Article Text

Abstract
Background Chimeric antigen receptor T-cell (CAR-T) infusion is associated with early toxicity. Yet, whether early toxicity development holds ramifications for long-term outcomes is unknown.
Methods From a large cohort of consecutive adult patients treated with CAR-T therapies for relapsed or refractory lymphomas from 2016 to 2019, we assessed progression-free survival (PFS), by toxicity development (cytokine release syndrome (CRS), neurotoxicity, or cardiotoxicity]. We also assessed the relationship of toxicity development to objective disease response, and overall survival (OS). Multivariable regression was utilized to evaluate relationships between standard clinical and laboratory measures and disease outcomes. Differences in outcomes, by toxicity status, were also assessed via 30-day landmark analysis. Furthermore, we assessed the effects of early anti-CRS toxicity therapy use (at ≤grade 2 toxicity) on maximum toxicity grade observed, and long-term disease outcomes (PFS and OS).
Results Overall, from 102 CAR-T-treated patients, 90 were identified as treated with single-agent therapy, of which 88.9% developed toxicity (80 CRS, 41 neurotoxicity, and 17 cardiotoxicity), including 28.9% with high-grade (≥3) events. The most common manifestations were hypotension at 96.6% and fever at 94.8%. Among patients with cardiac events, there was a non-significant trend toward a higher prevalence of concurrent or preceding high-grade (≥3) CRS. 50.0% required tocilizumab or corticosteroids. The median time to toxicity was 3 days; high grade CRS development was associated with cardiac and neurotoxicity. In multivariable regression, accounting for disease severity and traditional predictors of disease response, moderate (maximum grade 2) CRS development was associated with higher complete response at 1 year (HR: 2.34; p=0.07), and longer PFS (HR: 0.41; p=0.02, in landmark analysis), and OS (HR: 0.43; p=0.03). Among those with CRS, relative blood pressure (HR: 2.25; p=0.004), respectively, also associated with improved PFS. There was no difference in disease outcomes, or maximum toxicity grade (CRS, neurotoxicity, or cardiotoxicity) observed, based on the presence or absence of the use of early CRS-directed therapies.
Conclusions Among adult lymphoma patients, moderate toxicity manifest as grade 2 CRS after CAR-T infusion may associate with favorable clinical outcomes. Further studies are needed to confirm these findings.
- receptors
- chimeric antigen
- hematologic neoplasms
- immunotherapy
Data availability statement
Data are available upon reasonable request. All data relevant to the study are included in the article or uploaded as supplementary information. All data relevant to the study are included in the article or uploaded as supplementary information. Furthermore, any additional data are available upon reasonable request.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Chimeric antigen receptor T-cell (CAR-T) therapies have rapidly transformed the treatment of advanced hematologic malignancies.1–7 Initially FDA approved in 2017, anti-CD19 CAR-T therapies are associated with dramatically improved outcomes among patients with relapsed and refractory lymphoid malignancies, where prognosis was previously poor.1–8 Due to mounting evidence of preclinical and clinical efficacy suggesting benefit even beyond lymphoid malignancies, >550 additional clinical trials are currently ongoing.9
Despite CAR-T’s benefits, a significant number of patients do not see sustained response, with only 40% seeing longer-term progression-free survival (PFS).1–5 10 Available data have also shown variation in objective disease responses, with 54%–90% of patients responding depending on the population studied, and to date, there are no known early predictors of response.1–5 This is compounded by the common nature of immune-related toxicities, including cytokine release syndrome (CRS), neurotoxicity, and cardiotoxicity.10–13 Yet, currently there is limited evidence to guide the prognostication of outcomes after treatment with CAR-T therapies.
Recently, a growing number of reports have suggested potential relationships between the development of early immune-related toxicities and long-term therapeutic efficacy after the initiation of several immunomodulatory therapies.14–19 This is particularly true of therapies with broad T-cell modulatory and/or autoimmunity-related effects.17 Among patients treated with approved CAR-T therapies, with T-cell engagement and subsequent broad immunomodulatory effects, over 80% experience at least one low-grade adverse event within days of infusion.1–5 Yet, whether the development of CAR-T-related toxicities has ramifications on longer term outcomes after treatment initiation remains unknown.
Methods
From consecutive patients treated with CAR-T therapy for relapsed/refractory diffuse large B-cell lymphoma (DLBCL), follicular lymphoma, or mantle-cell lymphoma at The Ohio State University’s Comprehensive Cancer Center from January 2016 through Decemeber 2019, we evaluated the profile and outcomes of incident CRS, after Institutional Review Board approval. We also assessed the incidence of non-CRS toxicities, including cardiotoxic and neurotoxic events. Study patients included adults≥18 years of age treated with CAR-T for a relapsed or refractory lymphomas. Patients with incomplete medical records for the cancer, cardiovascular, or neurologic variables of interest were excluded. Patients treated with clinical trial combinations of CAR-T and other immunomodulatory experimental therapies were excluded. Baseline characteristics, including medical history, and previous treatment were evaluated. Disease bulk was evaluated and bulky disease defined as having a primary mass that is ≥10 cm, based on imaging (ie, positron emission tomography (PET) or CT). Incident (new) CRS was defined according to consensus criteria.20 Toxicities included those events meeting established Common Terminology Criteria for Adverse Events (CTCAE) within 30 days of infusion.21 We specifically considered cardiotoxicity given increasing evidence of potential profound or limiting events.12 13 Baseline and preceding anticancer therapies were also recorded. Disease response was assessed per Lugano consensus criteria.22 Moreover, we manually searched all subject charts for incident adverse events, inclusive of CRS, neurotoxicity, and cardiotoxicity, in addition to cardiovascular or all-cause death.
