Article Text

Abstract
Immunotherapy for cancer is now a standard pillar in the armamentarium of treatments for many cancers. Immune checkpoint inhibitors, in particular, have resulted in significant therapeutic benefit and prolongation of survival in solid organ cancers, such as melanoma and lung cancer. However, the extent of benefit is not uniform. There are several groups studying predictors of benefit from these therapies. Recently, there has been a burgeoning interest in studying predictive biomarkers from the blood. These markers include circulating tumor DNA, circulating tumor cells, lymphocyte subpopulations, exosomes and metabolites to name a few. The logistics involved in such biomarker work are complex and rigorous with potential to impact a given study. Such pre-analytic components include development of a rigorous protocol, standard operating procedures for collection and storage of various blood components, ethics of patient consent, personnel involved as well as budget considerations. In this primer, we lay out representative aspects of each of the aforementioned components as a guide to blood-based biomarker research for immunotherapy studies in cancer.
- immunotherapy
- biomarkers
- tumor
Data availability statement
There are no data in this work.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
In the last decade, immunotherapy has transformed cancer care. Immune checkpoint inhibitors (ICIs) are now Food and Drug Administration (FDA) approved in more than half of human cancers, and are capable of curing patients with metastatic cancer.1 Despite the success of ICI, the majority of patients do not derive significant clinical benefit from treatment.2 It is critical to develop robust biomarkers which can identify those patients who will most benefit from ICI as well as those patients who are most at risk of developing immune-related adverse events.
Preclinical and translational studies have nominated a number of biomarkers which may predict outcomes to ICI. The most widely used biomarker is programmed cell death ligand (PD-L1) expression on tumor tissue.3 For non-squamous cell lung cancer, studies have shown greatest survival benefit with PD-L1 expression over 50%.4 Limitations to PD-L1 utilization include limited negative predictive value, arbitrary cut-offs within and across diseases, and a lack of understanding if tumorous or tumor microenvironmental PD-L1 is critical.5 Additionally, tumor mutational burden (TMB) has been nominated as a predictive biomarker with higher TMB correlating with higher response rates and progression-free survival (PFS).6 There are limitations to TMB.7 TMB calculation is often higher in cancers associated with strong mutagens (melanoma, non-small cell lung cancer)8; yet the adopted threshold for cut-off is the same across solid tumors; furthermore, TMB may be overestimated if tumor only (without germline/normal tissue) sequencing is performed.9 These aforementioned biomarkers are generally collected via tumor tissue sampling, which oftentimes can be difficult to obtain, expensive, invasive, lead to complications, and delay treatment.10 A simple, less expensive, non-invasive method would be the identification of peripheral blood biomarkers, otherwise known as ‘liquid biopsy’. Solid tumors are constantly shedding materials into the blood stream; analysis of such trace tumor materials, including circulating tumor cells, circulating cell free DNA and extracellular vesicles (EVs), provides crucial information about the solid tumor.11 Meanwhile, blood-based biomarkers may allow prediction of patient response to ICI: these biomarkers include, but not limited to, determination of human leukocyte antigen type, evaluation of T cell phenotypes and genotypes, enumeration of myeloid-derived suppressor cells, natural killer cells, and circulating tumor cells, quantification of circulating tumor-derived nucleic acids and exosomes.6 12 13 Peripheral blood-based markers can also be used throughout the duration of treatment and provide monitoring and potentially early detection of treatment failure and/or disease progression as collection is minimally invasive.
Despite the studies suggestive of evidence of peripheral biomarkers aiding in ICI treatment decisions, there are currently no validated or approved peripheral blood-based biomarkers that inform cancer immunotherapy in the clinic.10 Lack of standardization of pre-analytic variables has been identified as a major challenge in reproducing biomarker results.14 15 Pre-analytic factors include specimen collection, preparation, and storage as well clinicopathologic annotation to enable multivariate modeling. Recently, the Reporting Recommendations for Tumor Marker Prognostic Studies (REMARK) guidelines were developed to improve high-quality result reporting for prognostic markers.16 As such, the REMARK checklist provides a comprehensive checklist outlining the information which should be included in reports evaluating prognostic biomarkers.17 18 Herein, we provide a complimentary comprehensive evaluation of the information required for blood-based predictive biomarker studies. In order to improve this area of research and implement the use of peripheral biomarkers, it is imperative that a standardized set of guidelines be created. Furthermore, there are significant resources that need to be allocated for successful initiation and completion of blood-based biomarker studies. We aim to provide practical insights into performing blood-based biomarker studies, including protocol and budget development, resource allocation, and data management.
Suggested guidelines for protocol development
Biomarkers can be integrated as defined endpoints on interventional clinical trials examining ICI or can also be analyzed during routine clinical care with ICI. In the latter situation, a non-therapeutic protocol needs to be developed. Below are some considerations that should be addressed by a protocol written for blood biomarker studies relevant to immunotherapy. Refer to online supplement section 1 for an example table of contents for a study protocol.
Supplemental material
Significance
Question: What is the clinical context which will be informed by this biomarker and what is the unmet need for the development of new biomarkers?
The first step to initiating a study protocol on blood-based biomarkers is identifying the significance of the proposed biomarker of interest and assessing if there is prior research to support its relevance. In order to determine the answer to the above questions, it is imperative to do a thorough review of literature using database search engines such as PubMed, EMBASE, and Cochrane Library. This will allow investigators to discover areas where a knowledge gap exists and provide opportunity to fill those gaps with the proposed study.
