Article Text

Abstract
Background Anti-CD19 chimeric antigen receptor T cells (CART-19) frequently induce remissions in hemato-oncological patients with recurred and/or refractory B-cell tumors. However, malignant cells sometimes escape the immunotherapeutic targeting by CD19 gene mutations, alternative splicing or lineage switch, commonly causing lack of CD19 expression on the surface of neoplastic cells. We assumed that, in addition to the known mechanisms, other means could act on CD19 to drive antigen-negative relapse.
Methods Herein, we studied the mechanism of antigen loss in an in vivo CD19-negative recurrence model of chronic lymphocytic leukemia (CLL) to CART-19, established using NOD-scid IL2Rgnull mice and HG3 cell line. We validated our findings in vitro in immortalized B-cell lines and primary CLL cells.
Results In our in vivo CLL recurrence model, up to 70% of CART-19-treated mice eventually recurred with CD19-negative disease weeks after initial positive response. We found that the lack of CD19 expression was caused by promoter DNA hypermethylation. Importantly, the expression loss was partially reversible by treatment with a demethylating agent. Moreover, this escape mechanism was common for 3 B-cell immortalized lines as well as primary CLL cells, as assessed by in vitro coculture experiments.
Conclusions Epigenetically driven antigen escape could represent a novel, yet at least partially reversible, means of CD19 loss to CART-19 in B-cell tumors.
- antigens
- B-lymphocytes
- hematologic neoplasms
- receptors
- chimeric antigen
- translational medical research
Data availability statement
Data sharing not applicable as no datasets generated and/or analysed for this study. Data are available upon reasonable request. All data relevant to the study are included in the article or uploaded as supplementary information. Publication-related data are available upon request from the corresponding authors.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Background
CD19 is a transmembrane protein present on the surface of all B lineage cells except plasma cells. Likely due to its role in B-cell receptor signaling, CD19 expression is rarely lost upon B-cell neoplastic transformation, and CD19 immunotherapeutic targeting has been proposed decades ago.1 Indeed, spectacular results have been achieved by anti-CD19 chimeric antigen receptor T cells (CART-19), which currently represent the most advanced personalized cell-based immunotherapeutic tool.2–6 Depending on the cancer type, the initial remission rate induced by CART-19 in patients with B-cell cancer can be as high as 90%.2 3 However, after an initial positive response, 30%–60% of CAR-treated patients will relapse. While this is mostly caused by suboptimal persistence of CART-19 cells, up to 20% of these relapses are due to antigen loss on the surface of neoplastic cells.7 8
Some of the possible means of CD19-negative tumor escape are already known. First, a dramatic upregulation of alternatively spliced CD19 transcripts lacking the epitope targeted by CART-19 was described.9 Later, direct CD19 mutations leading to a truncated protein with non-functional or absent transmembrane domain10 extended the landscape of genetic means of antigen loss. Occasionally, some of the genetic mechanisms might be at play already prior to CART-19 treatment, leading to a small CD19-negative population already detectable at baseline.6 11 Such CD19-negative clones are selected under the treatment pressure, expand and drive the subsequent relapse. The immune pressure, triggered by CART-19 treatment, combined with the inherent B-cell lineage’s plasticity can also induce antigen-negative relapse by lineage switch,12 13 for example, from lymphoid to myeloid. Finally, trogocytosis has recently emerged as a cause of invertible antigen loss provoked by CAR T cells.14 Herein, using one in vivo and various in vitro disease models of chronic lymphocytic leukemia (CLL), we show that CD19 epigenetic repression can also cause antigen escape, and this mechanism is partially reversible using demethylating agents.