Toxicity definitions and grading
Toxicity was considered as events within 30 days of CAR-T infusion or during index-admission. Individual CRS components were assessed according to consensus (Penn) criteria.20 Specifically, fever, coagulopathy, hypoxia, hypotension, and end organ toxicity (eg, renal failure) were included as criteria for incident or worsening CRS (online supplemental table 1). Fever was defined as oral temperature >38°C, and hypoxia was considered oxygen saturation <90% on room air or need for supplemental oxygen, while hypotension was considered as minimum systolic blood pressure (SBP) of <105 mm Hg. We selected <105 mm Hg due to the desire to capture even those with probable relative hypotension. Neurotoxicity grades included encephalopathy, seizure, dysphasia, tremor, headache, confusion, depressed level of consciousness, and cerebral edema.20 Furthermore, CVD events were defined as myocarditis, congestive heart failure, stroke, myocardial infarction, symptomatic arrhythmia (eg, atrial fibrillation ventricular tachycardia), or cardiac death.12 13
Supplemental material
Clinical laboratory measures
Available clinical laboratory measures, including c-reactive protein (CRP), ferritin, complete blood counts with differential, troponin, alanine aminotransferase, and lactate dehydrogenase (LDH) were also considered. These were widely available and routinely measured among treated patients, including those with concern for toxicity.23 24 The maximum value was included where multiple measures were obtained.
Outcomes
The primary outcome was the development of cancer disease progression or death (ie, PFS) after CAR-T initiation. The secondary outcome was the occurrence of partial or complete response at 1 year, and overall survival. Follow-up began from the time of CAR-T initiation. Toxicity events were graded using CTCAE V.5, and Lugano consensus criteria. Moreover, we assessed the incidence and outcomes CRS, by CAR-T product employed (axicabtagene ciloleucel (axi-cel), and tisagenlecleucel (tisa-cel), respectively). Axicabtagene ciloleucel and brexucabtagene autoleucel were considered together as axi-cel, as the two products are clinically identical, with some difference in T-cell enrichment/processing.
Statistical analysis
Descriptive statistics were used to summarize patient characteristics, using mean±SD or median (IQR) for continuous variables and frequency counts with percentages for categorical variables. Time-to-event analysis methods were used to summarize PFS and evaluate associations with these outcomes. Patients without progression or death were considered censored at the last follow-up date. PFS was estimated and displayed by Kaplan-Meier curves. Cox proportional hazards models were used to assess association of patient factors with PFS. Univariable models were fit using maximum CRS grade as the independent variable, followed by using individual CRS components, neurotoxicity, and patient laboratory values. Multivariable Cox proportional hazards models were then fit using potential risk factors identified in the univariable models. To avoid overfitting, we considered the number of PFS events when evaluating how many independent variables to include in the models. Log-rank tests and Kaplan-Meier curves were also used to compare PFS by product type received. Furthermore, χ2 tests were used to assess differences in CRS grade, and its’ individual components (eg, hypotension, etc.) between patients with complete (disease) response at 3, 6, and/or 12 months, versus those not seeing complete response. Multivariable logistic regression was used to account for potential confounders in 12-month disease response. To further delineate the impacts of toxicity, we performed additional landmark analysis of time to PFS, beginning at 30 days after CART initiation. This analysis included only those patients who survived to 30 days without disease progression, stratified by toxicity grade.
In order to understand the potential contribution of specific components of CRS in association with clinical outcomes, individual components of CRS, as well as the presence of neurotoxicity, and cardiotoxicity, respectively, were considered. Blood pressure variables were included alternatively as hypotension (SBP <80 mm Hg) and percent SBP decrease ranges; these were not included together in any model so as to avoid multicollinearity issues. A univariable model for overall survival was also fit with maximum CRS grade as the independent variable. We also assessed the effects of the antitoxicity interventions (tocilizumab and steroids, respectively, at grade ≤2 toxicity) on the maximum grade observed. All statistical tests were two-sided and evaluated at the α=0.05 type-I error rate, with no adjustments for multiple comparisons or evaluation of multiple cut points. All analyses were performed with SAS Software V.9.4.
Results
Overall, from 102 CAR-T-treated patients, 90 were identified as treated with single-agent therapy, including 58 (64.4%) treated with axi-cel, and 32 (35.6%) with tisa-cel, respectively. The patients had a mean age of 61.0±10.9 years and 57.8% were male; and 92.2% had an Eastern Cooperative Group performance status of 0–1; while 94.4% had relapsed/refractory DLBCL. Most patients were heavily pretreated, with a median number of prior treatments of 3±1.3 (range: 0–8), including 33.3% (30) who underwent prior autologous stem cell transplantation. Furthermore, 16.7% (15) of patients were treated within early trials, while 83.3% (75) received CAR-T as part of standard of care. Additional baseline and disease-specific characteristics are described in table 1, including stratification by the degree of CRS.