Example: PD-L1 is a commonly studied biomarker for multiple different types of cancer and is the only validated predictive biomarker in immunotherapy cancer treatment. However, despite FDA approval of its use, this tissue-based biomarker is not perfect.19 Several biomarkers, including the use of tumor mutation burden, have been proposed in conjunction with PD-L1 to build a better predictive model.20 To determine the knowledge gaps present, investigators can use Medical Subject Headings terms such as ‘immunotherapy’, ‘cancer’, and ‘biomarker’ to search through medical databases. The investigator must justify the study of their proposed biomarker in the context of the current literature.
Innovation
Question: How is it different from other studies of the same biomarker in the same population? Does this represent a methodologic or analytic advancement?
Once the significance of the proposed biomarker is determined, the next step is assessing the level of innovation of the study. It is important to clearly state what specific aspects of the suggested study are novel compared with the existing body of scientific knowledge. This offers protocol readers perspective on how the study will benefit the scientific community and further advance predictive biomarkers for ICI efficacy.
Example: A one-time assessment of tumor PD-L1 is not an accurate representation of the patient’s tumor (tumor heterogeneity) at baseline or through the course of therapy. Innovative methods to assess PD-L1 may include measuring expression of PD-L1 in circulating tumor cells in the blood over the course of therapy as a way of obtaining repeated measurements.21
Approach
Question: What are the steps involved and is there justification for these steps? What are the study goals? Will this be an integral or integrated biomarker study? What next steps will the results inform?
This section of the study protocol outlines the logistical plan for conducting the study. It involves first stating the primary and secondary goals of the study and then describing the steps required to achieve these goals. The steps listed should include approach for pre-analytic, analytic, and post-analytic factors, along with justification for why the approach was picked. In clinical trials, biomarker studies can be conducted either as an integral or integrated study. During integral studies, the biomarker being assessed is imperative for the continuation of the trial and the primary study objective is designed around the biomarker.22 While in integrated studies, tests are conducted to identify or validate biomarkers that can be used in future trials and a hypothesis is tested as a secondary study objective.22 This portion of the protocol should conclude with how the results obtained will inform future investigations.
Example: Determining if repeated measurements of PD-L1 via circulating tumor cells throughout immunotherapy treatment may better predict durable benefit from ICI. Pre-analytic factors to consider include: blood sample collection, processing (how circulating tumor cells are obtained from a blood sample), and storage. Analytic factors involve the actual testing of the sample (ie, which assay will be used to test for PD-L1). Post-analytic factors include how the data will be recorded and interpreted. The study is classified as an integrated biomarker study as it will be measured in all participants and will not determine study eligibility or treatment received. The results from this study will shed light on how repeated measurements of PD-L1 during therapy will influence its use as predictive biomarker throughout treatment.
Investigators
Question: Are the principal investigator and the team adequately qualified?
The roles of each team member are described in the Statement of work for study coordination section of this paper. In addition to this, there should be an explanation regarding the study principal investigator (PI) qualifications for undertaking the project. The investigators must have technical and clinical expertise to perform the study. There are often institutional requirements as well in regard to training for handling patient information, working with human specimens, and remaining compliant with the Healthcare Insurance Portability and Accountability Act (HIPAA).
Example: Experience in the circulating tumor cell field is desirable if the blood-based biomarker involves serial measurement of PD-L1 in circulating tumor cells (CTCs) over time. Investigators involved in the study must undergo research, ethics, and compliance training and obtain certificates of successful completion in order to participate.
Environment
Question: Are there adequate resources to carry out the study to completion?
In order to successfully complete blood-based biomarker studies, a wide variety of research personnel are required—research coordinators, data managers, and regulatory staff. Further, sufficient laboratory staff is needed for data acquisition and analysis. Additionally, access to equipment or necessary supplies should be assessed as well. In this section, inclusion of a budget may be useful and is further expanded on in the Budget requirements section.
Example: It is strongly recommended to process fresh blood in a timely fashion for circulating tumor cell isolation. Proper coordination between research labs and personnel is needed to make sure essential equipment is ready and sample is properly dropped off. In the instance of storing processed blood components, appropriate refrigeration and freezing conditions are critical.
Databases
Question: How will the data be recorded? Who will maintain the codes for the patient data? What type of output will be generated?
After data is collected, it must be stored securely and specific members of the research team should be identified to maintain a separate data sheet associating each patient to a unique code. Database management can be done in a number of ways and is further addressed in the Database management section. Options include storing the data on a secure institutional network drive, controlling access with encryption, or password-protected cloud storage. It is crucial to have the data backed up as well. Determining what additional data to collect, such as demographic and clinical information, is reviewed in the Standardized documentation of demographics and clinical information section.
Example: If evaluating the role of PD-L1 on CTCs as a novel biomarker, each study participant will be assigned a unique patient identifier, which will be maintained on a password-protected database. Clinical, pathologic, and biomarker data collected from the study will be inputted into a separate password-protected database which is linked only through the use of the unique patient identifier.
Statistical methods
Question: How will the statistical analysis be designed, analyzed, and validated?
Considerable thought must be given as to how many replicates are needed to reduce the measurement error (eg, reducing batch effect and minimize assay-to-assay variability). To associate the biomarker to the clinical outcome, the type of the outcome must be selected. These include association of the biomarker of interest with ICI response or time-to-event variable, such as PFS. A benefit of using continuous data analyses (with appropriate data transformation, such as log) is that all information is retained. Based on this endpoint, sample size calculations are needed to ensure the study has adequate power to test the hypothesis.
Example: In order to determine relationship between PD-L1 in CTC as a predictive biomarker of PFS, ensure that a multivariate analysis is performed, missing data are minimized and adjusted for, and validation studies are planned.