Methods
Establishment of CD19-negative and CD19-positive cell lines from recurrent tumors
CAR T cell preparation, quality assessment and in vivo studies were previously described.15 Briefly, T cells were isolated from buffy coats of healthy donors purchased from the Department of Transfusion and Tissues of University Hospital Brno using the RosetteSep Human T cell enrichment Cocktail (StemCell). Immediately following isolation, T cells were activated using IL-2 (50 U/mL; Miltenyi Biotec) and Dynabeads Human T-activator CD3/CD28 (ratio 1:3 bead:cell; ThermoFisher Scientific). After 24 hours, cells were transduced using anti-CD19 CAR lentivirus and a spinoculation protocol.
Eight-week-old to twelve-week-old male and female NOD-scid IL2Rgnull (NSG) mice were injected with 2×106 HG3 cells via tail vein. Control or CAR T cells were injected intravenously 6 days later, and mice were closely followed for manifestation of the disease. Subcutaneous relapse tumors (figure 1A) that developed in CAR-treated mice were excised, homogenized and the cell suspension stained with the following antibodies: eFluor450-conjugated CD3 (OKT3; eBioscience), APC-conjugated CD19 (4G7; CapricoBio) and PE-conjugated CD45 (HI30; Invitrogen), followed by cell sorting of CD19-negative and CD19-positive cells using BD Aria Fusion. Cell viability was monitored with 7-aminoactinomycin D (7-AAD; Invitrogen). The gating strategy for sorting of CD19-negative B cells was 7-AAD-, CD45+, CD3-, CD19- while for CD19-positive cells 7-AAD-, CD45+, CD3-, CD19+ were selected. Following sorting, cells were incubated in RPMI with 10% fetal bovine serum (FBS; 37°C, 5% CO2).

Establishment of CD19-negative in vivo recurrence model to CART-19. (A) Setting up of the recurrence disease model in NOD-scid IL2Rgnull mice (NSG). Timing of B and T cells’ injection is depicted in the upper panel. Mice were closely followed for disease manifestation (by regular weighing depicted in the middle panel). Around 2 weeks after tumor cell injection, mice started to show signs of leukemia and subsequently all control and some CAR-treated mice had to be sacrificed due to severe illness (~day 20). At this point, a clear population of CD19-positive B cells as well as CD3-positive T cells could be found in disease-affected organs by flow cytometry (lower panel, representative sample depicted). Weeks after initial response, up to 70% of CART-19-treated surviving mice relapsed with subcutaneous tumors that lost CD19 antigen (lower panel). (B) Immunohistochemistry staining for CD19, CD20 (both markers of tumor B cells) and CD3 T-cell marker in one representative recurring tumor showed loss of CD19 antigen in tumor cells. B cells from this tumor were sorted into CD19-negative and CD19-positive population and seeded in vitro. (C) CD19 expression was gradually regained in in vitro culture over time as shown by flow cytometry. Freshly sorted CD19-negative cells (in gray), CD19-negative cells cultivated for 22 days (in red) and CD19-positive cells (in black) were assessed. (D) Freshly sorted CD19-negative cells did not express CD19 mRNA as assessed by qRT-PCR. (E) Alternative splicing is likely not driving the antigen-negative escape in assessed samples, as no truncated cDNA forms were detected by PCR. Primers used in the assay were described previously9 and encompass the region of CD19 exons 1–5, exons 4–8 or exons 1–4. Arrows indicate the expected size of full length (FL; green) or truncated cDNA product (red). CD19-(D22)=CD19-negative cells cultivated for 22 days; ∆ex2=skipping of exon 2; ∆ex5−6=skipping of exons 5 and 6. CART-19, anti-CD19 chimeric antigen receptor T cells.
Immunohistochemistry
One part of the recurring tumor was fixed in 10% neutral buffered formalin immediately following mouse sacrification and embedded in paraffin. Sequential paraffin-embedded complete sections were used for evaluation of B-cell markers CD19 and CD20 as well as the T-cell marker CD3. Monoclonal antibodies against CD19 and CD20 were purchased from Dako (LE-CD19 and L26, respectively); anti-CD3 polyclonal antibody was purchased from Sakura. Immunohistochemistry (IHC) staining and evaluation was performed at the Department of Pathology of the Faculty Hospital Brno.