Baseline characteristics
Incidence and severity of CRS, and other toxicities
Following CAR-T infusion, 88.9% (80) of patients developed CRS (figure 1), with a median time to CRS onset of 2.5 days (IQR: 1–5 days; range: 0–9 days). This included 98.3% (57/58) with CRS after axi-cel, and 71.9% (23/32) after tisa-cel infusion. Of those patients who developed CRS, 38.7% (31) saw a maximum severity of grade 2, and 16.3% (13) reached at least grade 3; with hypotension (87.8% (79)), followed by fever (86.7% (78)) being the most common manifestations (online supplemental figure 1; table 2); 52.2% patients experienced a minimum SBPs of ≤90 mm Hg. There were no specific clinical or disease-related factors associated with the development of CRS. Neurotoxicity occurred in 41 (45.6%) patients, including 34 (37.8%) reaching at least grade 2 (online supplemental figure 2). Furthermore, all patients with neurotoxicity experienced CRS within the same admission. In addition, 17 (18.9%) patients developed a CVD event within days of infusion, including 11 (12.2%) with arrhythmias, 2 (2.2%) with myocarditis, 1 (1.1%) heart failure, and 3 (3.3%) with other CVD events (online supplemental figure 2). Among those with cardiac events, there was no difference in prevalence of concurrent or preceding high-grade (≥3) CRS (p=0.3).
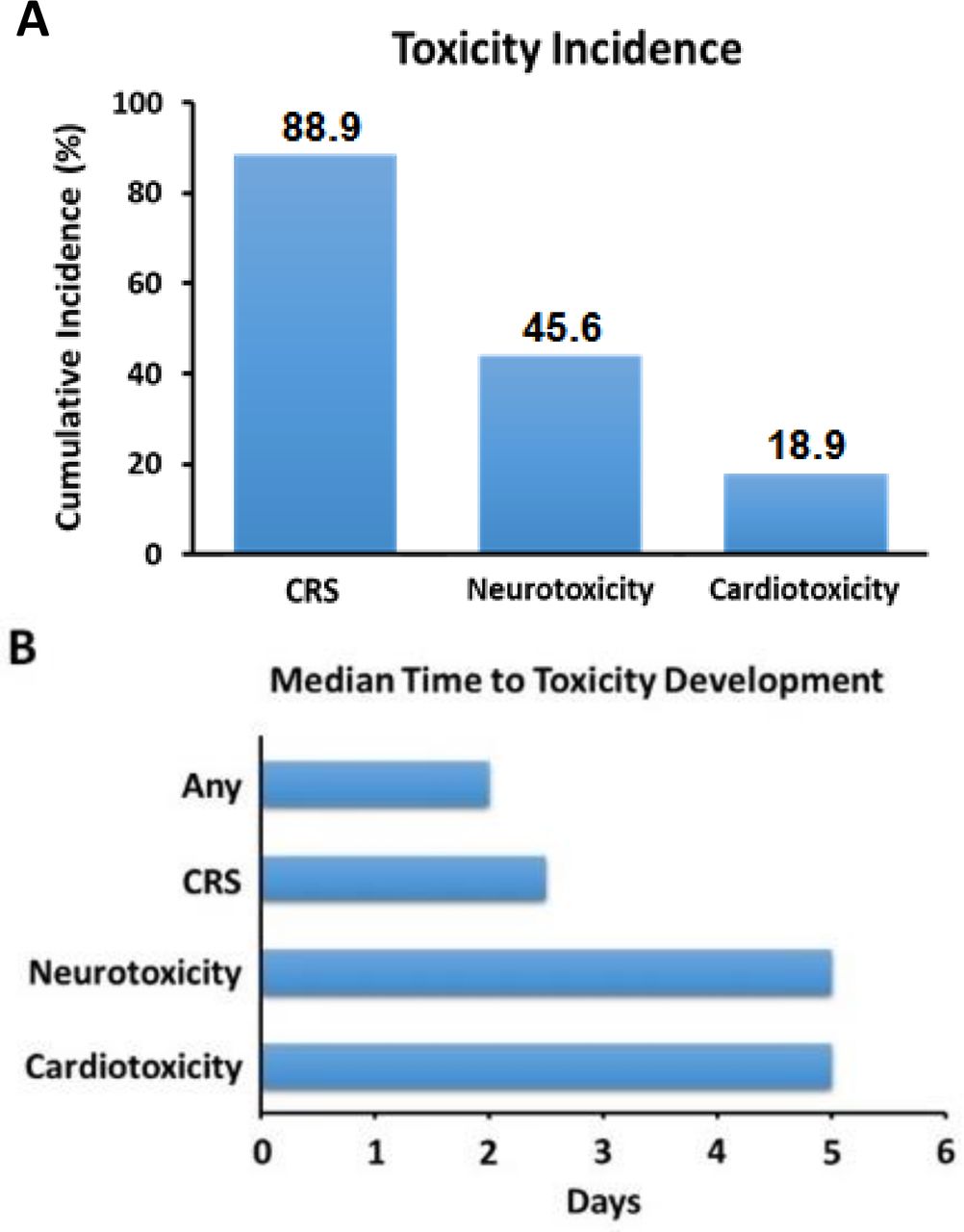
Cumulative incidence of early toxicities (A), and median time to toxicity development (B), after CAR-T cell infusion. CAR-T, chimeric antigen receptor T-cell; CRS, cytokine release syndrome
CRS, neurotoxicity, and cardiotoxicity characteristics after CAR-T cell infusion
Effect of drug product on toxicity occurrence
The use of axi-cel was associated with a higher incidence of CRS (98.3% vs 71.9%, p<0.01; online supplemental table 2). The median time to CRS onset differed by product (3.0 vs 3.4 days for axi-cel and tisa-cel, respectively; p=0.45), as did maximum CRS grade observed (1.93±1.1 vs . 1.15±1.08 for axi-cel and tisa-cel, respectively; p=0.005). Neurotoxicity was more common after axi-cel (63.6% vs 20.3%, p=0.001). There was no difference in cardiotoxicity development, by CAR-T product. There was also no difference in PFS or mortality, by product.