Statistical design
Sample size calculation
Potential risks of qualifying a biomarker should address the consequences of incorrect decision-making or harm to patients if the correlation between the biomarker and the outcome of interest is spurious. This requires accurate estimation of false positive fraction (FPF) and true positive fraction (TPF). Sample size should be determined to provide sufficient power to gain enough accuracy for the estimated FPF and TPF.23
Minimize bias from confounders
To facilitate subgroup discovery and minimize bias introduced through confounders, all necessary participant data should be collected, including demographics (eg, age, sex, race), medical history (eg, medications, comorbidities), oncologic history (disease stage, disease sites, prior local and systemic interventions; see the below list noted in the Standardized documentation of demographics and clinical information section).
Minimize missing samples
Each study design should include strategies to minimize and account for the effect of missing data and avoid collection of biomarker data only from a subset of clinical sites, groups, or treatments arms.
Statistical analysis
Data cleaning and normalization.
Understanding and addressing missing data via methods such as multiple imputation or doubly robust methods.
Determining the appropriate statistical approach, identifying and adjusting confounding factors via statistical analyses. For a binary response outcome, we can consider a logistic regression or a Cox regression for a time-to-event outcome, such as PFS. The goodness-of-fit of the model should be checked and the appropriate transformation should be used on the biomarker data (such as log) to ensure a good fit of the model.
Evaluating sampling or selection bias in the observational study and developing propensity score models to correct for such potential bias.
Appropriately addressing the multiple comparison issue if multiple biomarkers are tested simultaneously. For example, the Bonferroni procedure and Tukey’s procedure can be used to control for the family-wise type I error.
Avoid dichotomizing the continuous biomarker measurement for analysis to retain statistical power unless there is a clinical meaningful threshold commonly used for the biomarker. Repeated or arbitrary methods that create many possible thresholds for a cut-off to test for predictive ability or the lowest p value are discouraged and have been criticized for their ad-hoc nature, potential for overfitting, and lack of correction for multiple comparison.24 25
Statistical validation
To evaluate the predictive ability of the biomarker, the area under the receiver operating characteristic (ROC) curve can be computed to measure the ability of the model to correctly classify responders and non-responders.26 For the survival outcome, such as PFS, the c-index and time-dependent ROC curve27 28 can be used for evaluation of the discriminative ability of the model, and Brier score29 for evaluation of the predictive accuracy of the model. All the calculations should be based on a 5-fold or 10-fold cross-validation. Moreover, the added predictive ability of a new biomarker can be assessed using method described by Pencina et al.30 If the sample size allows, the biomarker should be validated in a separate cohort.
Obtaining study approval
Federal institution
For studies conducted at a federal institution, such as the Veterans Health Administration (VHA), the approval process starts with submitting a project application/proposal to the Research and Development (R&D) committee by the study coordinator. After submission, the R&D undergoes an initial review of the proposal and grants conditional approval of the study pending further approval by subcommittees. These subcommittees include the Institutional Review Board (IRB), Information Security Officer (ISO), Privacy Officer (PO), and Subcommittee for Research Safety (SRS). Full committee review and approval by the IRB is required. The SRS reviews laboratory and human subject safety. The ISO/PO must provide written approval. After approval is granted by these subcommittees, the project is sent back to the R&D to obtain final approval. The PI and study coordinator can then conduct research. As the study progresses, file amendments, continuing reviews, and reporting of adverse events are provided to the IRB and R&D. Figure 1 depicts the above described approval process. The VHA Office of Research released a guidance manual with full details regarding research process at their federal institutions.31

Process of obtaining study approval at a federal institution. ACOS, associate chief of staff; PI, principal investigator; R&D, Research and Development; RDC, Research Data Center; SC, study coordinator; VA, Veterans Affairs.
Non-federal institution
In studies conducted at non-federal institutions, such as universities, the approval process begins with the PI or study coordinator submitting the research application to the institution’s regulatory review system. At our institution, there is a web-based eResearch Regulatory Management system that centralizes the sequential review and approval process for Human Subjects Research Applications. This system ensures that the university is conducting ethical research and adhering to regulations. Once the application is submitted into this system, it may be reviewed by the Protocol Review Committee for approval. The application is also routed to the appropriate ancillary committees, which includes Conflict of Interest, Clinical Research Calendar Review and Analysis Office, Privacy Board, and Institutional Biosafety Committee. The IRB committee will also review the application and provide approval. Once these steps are completed and full approval is obtained, the PI or study coordinator may begin conducting research, reporting adverse events, submitting amendments, and requesting continuing review.
Standardized guidelines to obtain informed consent from patients
The specific ethical guidelines as outlined by the local IRB must be adhered to by all research personnel. It is the PI’s duty to ensure the study is designed in a way to minimize the amount of harm experienced by patients. The IRB at each study site should review the participant consent forms and approve them prior to any patient enrollment. Additionally, prior to study enrollment, potential participants should be given all information necessary for informed consent through written and oral formats. It is very important that this information is provided as it allows participants to fully understand the study, extent of their involvement, and any future study plans. These forms should include information on study purpose, visit structure, procedures, risks and benefits, alternative options, and ability to withdraw participation at any time. Participants should also be told how researchers will keep information confidential, handle samples, and how financial and legal consequences may occur with accidental information release. Online supplement section 2 includes sample authorization for use and release of identifiable health information form.
Notably, patient samples can frequently be used for the initially proposed study as well as for other secondary study purposes. If the researcher is attempting to create a blood biobank, then they may elect to have participants allow the use of their samples in multiple different studies as well as agreeing to sending the samples to other institutions for additional analysis. Participants may be asked to consent to allowing access to stored samples without requiring further consent or provide permission to be contacted in the future regarding use of sample. It is imperative to include language that allows patients to opt in or out, as well as further information on protection by law from discrimination based on their genetic information (Genomic Information Non-Discrimination Act).