Promoter methylation and gene expression analyses
Viable CD19-negative and CD19-positive cells from recurrent tumors were sorted into TRIzol (ThermoFisher) and RNA and DNA isolated following TRIzol manufacturer’s recommendations. DNA was bisulfite-converted using Fast Bisulfite Conversion Kit (Abcam). Region (chr16:28931659–28932127) containing 8 CpG sites and spanning the start of exon 1 of CD19, which was previously assessed with regard to gene regulation,9 was selected, amplified through nested PCR and sequenced as previously described.16 CD19_prom_FW1 (5′-TTTGAGAAGGAGTTTATGTGT-3′) and CD19_prom_RV1 (5′-ACTCAACCAAAAACACCCAA-3′) primers were used in the first PCR, while CD19_prom_FW2 (5′- GTATTGTTTTAGAGTATTTGTAA-3′) and CD19_prom_RV2 (5′-CCAACCTCAATCCCTTCC-3′) primers were used in the second PCR. PCR amplification was carried out using KAPA2G Robust HotStart ReadyMix (Sigma).
Additionally, 250 ng of total RNA were reverse-transcribed using Superscript II (Invitrogen) and random hexamers following manufacturer’s instructions. The amounts of PAX5, CD19 and GAPDH mRNA were quantified by real-time PCR with QuantStudio 12 Flex Real-Time PCR system (Applied Biosystems). Negative controls were included, and assays were carried out in triplicate. The ΔΔCt method was used for the calculation of mRNA content. Moreover, presence of alternatively spliced CD19 mRNA species was assessed by PCR using primers described previously.9 PCR amplification products were resolved on 1% agarose gel stained with MidoriGreen (Nippon Genetics).
5-aza-2′-deoxycytidine treatment in vitro and in vivo
Freshly sorted 1×106 CD19-negative cells were incubated with either 1 µM or 5 µM 5-aza-2′-deoxycytidine (Sigma) or DMSO (Sigma; negative control) for 48 hours in vitro. CD19 expression was then assessed by flow cytometry, and CD19 promoter methylation was studied as described above. Additionally, CLL was established in NSG mice as described above and the mice were injected with CAR T cells 5 days later. Next day, mice started to be treated with 5-aza-2′-deoxycytidine (2.5 mg/kg) or phosphate-buffered saline (PBS; vehicle control) subcutaneously daily until the end of the experiment. To assess the effect on CD19 expression, mice were killed when disease progression became apparent (defined as >20% loss of body weight or hind-limb paralysis). Spleen and liver were harvested, mechanically homogenized and leukemic cells analyzed by flow cytometry.
Human primary cells, cell lines and culture conditions
Two primary CLL samples (termed CLL1 and CLL2) were obtained from the biobank of the Department of Internal Medicine–Hematology and Oncology, University Hospital Brno. These samples were taken after written informed patient’s consent in accordance with the Declaration of Helsinki, under protocols approved by the hospital ethics committee. The selected patients fulfilled the iwCLL/NCI diagnostic criteria for CLL.17 Collected peripheral blood samples were processed and genetically screened as described previously15 and vitally frozen. After thawing, both samples showed high viability (>85%) and were kept in AIM-V medium (ThermoFisher Scientific).
HG3 (a generous gift from Dr Rosenquist, Sweden) and Ramos (DSZM) cell lines were maintained in RPMI-1640 medium (Biosera), while MEC1 (DSZM) in IMDM (Biosera). All media were supplemented with 10% (v/v) heat-inactivated FBS (Biosera) and 1% (v/v) penicillin/streptomycin (MP Biomedicals).