Toxicity development and subsequent disease response
Over a median follow-up of 16.5 months (range: 1–54 months), the median PFS was 6.0 months (95% CI: 3.3 to 8.8 months), and the 25th percentile for overall survival was 9.6 months (95% CI: 5.1 to 14.9 months).Fifty-four (60.0%) patients experienced disease progression or death at 12 months (online supplemental table 3). Complete remissions was attained in 42 (4672%) patients at 3 months postinfusion, while nine (10.0%) had a partial remission, and 38 (42.2%) had progressive (unresponsive) disease; one (1.1%) had stable disease. At 12 months, complete remission remained in 30 (33.3%), with no partial remissions; and 54 (60.0%) had progressive disease. Overall survival among all patients was 90.0% at 3 months, 80.0% at 6 months, and 72.2% at 12 months, with corresponding PFS of 57.8% at 3 months, 47.8% at 6 months, and 40.0% at 12 months. Response rates were similar to the ZUMA-11 and JULIET2 trials.
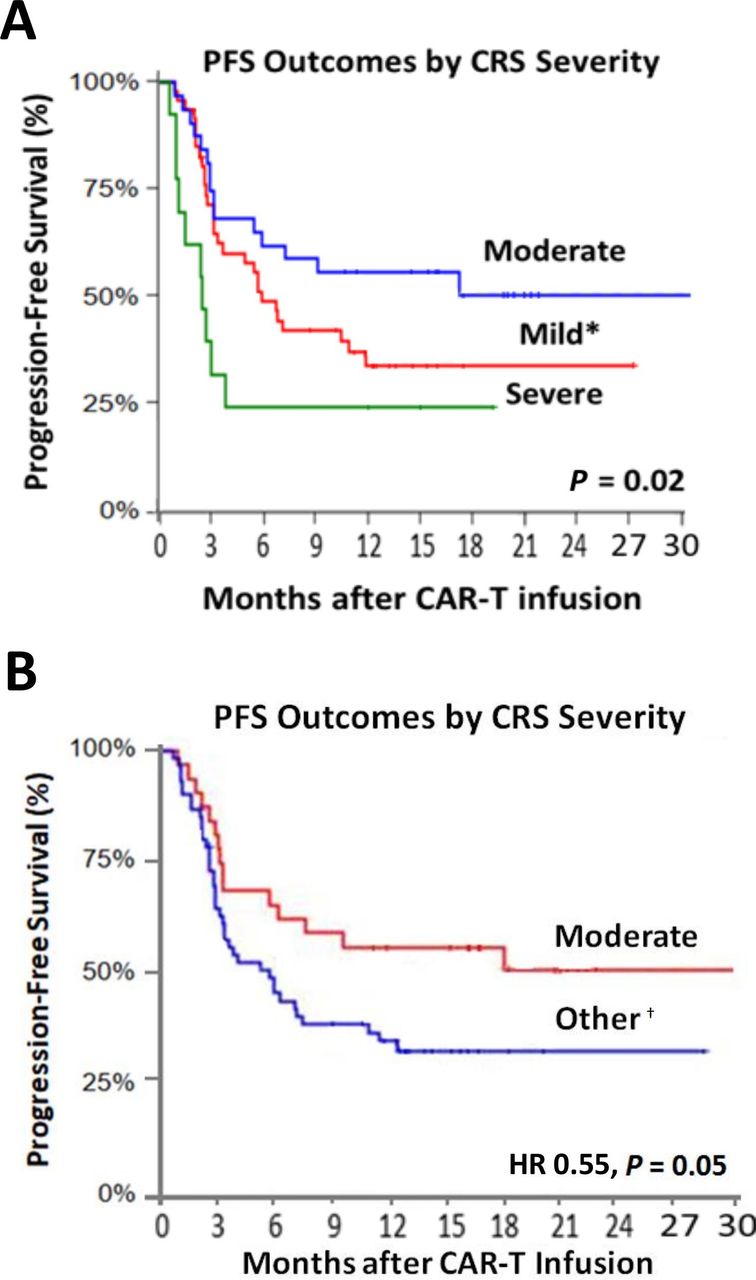
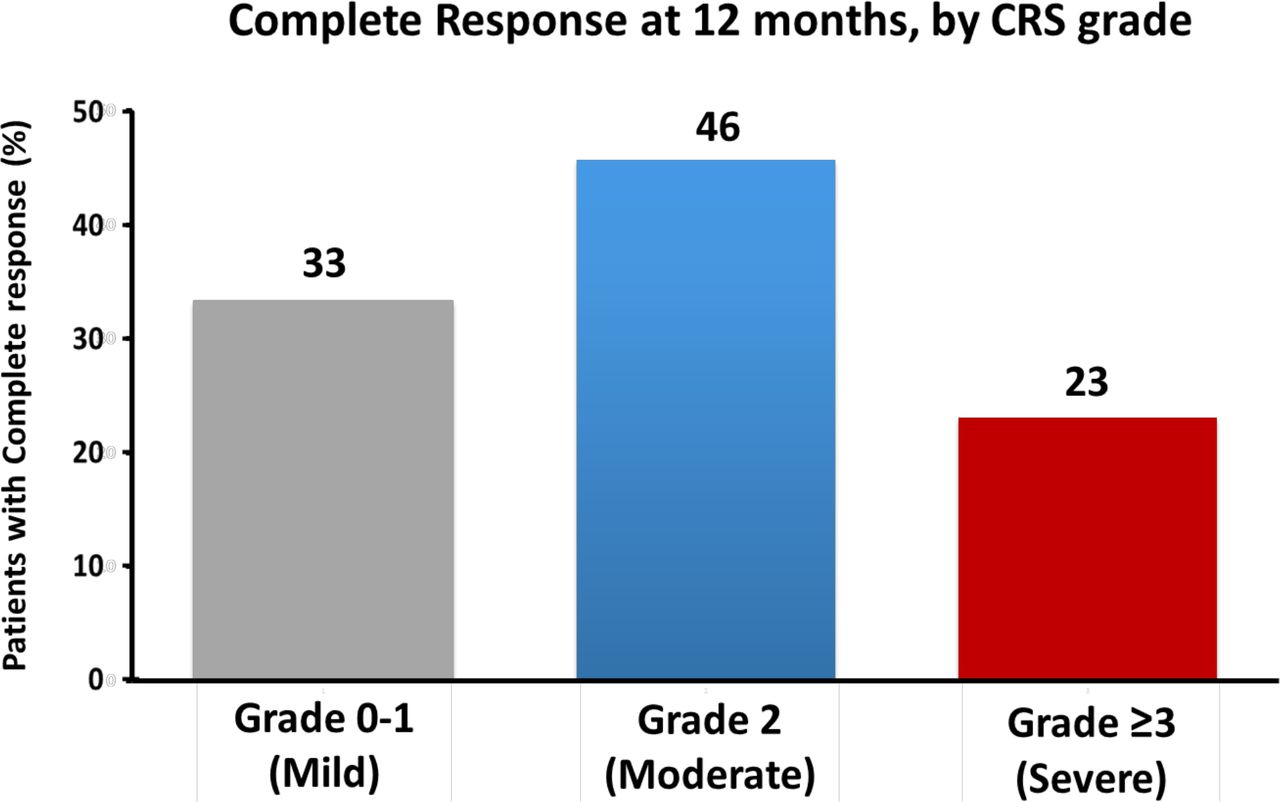