Obtaining informed consent
In order to obtain informed consent, investigators must present all pertinent information to patients then assess their understanding and allow time for questions. Information orally presented will also be available to patients on the written document. When reviewing this information with patients, it is important to explain the study in layman’s terms and avoid confusing patients with complex medical or scientific jargon by using language aimed at an 8th grade reading level. In order to provide informed consent, the patient must have medical decision-making capacity, which means they can demonstrate understanding of the risks and benefits, appreciate consequences of decision, show clear reasoning behind decisions, and effectively communicate their wishes.32 A good method to assess understanding is to have patients explain the information using their own words and provide reasoning behind their decision, also known as teach back or say back method. The consent form must provide comprehensive study explanation, study approval, methods, techniques, procedures, possibility of sample use in future studies, time commitment, travel requirements, costs, number of appointments, number of tubes of blood required (listed in mL) and duration of the project.33
All consent forms should include the ability to withdraw from the study at any time and without any consequences. If patients withdraw from the study, they should then be given the opportunity to also withdraw permission to use samples that were already collected. For some studies, researchers may have to obtain written consent on a yearly basis. Additionally, if there are new developments in risks, benefits, or alternative options then researchers are required to provide this additional information to participants. After assessing the new information, patients are then given the choice to continue onwards with the study or withdraw. A sample consent form can be viewed in online supplement section 3.
Obtaining informed consent for national or international collaborations
Biomarker research often includes working together with national and/or international groups to expand the data available. In these situations, participant consent forms should clearly request permission for use of biologic samples and data to be shared with the other organizations. Agreement to storing their samples in a biobank for any future research should also be included. Participant consent forms must be specific on the different types of specimens to be collected and shared with other institutions (ie, blood—red blood cells (RBCs), white blood cells (WBCs), serum, plasma, or whole blood).
Brief ethical discussion on informed consent
In the era of biobanking and the potential use of blood samples for future projects, it is difficult for participants to understand the full scope of sample use. Obtaining informed consent in these instances can be difficult as the future research questions and specific study information are often unknown at the time of initial sample collection. The question then arises if true informed consent is possible in these situations. Prior studies have shown that patients prefer some choice in how their data is being used for research.34 Continued transparency and further explanations on future sample use will be necessary moving forward.
Generation of standard operating procedures for various blood-based biomarkers
There are different requirements for collection, processing, and storage based on the stability of the marker being measured. Rigorous and standardized isolation protocols are required to generate robust and reproducible data. Notably, blood samples can be analyzed both retrospectively and prospectively. The advantages of retrospective analyses include more mature patient cohorts and increased sample size, but limitations include diminished sample integrity after prolonged storage. Prospective analyses are accompanied by high sample integrity, but it is unclear if future paired samples will be obtainable, nor is the patient’s outcome known at the time of collection.35 The AIDS network established an Immunology Quality Assessment Program to evaluate the processing/handling of patient blood samples in different immunologic labs and aimed to improve the comparability between these labs.36 Additionally, the use of centralized laboratories may be beneficial, especially for multicenter research, due to their standardization, high quality, reliability, and validation.36
Collection of blood sample
Variables that can potentially impact blood biomarker during sample collection include needle gage, needle type, collection container, and sample timing. Generally, a 21-gage needle can be used, and 30–45 mL of blood should be collected in order to establish a blood biobank. The age, weight, and health of individuals are considered when determining frequency and amount of blood that can be drawn. For adults weighing at least 110 lbs, the amount drawn should not be more than 10.5 mL/kg (550 mL) over 8 weeks and not exceed more than two times per week collection.37 38 In children, it has been suggested that 1% of total blood volume (0.8–0.9 mL/kg body weight) to 5% (5 mL/kg body weight) is safe in a single day and should not exceed 11% of total blood volume (9.5 mL/kg) during an 8-week time frame.39 The preferred site of collection is usually the median cubital vein given its easy access. All blood sample tubes should include a clearly marked label with the date, time of collection, sample description, unique patient identifier, and study identification number. The first set of samples should be collected at baseline prior to any receipt of ICI. This information should then be recorded on the sample log sheet and correlated to the matching demographical and clinical information on the institution’s secure, password-protected system.
Blood sample processing
Total blood
At least 5 mL of blood needs to be collected in a 13×75 mm vacutainer tube with K2EDTA. The blood should be mixed via inverting the tube 10 times at 90° in aliquots containing 1.5 mL low-binding protein Eppendorf tubes or cryovials with O-ring sealed screw leads. The sample must be stored frozen at −80°C until data collection. Figure 2 notes common liquid biopsy markers that are obtained from each blood component.