In vitro cocultivation assays
CAR/control T cells were mixed with 2×106 HG3, RAMOS, MEC1 cell lines and primary CLL cells in 3:1 ratio (effector:target) in AIM-V medium (ThermoFisher) and left undisturbed for 24 hours. CD19-negative and CD19-positive B cells were then sorted out, DNA was isolated, CD19 promoter methylation studied and CD19-negative cells were incubated with 5-aza-2′-deoxycytidine as described above. Besides, HG3 was mixed with CART-19 cells at 1:1 ratio for 12 hours before sorting CD19-negative and CD19-positive cells for CD19 promoter methylation analysis.
Results
Using HG3 cell line in NSG mice, we recently established an in vivo CLL disease model,15 which we treated with in house produced CART-19 or control T cells. Unlike the mice injected with control T cells, most of CART-19-treated mice overcame the progressive disease that developed after ~2 weeks of tumor cells’ injection and mostly affected spleen, liver and kidneys. However, up to 70% of the surviving mice recurred with subcutaneous tumors weeks after the initial positive response (figure 1A, online supplemental figure 1). Due to the striking difference in the neoplastic localization, we set out to characterize the malignant cells forming the recurring tumors. A prominent downregulation of CD19 expression on B cells isolated from these tumors was apparent both by flow cytometry and by IHC (figure 1A,B respectively).
Supplemental material
Importantly, we were able to separate CD19-negative and CD19-positive cells from two such tumors by fluorescence-activated cell sorting (FACS). Of note, only <0.2% of the originally infused HG3 cells were detectable in the CD19-negative gate used for sorting (online supplemental figure 2). Data from one of these tumors are depicted in the main figures, while results from the other are summarized in online supplemental figure 3. We cultivated the sorted cells in vitro for several weeks and noticed that CD19 surface expression was gradually reconstituted over time in the cell culture originally containing only CD19-negative cells (figure 1C; repeatedly observed in 2 rounds of CD19-negative cell FACS sorting). Importantly, while freshly sorted CD19-negative cells did not express CD19 mRNA (figure 1D, online supplemental figure 3A), its loss was only temporary, and apparently, it was not due to alternative splicing described previously9 (figure 1E, online supplemental figure 3B) or aberrant expression of PAX5, transcription factor known to regulate CD19 expression18 (online supplemental figure 4).
Supplemental material
Supplemental material
Supplemental material
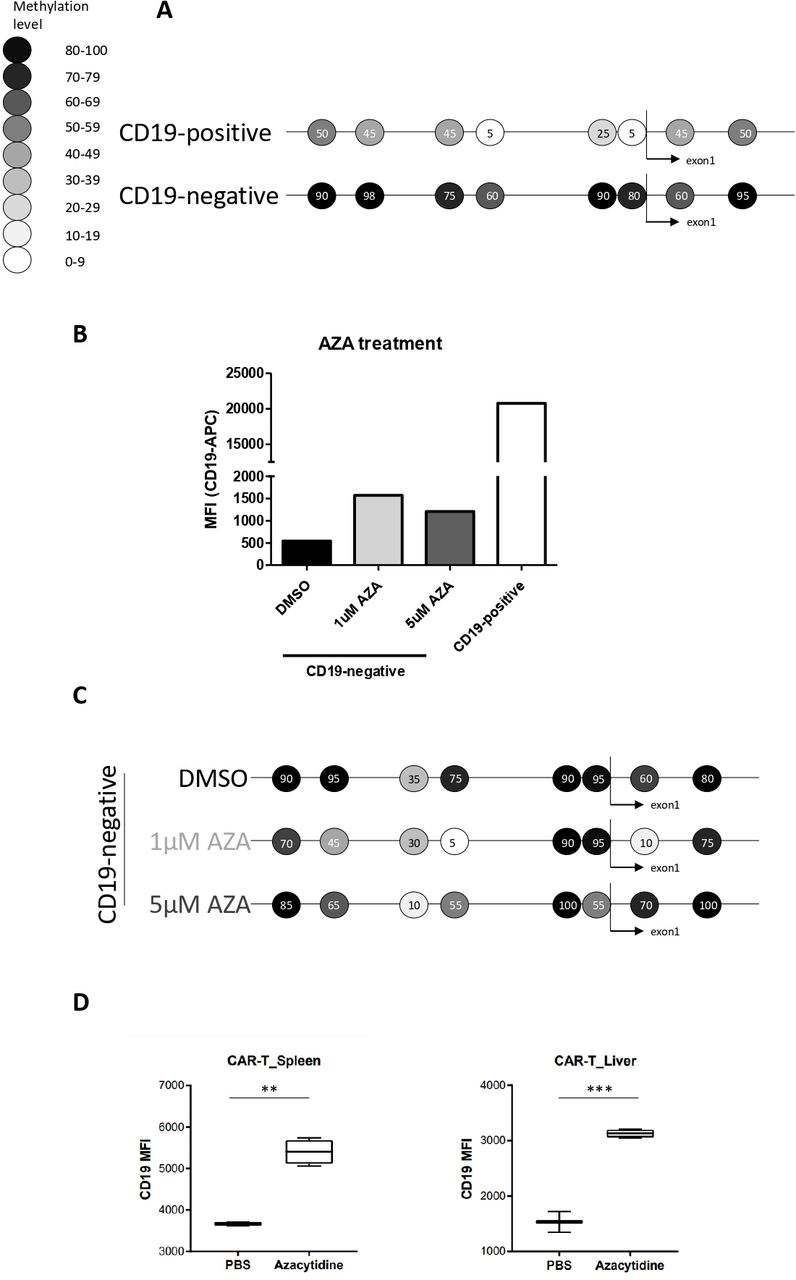