When stratified by CRS-status, maintenance of PFS at 3 months was observed among 70.0% of those with grades 0–1 CRS, and 55.0% with grade 3–4, and 80.0% in those with grade 2 CRS. This pattern remained through 12 months, with 36.0% maintaining PFS with grade 0–1, 33.0% with grade 3–4, and 54.0% with grade 2, respectively (figure 2). Similarly, complete disease response at 1 year was highest among those with moderate (grade 2) CRS (figure 3). These relationships remained even after accounting for other factors linked with disease and therapeutic outcomes (HR: 0.52; 95% CI: 0.19 to 0.88; p=0.03 in landmark analysis; table 3, online supplemental figure 3 and online supplemental table 4). Furthermore, similar differences in overall survival by CRS status were also observed, with moderate CRS seeing improved survival (HR: 0.43; 95% CI: 0.20 to 0.94; p=0.03; online supplemental figure 4 and online supplemental table 4).
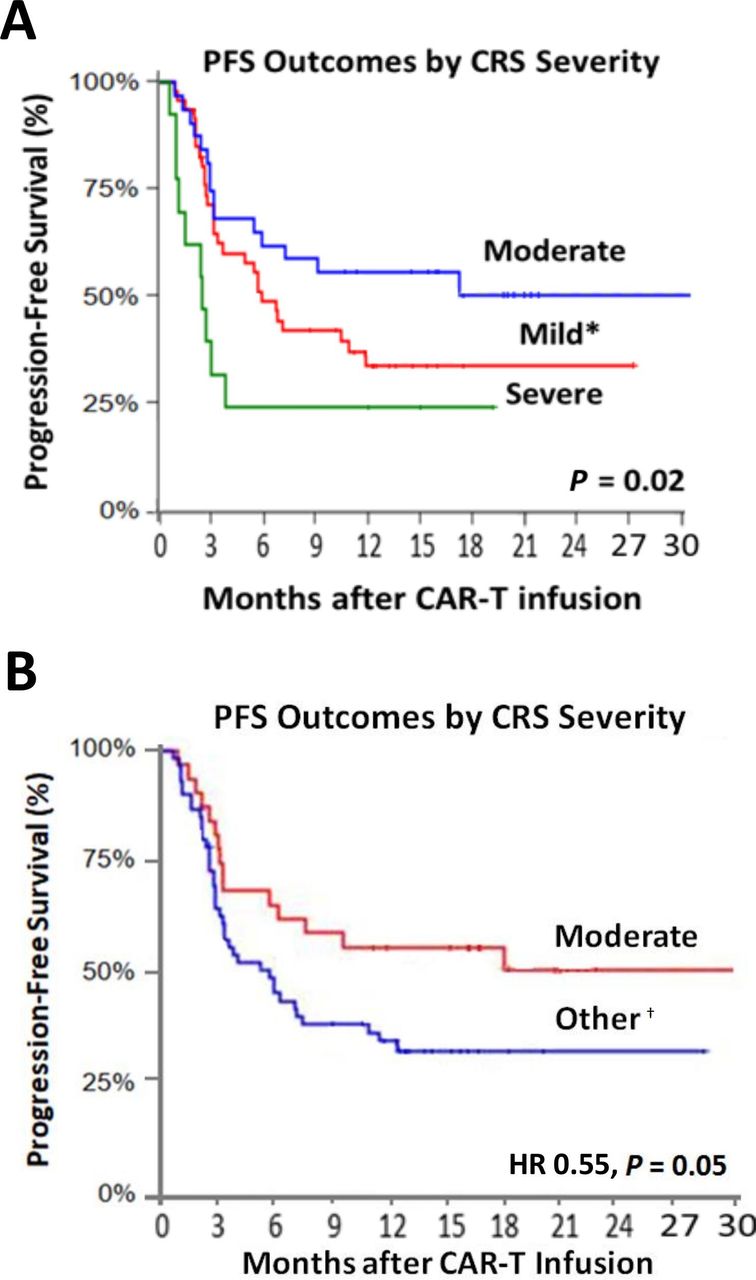
Kaplan-Meir curves with 30-day landmark analysis for progression-free survival across all CRS grades (mild, moderate, severe; A), and the presence or absence of moderate toxicity (B). *Reflects grades 0–1. †All grades except grade 2. CRS, cytokine release syndrome.