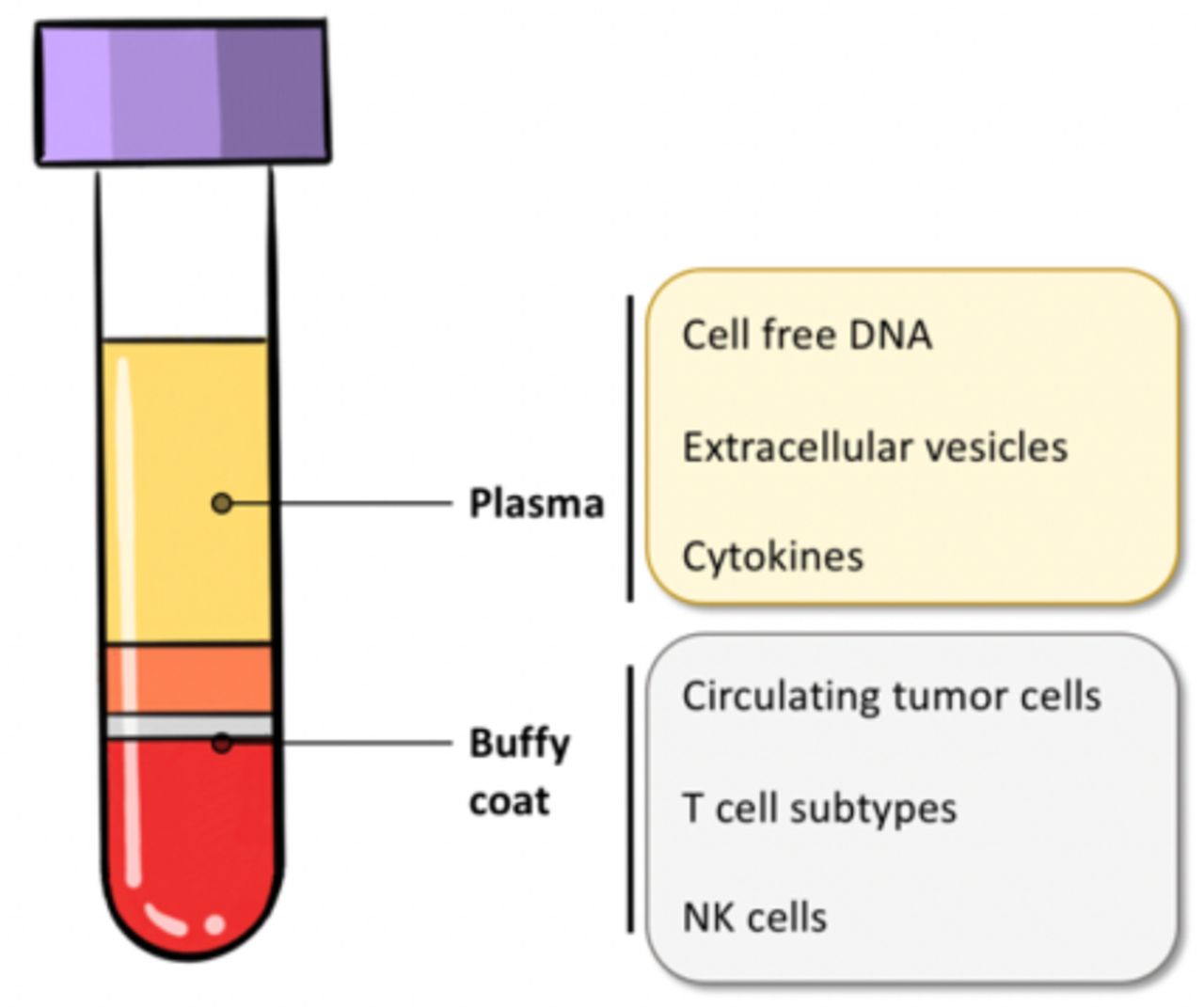
Common liquid biopsy markers origin with respect to blood components. NK, natural killer.
Plasma
At least 5 mL of blood needs to be collected in a vacutainer tube with K2EDTA. It is important to ensure it is properly filled as the additives are calibrated for optimum blood to additive ratio.40 The blood should be well mixed via inverting the tube four to six times at 90°. The samples are centrifuged for 10 min at 1300× g at 4°C. The plasma is removed from the top layer and aliquoted in 1.5 mL low-binding Eppendorf or cryovial tubes with O-ring sealed screw leads. The buffy coats (white layer) can be transferred to support aliquot tubes with the addition of 1.2 mL of ‘RNA Later’. Plasma should be stored frozen at −80°C until it is assayed. Refer to online supplement section 4 for sample standard operating procedure (SOP) for plasma processing.
Serum
At least 5 mL of blood needs to be collected in a vacutainer tube containing silica clot activator, polymer gel, silicone-coated interior, or equivalent. Allow sample to sit upright at room temperature for 30–60 min in order for clot formation to occur, may need to sit longer if patient was taking anticoagulant.40 It must be processed immediately or stored at 4°C (only up to 4 hours). In order to process the sample, it is centrifuged for 10 min at 1300× g at 4°C. The serum is removed from the top layer and aliquot 300 µL in 1.5 mL low-binding protein Eppendorf tubes or cryovials (decrease molecule of interest binding to tube surface) with O-ring sealed screw leads (prevent evaporation). Prior to assay, the serum is stored frozen at −80°C. Refer to online supplement section 4 for sample serum SOP.
White blood cells
Obtain 10 mL of blood in vacutainer tubes that are EDTA coated. The blood should be mixed well via inverting the tube 10 times at 90°. Using two 15 mL falcon tubes (or one 50 mL tube), layer 5 mL of blood on 5 mL of density gradient medium in each tube (ie, Ficoll). These tubes should be centrifuged for 40 min at 400× g at room temperature. Remove the WBC layer in the middle of the tube with a long pipette tip and place in a different 15 mL conical tube that contains 10 mL of phosphate buffered saline (PBS), pH 7.4. Then centrifuge the tubes again for 10 min at 400× g at room temperature. The WBC pellet is then washed by removing the supernatant and resuspending the pellet in 10 mL of PBS. This is again centrifuged for 10 min at 400× g at room temperature and the same washing process is repeated. Using a hemocytometer, the cells are counted and then added to freezing media in order to store samples at 1×107 cells/mL. Aliquot into 1.5 mL cryovials. Prior to assay, the samples should be kept frozen at −80°C. If commercially available, complete cell preparation tubes containing anticoagulant, liquid density medium, and inert gel barrier are used, then processing of the samples can start with centrifuge with subsequent sample washing and freezing media.
