the median, and the whiskers extend from the minimum to maximum values. Wilcoxon ranked sum test was used to assess significance. hTERT, human telomerase reverse transcriptase.
As the upregulation of CD38 on both CD4+ and CD8+T cells specific for hTERT was observed following immunotherapy, we explored the clinical relevance of this marker in relation to survival benefit for these patients. An in-depth immune analysis including the additional activation markers CD69 and PD1 (figure 5B) was performed on pancreatic cancer patients with available sample (n=12; 8 alive, 4 deceased). The expression of CD38 continued to be an important marker of immune activation relative to survival in this analysis. In particular, the majority of pancreatic cancer patients who were alive at the last follow-up visit exhibited an increase post immunotherapy in the frequency of hTERT-specific CTLs expressing granzyme B and perforin within the population of CD8 +T cells expressing CD38, CD69 and PD1 (figure 5C). The mean frequency of these cells was 0.58% at baseline and 7.29% following immunotherapy in the group of surviving patients representing an overall absolute increase of 6.71%, or a >12-fold increase from study start. Conversely, the patients who were deceased as of last contact exhibited a marked reduction in the frequency of these cells from 2.38% to 1.13% (absolute difference of −1.25%, a 0.47-fold change) (figure 5C). Overall the increase in CTLs expressing granzyme B and perforin within the population of CD8 +T cells expressing CD38, CD69, and PD1 above baseline in surviving patients (mean, 6.71%; median, 4.13%) was higher than the change in deceased patients (mean, −1.15%; median, −0.91%) (p=0.028) (figure 5D). Taken together, these findings demonstrate that INO-1400/INO-1401±INO-9012 induces hTERT-specific T cells that correlate with survival in some patients with cancer.
Conclusion
In this study, we used a DNA plasmid encoding a full-length optimized hTERT sequence (INO-1400 or INO-1401) with or without IL-12 DNA plasmid (INO-9012) to generate cellular responses to hTERT in high risk solid tumor patients with NED after local resection and standard neoadjuvant or adjuvant therapy. We demonstrated that INO-1400 or INO-1401, given with or without INO-9012, demonstrated an acceptable safety profile in patients with solid tumors. The majority of related AEs were secondary to administration of the study drug and low grade.
We found that hTERT immunotherapy induced a de novo cellular immune response or enhanced pre-existing cellular responses to native hTERT in 96% (88/92) of patients with various cancer types. It is unclear if we are rescuing exhausted cells, making new cells, or both. Treatment with INO-1400/INO-1401±INO-9012 drove hTERT-specific IFN-γ production, generated hTERT-specific CD4+ and CD8+T cells expressing the activation marker CD38 and induced hTERT-specific activated CD8+CTLs as defined by cells expressing perforin and granzymes. A post hoc analysis of the dose of hTERT showed similar cellular immune responses in patients receiving either the low-dose or high-dose immunotherapy, with no clear advantage afforded by the higher dose. The addition of plasmid IL-12 adjuvant elicited higher magnitudes of cellular responses including IFN-γ production, activated CD4+ and CD8+ T cells, and activated CD8 +CTLs. Notably, cellular responses in both the ELISpot and LGL assays were observed in pancreatic and ovarian cancer patients, whereas lung cancer patients had responses in the ELISpot assay only. Furthermore, the increased frequency of activated hTERT-specific CTLs expressing CD38, CD69 and PD-1 following INO-1400/INO-1401 immunotherapy correlated with a survival benefit in patients with pancreatic cancer. Future studies to confirm these findings are warranted.
A number of clinical studies have targeted hTERT as a therapeutic target for various cancer types using a multitude of platforms including synthetic peptides, pulsed dendritic cells, transfected B cells or DNA delivery.24 The overwhelming majority of these studies focus on delivery of peptides, an approach dependent on HLA type and prediction of proper binding to the MHC molecule, whereas DNA delivery allows for host cell processing and presentation of multiple epitopes independent of HLA type. One DNA plasmid platform, INVAC-1, was previously reported to be safe and elicit CD4 and CD8 +T cell responses as measured by IFN-γ ELISpot when delivered intradermally to patients with relapsed or refractory cancers.41 In that study, CD4 specific T cell responses were shown to correlate with overall survival; however, cytotoxic T cells were not specifically assessed. In the current study, we assessed the combination of CD4+ and CD8+T cell responses by IFN-γ ELISpot, and also report the induction of CTLs following immunotherapy using flow cytometry.
Our study has several limitations. It was designed to study the safety of INO-1400/INO-1401 and was not powered to assess the impact of DNA dose or inclusion of INO-9012 on immunological endpoints. The populations selected for this study, although all at high risk of relapse, were made up of nine different solid tumors, and each patient entered the study at a different time point in her or his clinical course, making interpretation of DFS more challenging, and making an estimation of immunological endpoints on survival more difficult. The data provided by our analyses nevertheless suggest a possible survival benefit in those patients who manifest an anti-hTERT response via INO-1400/1401; further research is required to confirm this observation. Based on these results, INO-1401 is included as an important component in a new combination immunotherapy, INO-5401, along with plasmids encoding for other tumor-associated antigens Wilms Tumor-1 and prostate specific membrane antigen in an ongoing study in subjects with known germline mutations in BRCA1/2 with, or at high risk for developing, cancer (NCT04367675). Taken together, these data support future examination of INO-1400/INO-1401 and INO-9012 as an immunotherapy in pancreatic cancers as well as other tumor types overexpressing hTERT.
Data availability statement
All data relevant to the study are included in the article or uploaded as online supplemental information.
Ethics statements
Ethics approval
The study protocol conformed to the ethical guidelines of the 1975 Declaration of Helsinki and was reviewed and approved by each site’s Institutional Review Board.
Acknowledgments
R.H.V. is supported by grants from the Breast Cancer Research Foundation and the Basser Center for BRCA.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors RHV, KAK, MPM, JP and JMS drafted the manuscript. RHV is the lead principal investigator for the trial. KAK, JP, AJS, MPM performed and supervised all clinical analytical analyses. TM was responsible for statistical analyses. AFS, AJM, JMJ, WS and AVC were clinical investigators for study. RS managed the clinical operations of the study. JDB, DW and LH provided guidance for immune assays and data, and contributed to overall aspects of study related activities. JJK supervised all clinical and other study related activities
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests RHV is an inventor on licensed patents relating to cancer cellular immunotherapy and cancer vaccines, and receives royalties from Children’s Hospital Boston for a licensed research-only monoclonal antibody. KAK, JP, AJS, TM, RS, JJK, JDB, MPM, LH and JMS are employees of Inovio and receive compensation in the form of salary and stock options. AFS, AJM, JMJ, WS and AVC report no conflicts of interest to disclose.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.