{kind=link}
{kind=link}
{kind=link}
Maintenance of complete clinical disease response (reflected by absence of progression or malignancy related death) by 1 year after CAR-T cell infusion, according to CRS grade (severity). Responses were confirmed and assessed according to the 2014 Lugano classification for non-Hodgkin’s lymphoma. CAR-T, chimeric antigen receptor T-cell; CRS, cytokine release syndrome.
Multivariable predictors of progression-free survival after CAR-T cell infusion, following 30-day landmark; by the Lee criteria*
Multivariable predictors of progression-free survival after CAR-T cell infusion, by toxicity type and component*
Multivariable predictors of complete response at 12 months after CAR-T cell infusion*
In univariable analysis of CRS components, hypoxia (HR: 1.88; 95% CI: 1.02 to 3.47; p=0.044), coagulopathy (HR: 11.62; 95% CI: 4.26 to 31.67; p<0.01), organ toxicity (HR: 3.99; 95% CI: 1.92 to 8.33; p<0.01), and profound hypotension (minimum SBP <80 mm Hg; HR: 2.07; 95% CI: 1.11 to 3.87; p=0.02) were linked with worse PFS (online supplemental table 3). Yet, in a multivariable model, only coagulopathy remained associated with worse PFS (HR: 4.72; 95% CI: 1.22 to 18.23; p=0.024; table 3). On stratification by temperature, there were no differences in outcomes. However, on stratification by the degree of SBP reduction, those with a minimum SBP of 80–104 mm Hg saw PFS at 3 and 12 months of 74.0% and 50.0%, respectively, compared with 70.0% at 3 months and 11.0% at 12 months in those with minimum SBPs of ≥105 or the 50.0% at 3 months and 18.0% at 12 months in those with SBPs<80 mm Hg; online supplemental figure 5. Furthermore, by the degree of SBP change postinfusion (≤80, 81–104, or ≥105 mm Hg SBP after CAR-T treatment blood pressure, and absence of active infection), SBP of ≤80 or ≥105 mm Hg was associated with higher PFS, which remained in multivariable analysis (HR: 2.25; 95% CI: 1.29 to 3.91; p<0.01; online supplemental table 5). Relative percent fall (≤10%, >10%–40%, or >40% decline in SBP from pretreatment blood pressure, and absence of active infection) in SBP of ≤10% or >40% was associated with higher PFS on univariate analysis, but not in multivariable analysis. There was no association between the development of other cardiovascular or neurologic events and PFS.
Laboratory markers and disease response
Where measured, the presence of CRP (≥50 mg/L), ferritin (≥500 ng/mL), and LDH (>280 units/L) elevation, respectively, was associated with lower rates of PFS (online supplemental figure 5). However, following multivariable adjustment, outside of ferritin, no associations with outcomes were observed (online supplemental table 4). Similarly, there was no association between prothrombin time, d-dimer, or alkaline phosphatase and disease outcomes after CAR-T infusion. Furthermore, baseline CRP was known in the majority (72.2%) of patients, with an average delta (Δ) peak increase in CRP of 24.0 mg/L, at a median of 3 days post-CART infusion. Among those with available measures, there was a correlation between Δ CRP and maximum CRS grade (r2=0.28, p=0.02).
Grading system selection and PFS outcomes
The effect of CRS grading system on PFS was assessed based on the three primary CRS grading criteria: Penn, Transplantation and Cellular Therapy(TCT), and Lee, respectively. The variance across the three scoring scales was small. The proportion of patients with grade 2 CRS was 34.4% by Penn criteria, 34.4% by Lee criteria, and 38.9% by TCT criteria. There was no difference in the relationship between grading system and PFS (p=0.9995, online supplemental figure 6). Furthermore, on multivariate analysis, a similar degree of significance was maintened between the relationship of b moderate CRS and PFS outcomes, by Penn, TCT, and Lee criteria (p=0.067, 0.067, and 0.033, respectively; online supplemental table 6); with axicel showing stronger associations (HR: 0.40; 95% CI: 0.18 to 0.87; p=0.02; online supplemental tables 7 and 8).