Red blood cells
Obtain 5 mL of blood in a 13×75 mm vacutainer tube with K2EDTA. The blood should be mixed well via inverting the tube 10 times at 90°. Then for 10 min, the sample is centrifuged at 1000× g at 4°C. The top layer (plasma) is removed using a pipette. Next, the white buffy layer containing leukocytes is removed. The RBCs are then lysed in 4 volume of ice cold high-performance liquid chromatography grade water and centrifuged for 15 min at 10 000× g at 4°C. The erythrocyte lysate (supernatant) is collected and placed in 1.5 mL low-binding Eppendorf tubes or cryovials. Prior to assay, the samples should be stored at −80°C.
Platelets
Obtain 5 mL of blood in a 13×75 mm vacutainer tube with K2EDTA. The blood should be mixed well via inverting the tube 10 times at 90°. The sample should then be centrifuged for 15 min at 160–200× g at room temperature. The supernatant-containing plasma rich in platelets is then collected and placed into a new tube. This supernatant is then centrifuged at 700× g for 15 min at room temperature. Remove the supernatant and resuspend the pellet with the addition of PBS. Complete the rinse two more times and then resuspend the pelleted platelets in PBS or media with EDTA at 2 mM. Using a hemocytometer, count the platelets and adjust concentration to 1–3×108 platelets/mL in PBS or media. Prior to assay, the samples can be stored at −80°C in low-binding protein Eppendorf tubes or cryovials.
DNA
Obtain 5 mL of blood in a 13×75 mm vacutainer tube with K2EDTA. The blood should be well mixed via inverting the tube 10 times at 90°. Aliquot 1 mL of blood into 1.5 mL low-binding protein Eppendorf tubes or cryovials with O-ring sealed screw leads. Prior to assay, the sample should be stored at −20°C or −80°C.
RNA
Obtain 2.5 mL of blood in a 16×100 mm RNA-stabilizer blood RNA tube. The blood should be mixed well via inverting the tube 10 times at 90°. Prior to assay, the sample should be stored at −20°C (<6 months) or −80°C (>6 months to years). In order to collect blood for RNA analysis, a blood collector set should be used, not a syringe. When collecting other specimens at the same time, ensure that RNA is the last specimen collected. If only RNA is being collected, then a small amount of blood is drawn into a ‘discard tube’.
Circulating cell free DNA
Cell free DNAs are usually isolated from plasma or serum instead of whole blood. Minimal time delay between blood draw and sample processing is preferred, as DNA concentration increases with more blood cell lysis.41 K2EDTA tubes are fine for short interval blood processing. If longer storage time is required between blood draw and processing, preservative blood collection tubes are recommended (ie, Streck Cell Free DNA Blood Collection Tubes).
Circulating tumor cells
Sample requirements vary among the numerous isolation techniques for CTCs. Generally, at least 5 mL of blood is needed in vacutainer tube with spray coated, or equivalent K2EDTA (tube size 13×75 mm). For studies involving CTCs, to avoid the potential skin/epithelial cell contamination by needle aspiration during the blood draw, the first few millimeters of blood needs to be discarded. It is strongly recommended that the sample is processed freshly, within 4 hours after blood draw to minimize cell loss. If shipping or storing is needed, obtain samples in blood preservative collection tubes such as CellSave tubes (Veridex) and BCT tubes (Streck). Studies have shown tubes with preservatives were able to retrieve comparable results from CTCs up to 96 hours at room temperature.42 The blood should be mixed well via inverting the tube 10 times at 90° prior to processing.
EVs and exosomes
EVs are usually isolated from plasma or serum instead of whole blood, in order to minimize vesicle secretion after blood draw. Samples should be stored in −20°C (short term) or −80°C (long term) prior to isolation. One of the most commonly used isolation methods is differential ultracentrifugation. Specifically, centrifuge and collect supernatant at 12,000× g for 20 min. Then, dilute and balance samples using sterile PBS pH 7.4, centrifuge at 100,000× g for 90 min.43 Replace the supernatant with sterile PBS and repeat centrifuge at 100,000× g for 90 min. Finally, EVs should then be suspended in 100 µL PBS pH 7.4 or Radioimmunoprecipitation assay buffer (RIPA buffer) with protease inhibitor cocktail, depending on downstream applications. Notably, the process for exosome collection is identical to EV collection and historically researchers have referred to lipid bilayer delimited particles released from a cell as either exosome or EV.44 Figure 3 notes how blood samples are processed for each biomarker being tested.

Blood sample processing. CTC, circulating tumor cell; EVs, extracellular vesicles; PBS, phosphate buffered saline.
Peripheral blood mononuclear cells
Add 5 mL of Lymphocyte Separation Medium to the bottom of a 15 mL centrifuge tube and transfer 10 mL of blood using a pipette into the tube. Place tubes in the centrifuge at 1600 rpm with brake off for 30 min. Once completed and separation has occurred, remove the interface containing the lymphocytes to a different 15 mL centrifuge tube, fill it with PBS, and mix by inverting tube at 90° angle. Place back into centrifuge for 10 min at 1350 rpm with brake on low. Remove tubes and decant supernatant into waste container and resuspend the cells. Fill with PBS and spin down at 1250 rpm with brake on high. Following this, wash and perform hypotonic lysis by decanting and resuspending the cells again, add 2 mL sterile water and gently mix then add 10 mL of PBS within 30 s. Centrifuge at 1250 rpm for 10 min with brake on high. Resuspend the cells and add 10 mL of PBS. Mix by inverting the tube at 90° angle a few times. Remove 50 µL and place into a 96 well plate and add 50 µL trypan blue to count. Final count should be 0.2×106. To store, add freezing medium (90% AB serum plus 10% dimethylsulfoxide) to the cells and place into cryovials. Store at −80°C. Refer to online supplement section 5 for PBMC processing SOP example.