Based on these results, we suspected the mechanism driving antigen-negative escape to be more flexible than those described previously, likely involving epigenetic orchestra. In this regard, CD19 promoter does not contain a CpG island per se, but there are several CpGs spanning the transcription initiation site. Indeed, when we studied the CD19 promoter methylation of CD19-negative and CD19-positive cells sorted from a relapsed tumor by direct bisulfite sequencing, we found that the entire assessed region was affected by hypermethylation in the CD19-negative cells (figure 2A, online supplemental figure 3C). To confirm the causal character of this methylation, we treated freshly sorted CD19-negative cells with demethylating agent 5-aza-2′-deoxycytidine (1 µM or 5 µM) in vitro. After 48 hours, increased CD19 expression on the surface of treated cells was apparent by flow cytometry (figure 2B, online supplemental figure 3D), and this was accompanied by a decrease in methylation of some of the CpGs assessed by the promoter methylation bisulfite sequencing (figure 2C). We next assessed whether 5-aza-2′-deoxycytidine treatment could prevent the loss of CD19 triggered by CART-19 in our NSG mouse model. Notably, dual treatment of CART-19 combined with 5-aza-2′-deoxycytidine in vivo indeed led to increased CD19 expression on leukemic cells compared with CART-19 monotherapy (figure 2D).

CD19 promoter DNA methylation can drive tumor escape and it is reversible with 5-aza-2′-deoxycytidine in vitro and in vivo. (A) Bisulfite sequencing of a region spanning CD19 promoter including 8 CpG sites in sorted CD19-negative and CD19-positive cells from one recurring tumor showed an inverse correlation between DNA methylation and CD19 expression. Results representative of two technical replicates are shown. Methylation levels’ color code is maintained throughout the figure and represents percentage of methylation. (B) Freshly sorted CD19-negative cells from one recurring tumor were treated with 5-aza-2′-deoxycytidine (AZA; 1 µM or 5 µM) or DMSO for 48 hours. Then, CD19 expression levels were assessed by flow cytometry. Results representative of two biological replicates are shown. (C) Bisulfite sequencing of CD19 promoter of samples assessed in (B) is shown. (D) NSG mice with established CLL were treated either solely with CART-19 (PBS) or by combination of CART-19 and 5-aza-2′-deoxycytidine (azacytidine). CLL cells harvested from these mice showed markedly different levels of mean fluorescence intensity (MFI) of CD19. **p value 0.0015, ***p value 0.0002. CART-19, anti-CD19 chimeric antigen receptor T cells; CLL, chronic lymphocytic leukemia.