Impact of toxicity reducing interventions
Tocilizumab, an interleukin-6 inhibitor, was used in 36 (40.0%) patients due to worsened CRS, with a mean CRS grade of 1.87±0.63 (median grade of 2) at the time of initiation. There was no difference in mortality (p=0.91), CRS worsening, cardiotoxic or neurotoxic events, by tocilizumab-status (p=0.99). Furthermore, steroids were used in 39 (43.3%) patients due to neurotoxicity (and three patients received steroids for CRS), with a mean toxicity grade at the time of initiation of 2.17±0.71 (median grade 2). Similarly, there was no difference in mortality (p=0.538), including in those with concurrent CRS (p=0.573); or the likelihood of neurotoxicity worsening (p=0.594), or cardiotoxic events (p=0.99). Furthermore, there was no difference in PFS or disease response based on the presence or absence of tocilizumab or steroid use, respectively (online supplemental table 4).
Discussion
In this evaluation of the prognostic implications of toxicity after CAR-T infusion, more than 89% of patients developed CRS or other toxicities. Those patients who developed moderate CRS (grade 2) saw marked improvement in the outcomes of PFS, objective disease response, and overall survival compared with those with no/minimal or severe CRS. This pattern remained, even after accounting for the timing of CRS development, and pretreatment disease severity. Moreover, considering the specific components of CRS, the presence of post-therapy fever and hypotension strongly associated with higher therapeutic response and the avoidance of disease-progression. Furthermore, the development of a post-treatment intermediate hemodynamic response to therapy, reflected by a 10%–40% fall in SBP after infusion, was associated with a nearly threefold increase in the likelihood of longer term PFS. These observations are of particular importance, given the rapidly increasing prevalence of CAR-T use, and the lack of available early markers to guide the determination of likelihood of post-treatment long-term disease response.
The observation of improved anticancer efficacy after early treatment-related toxicity adds to a growing body of evidence linking cancer immunomodulatory therapy related adverse events with clinical disease outcomes.14–19 In prior studies of relapsed lymphoma patients treated with programmed cell death ligand inhibitor-1, nivolumab, the development of adverse events within weeks of treatment initiation was associated with a nearly twofold improvement in long-term objective disease control, and PFS.14 Similarly, the occurrence of early toxicity was associated with a two to fivefold improvement in PFS among nivolumab treated non-small cell lung and melanoma patients.15 16 This pattern of association between anticancer therapeutic efficacy and adverse events has also been noted following other immunotherapies, including interferon, interleukin-2, and talimogene laherparepvec therapy.17–19 Knowledge of these effects has dramatically altered the understanding of cancer cellular biology, anticancer therapeutic efficacy, toxicity, and disease-prognostication. This has generally pervaded the relative acceptance of some toxicities after the initiation of clinically efficacious therapies, particularly if linked with therapeutic effect. Yet, to the best of our knowledge, this is the first investigation linking toxicity with therapeutic efficacy after CAR-T infusion. The implications of these findings may bear weight on the understanding and interpretation of the adverse events after the initiation of CAR-T based therapies.
Knowledge of the indicators of CAR-T activity in patients with advanced malignant disease is of substantial importance. Currently, the primary available tools for monitoring the anticancer efficacy of CAR-T includes postdischarge PET disease activity at least 90 days after CAR-T infusion (in those with lymphomas).25 And although other techniques have been proposed, including cytokine profiling, enumeration of tumor-specific T-cells in peripheral blood, and invasive tumor biopsies, their use has been confounded by CRS syndrome and the need for invasive monitoring.26 Identification of more practical early tools, including the development and degree of toxicity itself, represent a step forward for clinicians tasked with decisions regarding subsequent timely treatment strategies. Given the high correlation between moderate CRS, a fall in SBP of 10%–40%, and subsequent PFS, it is plausible that this prognostic variable could be used to identify patients where early PET imaging (eg, 30 days) may be beneficial. Specifically, this may allow for the initiation of early alternative interventions targeted at enhanced augmentation of CAR-T activity, and the opportunity for improved longer term clinical responses. Nevertheless, with the high prevalence of CRS, initially described in the early phase trials, identification and potential interpretation of these common events may prove beneficial to the rapid assessment of therapeutic efficacy following the initiation of these therapies.
Despite a general awareness of the prevalent nature of toxicity after CAR-T infusion, variation in the reported incidence of CRS has been previously observed. In the pivotal ZUMA-1 trial, CRS was observed among 93% of patients after axi-cel therapy (using the National Institutes of Health consensus criteria), with 59% developing significant hypotension.1 In the JULIET trial, 58% of patients developed CRS using the Penn toxicity criteria, with 22% being grade 3 or higher, and 21% of patients developed neurotoxicity, with 12% being grade ≥3.3 This effect has also been seen with other emerging forms of CAR-T-based therapies.6 7 Moreover, in these studies, the occurrence of more significant (≥grade 2) events has been reported to be over 55%. However, due to the heterogeneity in CRS grading, and the innate differences between CAR-T constructs, adequate comparisons across studies have been limited. Given the high prevalence and the potential impact, these events may portend for disease response, consistent application of uniform criteria, including recent proposals by the American Society for Transplantation and Cellular Therapy to standardize CRS assessments may prove vital to the understanding and evaluation of CAR-T-related toxicity.27 The observation of stronger associations between toxicity and outcomes with axi-cel, accounting for some biologic T-cell activation difference, may also warrant prospective investigation to further delineate the potential impacts of product selection.