Proper storage and shipment of samples
Storage of samples
All patient samples should be stored at a secure facility that includes fridges and freezers that can be locked and are only accessible by approved research personnel. There must be the capability to store samples at −20°C and −80°C, as well as liquid nitrogen tanks. In case of power issues, there should be a backup generator or power plan in place, as well as alarms that indicate any issues with maintaining temperature or power. All of the fridges and freezers themselves should have careful temperature records, a CO2 backup system to maintain temperature as low as −70°C in case of a power outage, and a dedicated computer to store information. Investigators handling specimens should monitor and record storage conditions, number of freeze thaw cycles, and number of equipment failures (ie, temperature changes).45 For uniform temperature, validation of freezer units (ie, temperature mapping of freezer interior) and regular defrosting is recommended. An inventory system should be in place denoting location of each aliquot with documentation of specific box, shelf, and freezer number.
Shipment of samples
If researchers are working with other institutions or even international groups, then oftentimes samples must be shipped to other locations. When this is the case, researchers should keep in mind the shipping temperature, time, distance, weather, and transportation method. Ideally, samples should be sent overnight during the weekday, in order to decrease sample loss. In order to maintain frozen temperature, ensure that dry ice or liquid nitrogen is available. If using dry ice, ensure that all personnel handling have appropriate training per federal regulations and certification is on file. Due to temperature sensitivity of samples, a courier who is able to refill the dry ice should be used, especially if there are shipping delays. The package should also contain a device to constantly monitor temperature. Put filled cardboard boxes with samples in a plastic Ziploc bag along with absorbent pad and seal it. Place bagged boxes in the bottom of shipping box, these boxes must have Styrofoam insert surrounded by cardboard outer box. Fill shipping box with as much dry ice as possible (pelletized dry ice is ideal). Place Human Specimens Exempt label on shipping container. Ensure shipment is sent overnight on Mondays–Wednesdays only. Prior to shipping the samples, the sending institution should contact the receiving institution and ensure research personnel will be available to accept the package. Further paperwork should be included in the package that lists all samples by their identification number and description of all specimens. Once arrived at its destination, it must be promptly stored in appropriate conditions. Notably, the thawing method used is important in maintaining sample integrity. Using additives like human serum albumin, dextran, and fetal bovine serum was better than human AB serum; using medium pre-warmed to 25°C–37°C yielded better results than chilled medium.36
Standardized documentation of demographics and clinical information
Various patient factors such as age, gender, ethnicity, smoking status, alcohol use, and other illicit substance use can alter the blood-based biomarker measurements that are collected, thus standardized documentation of these factors can help decrease the effect they have on biomarker measurements. Other pieces of important clinical information to include for standardized documentation include the following:
Performance status at start of treatment (ie, Eastern Cooperative Oncology Group score).
Charleston Comorbidity Index.
Inflammatory comorbidities (categorical variable yes/no, if yes then indicate specific inflammatory disorder).
Autoimmune medical history (categorical variable yes/no, if yes then indicate specific autoimmune disorder).
Baseline medications—use of aspirin, non-steroidal anti-inflammatory drugs, corticosteroids, statins, antibiotics and immunosuppressive medications.
Date of diagnosis—determined based on date of biopsy.
American Joint Commission on Cancer Eighth Edition stage at diagnosis.
Histologic information.
Prior chemotherapy regimens.
Prior radiation therapy—including location and units in Gray.
Baseline sites of metastatic disease location prior to immunotherapy treatment (ie, lungs, liver, brain, bone, adrenal, lymph nodes and other).
Tumor molecular characterization (ie, PD-L1 expression, estimated glomerular filtration rate mutation, ROS rearrangement, ALK mutation, tumor mutational burden).
Tumor burden prior to immunotherapy—sum of unidimensional diameters of tumors in centimeters.
Baseline complete blood count with differential including neutrophils, lymphocytes, eosinophils, basophils, and monocytes.
Immunotherapy medication used—including date of treatment start, how many cycles were given, and date of last treatment cycle.
Date of progression—based on radiographic or symptomatic progression.
Date of last follow-up—including status at that time.
Date deceased.
Statement of work for study coordination
The PI
During a study, the PI has the responsibility of managing the study design, financial needs, study conduct, data submission and reporting, adherence to protocol requirements, patient safety and confidentiality, and maintaining regular communication with research coordinators. It is important to ensure the protocol is clear on data and specimen requirements and that the aims and objectives, as well as patient eligibility criteria, are clearly described. The PI must also ensure that all aspects of the research project are in adherence with departmental, institutional, and federal guidelines and policies. They will oversee the drafting of the informed consent and study recruitment materials. Study trials registered on ClinicalTrials.gov (overseen by the National Institute of Health) have the authorization per section 801 of the FDA Amendments Act and the Final Rule to enact monetary penalties if not in compliance with registering the trial or failing to submit study results within 1 year of study completion date.46 It is the PI’s responsibility to complete these tasks and includes submission of full protocol and statistical analysis plan, primary and secondary outcomes, adverse event information, demographic/baseline characteristics, and participant flow.46
Clinical research coordinators
Research coordinators have various roles throughout the duration of the project. Generally, they will be Society of Clinical Research Associates or Association of Clinical Research Professionals certified and have completed Good Clinical Practice training. Responsibilities of coordinators are clinically focused and described in the below section. Figure 4 details the percent of time spent for each aspect of the coordinator’s role.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Percent of time dedicated to each study aspect by research coordinators. AE, adverse event; HIPAA, Healthcare Insurance Portability and Accountability Act; IRB, Institutional Review Board; PHI, protected health information; PI, principal investigator; RDC, Research Data Center; SAEs, serious adverse events.