Finally, to establish if this epigenetic escape mechanism was solely specific to HG3 cells, we cultured B-cell lines (HG3, RAMOS, MEC1) as well as primary cells from 2 CLL patients with CAR/control T cells for 24 hours in vitro and sorted out CD19-negative and -positive cells for the promoter methylation study. All three studied immortalized cell lines, as well as primary CLL cells, employed the same epigenetic downregulation of target antigen according to our results (figure 3A,B respectively). Since MEC1 cells did not fully abrogate CD19 expression in response to CART-19 cells, we exposed them solely to CART-19 and sorted out the CD19-negative and CD19-positive fraction from this single coculture. Furthermore, we mixed HG3 with CART-19 at 1:1 ratio for only 12 hours, to achieve a partial response, and sorted the most CD19-negative and the most CD19-positive cell fraction. In both these approaches, CD19-negative cells clearly showed a strong hypermethylation of CD19 promoter in contrast to their CD19-positive counterparts (figure 3C). Of note, CD19 downregulation in these short-term coculture experiments was highly transient, as within 48 hours CD19 levels were readily restored likely due to the short-term character of the immunotherapeutic pressure enforced by CART-19 as opposed to the in vivo situation (figure 3D).
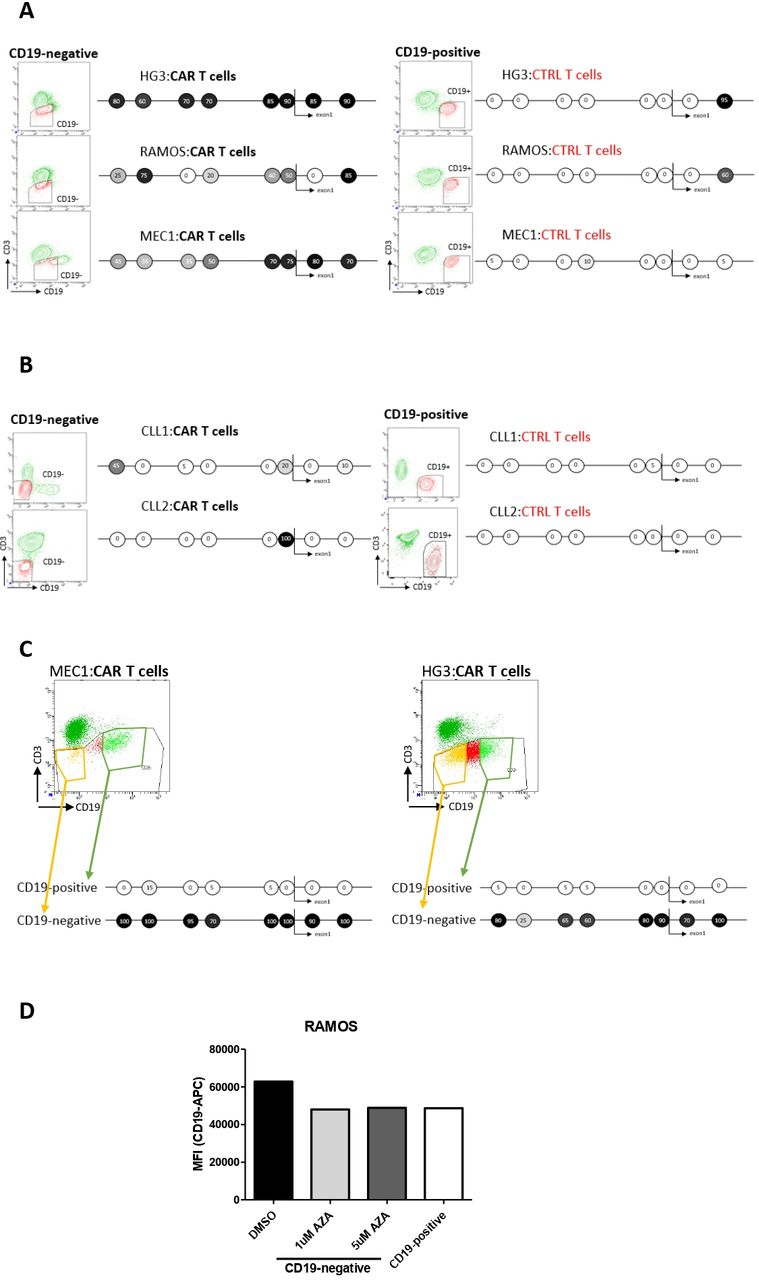
{kind=link}
{kind=link}
{kind=link}
Transient antigen-negative escape can be caused by CD19 promoter methylation in vitro. (A) HG3, RAMOS and MEC1 cell lines cocultured for 24 hours with CAR/control T cells were sorted into CD19-negative and CD19-positive B-cell populations and CD19 promoter methylation was studied with bisulfite sequencing. (B) Primary CLL cells were cocultured and analyzed as in (A). The CLL1 cells harbored a minor TP53 mutation (VAF <4%) and no recurrent genomic aberrations (eg, 13qdel, 11qdel, trisomy of 12), while CLL2 cells were wild-type for TP53, and harbored a deletion of 11q region. (C) MEC1 cells were mixed with CART-19 cells for 24 hours and both CD19-negative and CD19-positive proportion of cells were sorted out for CD19 promoter methylation analysis (left). HG3 cells were mixed with CART-19 cells at 1:1 ratio for 12 hours and then CD19-negative and CD19-positive fraction of cells was sorted out for CD19 promoter methylation analysis (right). (D) Freshly sorted CD19-negative RAMOS cells from (A) were treated with 5-aza-2′-deoxycytidine (AZA; 1 µM or 5 µM) or DMSO for 48 hours. Then, CD19 expression levels were assessed by flow cytometry. CLL, chronic lymphocytic leukemia; MFI, mean fluorescence intensity.