Although the exact reasons behind the association of early toxicity with CAR-T outcomes is unknown, several plausible mechanisms may underlie these findings. The level of CRS severity has been previously linked to higher pretreatment disease burdens.5 Accordingly, it is plausible that the occurrence of these phenomena may simply be due to the cellular expansion and activation of immune cell subsets with inflammation, and thus act as an early surrogate for longer term disease response. However, this consideration is limited by the lack of association between high grades (≥3) of CRS and reduction in the rate of continued disease remission. Similarly, a link between the development of other forms of toxicity, including cardiovascular and neurotoxicity was not seen. The occurrence of more moderate degrees of CRS may reflect a stable, but optimal level of immune response to CAR-T therapy, as observed with other immunotherapies.14–19 Given the early report of reasonable disease response with other investigational CAR-T therapies linked with less prevalent manifestations of CRS, it may be likely that the degree of clinical sequelae with established therapies may more clearly reflect the nature of the relationship of toxicity to disease response. Within the current analysis, hypotension was observed to critically predict future disease response. Vascular leak has been previously proposed to contribute to this clinical phenomena.26 CAR-T treatment may induce change in T-cell morphology, prompting abrupt cellular volume expansion and extracellular fluid loss and subsequent intravascular depletion. Yet, with the commonly used CD19-specific CAR-Ts, this relationship has not been observed. In conjunction with prospective evaluations of the prognostic role of early toxicity (including cardiac), additional mechanistic studies are needed.
Study limitations
Several limitations should be acknowledged. Given the retrospective nature of the study, available follow-up within the cohort was not uniform. Also, we could not exclude information bias. Uniform cardiac, neurologic, and correlative laboratory testing protocols were not in place near the initiation of programmatic CAR-T-based treatments, as many patients were in early phase clinical trials. Similarly, timely tocilizumab and steroid application was at the discretion of the treating clinicians and thus varied across time. CART management strategies shifted over time. Notably, the proportion of patients receiving tisa-cel increased after FDA approval in 2018. Furthermore, tocilizumab was utilized more frequently in practice than was initially outlined in the prior JULIET and ZUMA-1 trials.1 2 Despite these factors, the data represent real-world practice, and are thus generalizable to this population. Although we did not observed mitigation in maximum toxicity grade by intervention antitoxicity therapies, it is not known if treatment in combination with supportive therapies (eg, fluids), may have shortened toxicity durations,28 or prevented some patients from reaching high grades or death. We did not include children. Relationships with axi-cel were more robust. Keppra was not commonly used for seizure prophylaxis. The population consisted primarily of DLBCL patients, and it is unclear if the same relationships would be observed among non-lymphoma patients. The current study focused on relapsed and refractory lymphoma patients treated with axi-cel and tisa-cel, respectively, and did not include data from more recently available CAR-T products or other disease populations. Pooled product data were employed for most analyses, as sample size limited further product subgroup analyses. Yet, there was no difference in PFS by CAR-T product used. However, given the desire to best reflect commonly used CAR-Ts in contemporary clinical practice, we focused on these high-profile and more available therapies. Additionally, given that the risk of non-CRS toxicities (namely cardiotoxicity) was not known during the early experience with CAR-T therapy, several adverse events may have gone uncaptured despite extensive search.
Conclusion
CAR-T therapy is associated with an elevated risk of early toxicity. However, the occurrence of a moderate degree of this toxicity appears to associate with long-term disease response and PFS. This suggest that the recognition of early clinical events may enhance the early interpretation of the therapeutic effects of CAR-T-based therapy, and potentially provide the opportunity for the earlier identification of patients likely to need additional disease-specific interventions. Given the anticipated increase in CAR-T use, additional studies of the role of early toxicity as a potential prognostic biomarker, in the setting of severity limiting interventions are needed.
Data availability statement
Data are available upon reasonable request. All data relevant to the study are included in the article or uploaded as supplementary information. All data relevant to the study are included in the article or uploaded as supplementary information. Furthermore, any additional data are available upon reasonable request.
Ethics statements
Ethics approval
The study was approved under the Ohio State University Comprehensive Cancer Center Institutional Review Board.
Acknowledgments
The authors acknowledge and thank the patients and their families treated at the Ohio State University Comprehensive Cancer Center.
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
JEB and ZB contributed equally.
Contributors Concept and design: JEB, ZB, and DA. Acquisition, analysis, or interpretation of data: JEB, ZB, AK, MB, and DA. Drafting of the manuscript: JEB, ZB, KP, and DA. Critical revision of the manuscript for important intellectual content: All authors. Statistical analysis: KP, AG, VYO, and BB. Administrative, technical, or material support: JEB, SJ, and DA. Supervision: JEB and DA.
Funding This work was supported, in part, by National Cancer Institutes (NCI) grant P30 CA016058, and by National Institutes of Health (NIH) grants KL2-TR002734 (Dr Brammer), R01HL127442-01A1 (Dr Gumina), P30CA016058 (Dr Addison), K23HL155890 (Dr. Addison) and K12CA133250 (Dr Addison), and by American Heart Association‐Robert Wood Johnson Foundation (Harold Amos) grant (Dr Addison).
Disclaimer The manuscript’s content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.