Institutional start-up
If collaborating with a federal agency, an Intergovernmental Personnel Act agreement may be necessary. This allows for temporary assignment of employees between federal agencies and state and local governments, colleges, and universities. Additionally, there may be consideration of a Cooperative Research and Development Agreement, which allows federal labs to participate in collaborative work with non-federal institutions, such as universities. In these instances, data use agreements will be required in order to share data between collaborating agencies.
Project start-up
The initial pre-site selection meeting (determine if investigators and clinical site are adequately equipped to complete the study) and start-up meetings (occurs after clinical site meets all regulatory requirements and obtains IRB approval) are all arranged by the research coordinators. The project proposal is submitted by the coordinator to IRB and Research Data Center for local and regulatory approval. At this step, a draft regarding plan for sample collection is created.
Patient prescreening
Coordinators create a patient tracking system and proceed to screen patients presenting for clinic appointments for eligibility of enrolling into the study. IRB-approved recruitment materials including letters and brochures can be used. Once patients are identified, the coordinator communicates with the PI to confirm that the patient meets all eligibility criteria. At this time, the coordinator will introduce the idea of consenting to the study to patients and continue communication with follow-up telephone contact.
Patient screening and enrollment
Once patient is determined to be eligible, the study coordinator will meet the patient and further explain the project. The coordinator then proceeds ahead with obtaining informed consent from the patient and HIPAA authorization. The coordinator should log screen failures and successful enrollments. Once consented, the coordinator will help arrange and schedule all necessary tests and lab orders. They will also help with obtaining required demographic and survey data and drug reconciliation.
Sample collection and processing
The coordinator assists in the collection of patient blood samples and helps route them to the appropriate locations. They help label samples, maintain the sample database, and shipping log.
Central lab sample and shipping
If samples are being shipped to central labs or other collaborating institutions then coordinators help with preparing the shipping label, packaging (ie, dry ice), adhering to exempt human specimen standards, and following up with the receiving locations regarding sample arrival and results obtained.
Data entry and maintenance
Coordinators enter visit data into the patient database within a specified time frame. Prior to inputting data, they de-identify any patient imaging and protected health information. They also aid in monitoring and entering any adverse events (condition which appears or worsens after participant is enrolled) or serious adverse events (results in death or life-threatening problem leading to inpatient hospitalization) experienced by patients. During this time, coordinators also answer any questions that patients have.
Regulatory updates
Maintenance of accurate training logs and delegation is completed by the coordinator. The regulatory binder contains documents such as investigator curriculum vitae, delegation of authority/duty logs, IRB approvals, Human Subjects Protections training/certifications, and HIPAA training certification. Coordinators will also be in charge of completing the annual continuing review for safety and IRB approval, as well as provide any updates or amendments.
Patient long-term follow-up
As patients continue in the study, coordinators will assess patient survival, long-term follow-up clinic visits, and updating clinical data.
Budget required for study
The budget contains the expected expenses for each study. A variety of aspects must be considered and accounted for. This includes costs for research personnel, supplies, equipment, services, travel, and other indirect costs. It should be clearly stated what aspects are included to reach the total expense for each category. The research personnel category includes the salary for each person. The estimated cost per patient sample is approximately $500–$1200. This is calculated based on time spent, effort per patient, and phlebotomy blood draw costs. The costs for laboratory supplies, chemicals, and glassware should also be included. Travel costs include professional conferences, mileage for research participants, and any travel for consultants. Services to budget for are data storage, publication, graphics, and data analysis. Online supplement section 6 includes a sample suggested budget proposal.
Database management
While collecting clinical, demographic, and laboratory information from patients, all data collected must be kept confidential at all times. All of this information should be de-identified and each patient should be assigned a unique code that links them to their clinical information. The patient names and codes they are associated to will be kept in a separate secure database. Database tools to use include Research Electronic Data Capture to securely maintain protected data or Velos, which aids in clinical trial management by tracking budget/financial information, patient recruitment, and data. If information is collected via the electronic medical record, then all personnel involved should be trained per institution guidelines to handle personal information and be listed as an investigator on the protocol approved by the local IRB to have access to these records. Anytime data is shared among other research personnel, the information must only be de-identified data and always sent through an encrypted, password-protected file on a secure server. The data should be backed up in a password-protected folder within the research institution’s network. When the project is finished, the de-identified samples should be discarded based on state and federal rules and regulations. If creating a biobank and storing samples for an extended period of time, then appropriate assays should be integrated to assess samples’ integrity.
Conclusion and future direction
Peripheral blood biomarkers are rising to the forefront of immunotherapy cancer care as additional information is required for patient and therapy selection. Due to the lack of standardized guidelines for control of pre-analytic variables, robust and externally validated blood biomarkers have yet to be identified. Following best practices in biomarker selection and analysis ensures sample integrity and maximizes opportunities for data reproduction and validation. Future studies are needed to further standardize pre-analytic, analytic, and post-analytic procedures with the goal of enabling blood biomarker precision immuno-oncology.
Supplemental material
Data availability statement
There are no data in this work.
Ethics statements
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors CYJ—conception of work, collecting data/information, writing original draft, reviewing and editing draft. ZN—reviewing, writing, and editing portions of blood processing section, and creating blood sample and processing figures. MDG—reviewing and editing drafts. LZ—writing statistical analysis portion. SR—reviewing and editing drafts, creating figures for study approval and research coordinator efforts. BP—reviewing and editing drafts, creating figures for study approval and research coordinator efforts. DEB—reviewing and editing drafts. SN—reviewing, writing, and editing portions of blood processing section. NR—conception of work, collecting data/information, reviewing and editing draft.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.