DISCUSSION/CONCLUSION
Disease relapse represents the key limitation of CART-19 response durability. While optimized CAR designs, which would increase the specificity and persistence of effector cells, are undoubtedly critical to overcome this hurdle,19 it has become apparent that some tumors will still be able to escape through an antigen-loss mechanism.9 10 12–14 Herein, we have identified epigenetic repression as an additional means of CD19 downregulation. This mechanism was causing relapse in vivo in a recently established CLL disease model15 as well as in several in vitro models. According to our results, this could well be the first means of antigen-negative escape to CAR T-cell immunotherapy partially reversible with drugs to be discovered.
Until today, in vivo relapse models to CART-19 immunotherapy have been described only by a handful of studies.13 14 Of these, one uses limiting doses of CART-19 in acute lymphoblastic leukemia (ALL) mouse model to mimic relapse,14 while the other takes advantage of a syngeneic murine ALL model to induce lineage switch in CART-19 recipient mice.13 Herein, we had a unique opportunity to study CD19-negative recurrence in immunocompromised mice injected with a CLL cell line HG3. Despite the early potency and persistence of functional CAR T cells, up to 70% of CART-19-treated animals eventually developed a CD19-negative recurrence. To the best of our knowledge, this is the first in vivo antigen loss CLL recurrence model to CART-19 to be developed.
We were able to isolate and culture CD19-negative cells causing recurring tumors and perform some molecular analyses, which ruled out alternative splicing9 as the causal escape mechanism. On top of that, CD19 expression was steadily regained in in vitro culture. These were unexpected findings as they clearly contradicted all so far known mechanisms of target loss; neither genetic escape10 20 nor lineage switch12 13 were likely to be resolved so promptly during in vitro culture, and trogocytosis14 could not be at play since tumor-sorted B cells were cultured and re-sorted in absence of T cells.
Instead, we found an inverse correlation between CD19 promoter DNA methylation and its expression in the relapsed tumors, as well as in in vitro coculture experiments with B-cell immortalized cell lines and primary CLL cells. On top of that, when cells with methylated CD19 promoter were treated with a demethylating drug, an increase in the CD19 expression accompanied by a decrease in DNA methylation was observed, validating the repressive character of methylation. Interestingly, DNA methylation of the region studied herein had already been assessed once in CD19-negative relapse samples from patients with ALL, but no promoter methylation had then been found that would explain the loss of CD19 expression in the primary samples.9 However, ALL and CLL are two biologically distinct entities, and differences in gene regulation can be expected and have already been described.21 22
Finally, we cannot completely rule out the possibility of selecting a pre-existing CD19-dim or CD19-negative HG3 cell population at the time of in vivo recurrence as such event has already been described in the clinics.6 11 23 Nevertheless, it is highly unlikely that the entire starting CD19-positive target cell population would be replaced by CD19-negative cells within just 24 hours in in vitro coculture experiments, thereby undermining the selection theory. Notably, given how readily CD19 is reconstituted on surface of CD19-negative cells sorted after 24 hours of in vitro coculture, other epigenetic means (histone modifications, etc) could well be involved in stabilizing the effect of DNA methylation on CD19 repression in vivo.
These results indicate that at least a part of B-cell derived tumors could apply epigenetic silencing to escape CD19 immunotherapeutic targeting. Importantly, this mechanism could be at least partially reversed using demethylating agents. We propose that the use of agents like 5-aza-2′-deoxycytidine in combination with CAR T-cell therapy might prevent such CD19-negative relapses and thereby improve the response rate in these scenarios.
Data availability statement
Data sharing not applicable as no datasets generated and/or analysed for this study. Data are available upon reasonable request. All data relevant to the study are included in the article or uploaded as supplementary information. Publication-related data are available upon request from the corresponding authors.
Ethics statements
Ethics approval
The study was approved by the institutional review board, the animal study was additionally approved by the Ministry of Education, Youth and Sports (MSMT-25883/2017–2).
Acknowledgments
The authors would like to thank Karla Plevova, PhD, for selecting primary CLL cells for the study. We would like to also thank all medical doctors and nurses involved in collecting patients’ samples (Prof Doubek, Dr Brychtova) in the University Hospital Brno. We would additionally like to thank Sarka Pavlova, PhD, and Lenka Jurackova for extensive help with bisulfite sequencing. Finally, we would like to acknowledge the help of Eva Vojackova and the Department of Pathology of the Faculty Hospital Brno, which was instrumental in performing IHC.
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
AL, LD and VK contributed equally.
Contributors VM, LD and VK prepared CAR T cells, VM and ALe performed the in vitro co-cultivation experiments with cell lines and primary CLL cells and bisulfite sequencing. HP, ALa and LD conducted all other experiments. JV and MC supervised in vivo studies. TL supervised flow cytometry experiments. VM and MS designed the project, discussed and analyzed data. All authors contributed to drafting of the manuscript.
Funding This project has received funding from the European Union’s Horizon 2020 research and innovation program under the Marie Skłodowska-Curie actions and it is co-financed by the South Moravian Region under grant agreement no 665 860 (to VM). It also has been financed by the research grant MUNI/A/1595/2020, TACR (TN01000013) and Martina Roeselová Memorial Foundation (to ALe). The work was carried out with the support of the research infrastructure EATRIS-CZ (ID number LM2018133) funded by MEYS CR. The project "Modernization and instrumental upgrade of the National Center for Medical Genomics" (reg. No. CZ.02.1.01/0.0/0.0/18_046/0015515) is supported by the Operational Programme Research, Development and Education. This report reflects only the author’s view and the EU is not responsible for any use that may be made of the information it contains.
Competing interests None.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.