Article Text

Abstract
Background To provide pooled longer term data from three groups of a phase 2 study of cemiplimab in patients with advanced cutaneous squamous cell carcinoma (CSCC), and to determine duration of response (DOR) and impact on quality of life (QoL).
Methods Patients received cemiplimab 3 mg/kg every 2 weeks (group 1, metastatic CSCC [mCSCC], n=59; group 2, locally advanced CSCC, n=78) or cemiplimab 350 mg every 3 weeks (group 3, mCSCC, n=56). Primary endpoint was objective response rate (ORR) per independent central review (ICR). QoL was repeatedly measured at day 1 of each treatment cycle (groups 1 and 2: 8 weeks; group 3: 9 weeks).
Results Median duration of follow-up was 15.7 months. Overall, ORR per ICR was 46.1% (95% CI: 38.9% to 53.4%). Complete response (CR) rates were 20.3%, 12.8%, and 16.1% for groups 1, 2, and 3, respectively. Median time to CR was 11.2 months. Among patients with partial response or CR, the estimated proportion of patients with ongoing response at 12 months from the first objective response was 87.8% (95% CI: 78.5% to 93.3%), with median DOR not reached. Kaplan-Meier estimated probability of overall survival (OS) was 73.3% (95% CI: 66.1% to 79.2%) at 24 months, with median OS not reached. Global Health Status (GHS)/QoL improvements were observed as early as cycle 2 and were significantly improved and durable until last assessment. Kaplan-Meier estimate of median time to first clinically meaningful improvement for pain was 2.1 (95% CI: 2.0 to 3.7) months and was significantly improved in responders versus non-responders (p<0.0001).
Conclusions This is the largest (n=193) clinical dataset for a programmed cell death-1 inhibitor against advanced CSCC, confirming the sustained substantial clinical activity of cemiplimab in these patients, including new findings of improved CR rates over time, increasing DOR, and durable pain control and GHS/QoL improvement.
Trial registration number ClinicalTrials.gov Registry (NCT02760498), https://clinicaltrialsgov/ct2/show/NCT02760498.
- immunotherapy
- programmed cell death 1 receptor
- skin neoplasms
- clinical trials
- phase II as topic
Data availability statement
Data are available upon reasonable request. Qualified researchers may request access to study documents (including the clinical study report, study protocol with any amendments, blank case report form, statistical analysis plan) that support the methods and findings reported in this manuscript. Individual anonymized participant data will be considered for sharing once the indication has been approved by a regulatory body, if there is legal authority to share the data and there is not a reasonable likelihood of participant re-identification. Submit requests to https://vivli.org/.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Background
Cutaneous squamous cell carcinoma (CSCC) is the second most common skin cancer in the USA, with increasing incidence.1 Most cases are cured by complete surgical excision.2 3 However, a small but substantial number of patients present with or subsequently develop metastatic CSCC (mCSCC) or locally advanced CSCC (laCSCC) not amenable to curative surgery or curative radiation (collectively, ‘advanced CSCC’), which has a poor prognosis.4–6
Treatment of advanced CSCC, particularly CSCC with a primary site of head and neck, can lead to reduced quality of life (QoL).7–9 Surgery for CSCC can result in considerable morbidity, for example, some patients require orbital exenteration,10 which significantly reduces QoL, including increased anxiety and depression, difficulty driving, phantom pain, and hallucinations.11 12 Radiotherapy is associated with substantial toxicity, including fibrosis, lymphedema, skin necrosis, and functional deficits.13 14 Furthermore, pain is a common symptom associated with detriments to QoL, especially among those with CSCC for which curative surgery is not an option.15
Cemiplimab is a high-affinity, highly potent, human, IgG4 monoclonal antibody to programmed cell death (PD)-1.16 In primary analyses of the phase 2 data in patients receiving 3 mg/kg every 2 weeks (Q2W) with mCSCC (group 1, n=59) or laCSCC (group 2, n=78), cemiplimab demonstrated substantial antitumor activity, emerging evidence of durable response, and an acceptable safety profile.17 18 Furthermore, the primary analysis of patients with mCSCC receiving cemiplimab 350 mg every 3 weeks (Q3W) (group 3, n=56) and 11-month follow-up data of group 1 showed similar activity.19
Cemiplimab (cemiplimab-rwlc in the USA) is approved for treatment of patients with advanced CSCC in the USA and Europe, and is approved or under review by other health authorities.20–22 Additionally, cemiplimab is recommended for treatment of patients with laCSCC and mCSCC by the European Association of Dermato-Oncology, European Organisation of Research and Treatment of Cancer (EORTC), and National Comprehensive Cancer Network.3 23 Cemiplimab-rwlc is also indicated for patients with advanced basal cell carcinoma post hedgehog inhibitors (HHIs) or for whom HHIs are not appropriate.20
This manuscript provides additional pooled data from these three groups, including updated duration of response (DOR) and complete response (CR) rates, and describes for the first time the impact of cemiplimab on durability of pain control and QoL. This aggregation of combined long-term data from the original study groups represents the largest (n=193) experience with PD-1 blockade in advanced CSCC to date, with an overall follow-up of up to 36.1 (median 15.7) months.
Methods
Study design
The phase 2 study is an open-label, non-randomized, multicenter, international study of patients with advanced CSCC treated with cemiplimab. The study design has previously been published.17 19
Patients
Briefly, eligible patients were aged ≥18 years with histologically confirmed diagnosis of metastatic or unresectable laCSCC, an Eastern Cooperative Oncology Group performance status score of 0/1, adequate organ function, and at least one measurable lesion.
Treatment
Patients with mCSCC (group 1, n=59) or laCSCC (group 2, n=78) received cemiplimab 3 mg/kg Q2W for 96 weeks, and patients with mCSCC received cemiplimab 350 mg Q3W for 54 weeks (group 3, n=56), with the option to extend treatment to 96 weeks.
Assessments
The primary efficacy endpoint was objective response rate (ORR) per independent central review (ICR). Secondary endpoints included ORR per investigator review (INV), DOR and progression-free survival (PFS) per ICR and INV, overall survival (OS), CR rate per ICR, safety and tolerability, and QoL.
Durable disease control rate (DCR), defined as the proportion of patients with response or stable disease ≥105 days, was examined. An exploratory clinical activity analysis by prior systemic therapy was performed.
The EORTC Quality of Life Questionnaire Core 30 (QLQ-C30)24 was used to evaluate the impact of cemiplimab on symptoms and functioning. The QLQ-C30 includes Global Health Status (GHS)/QoL, functioning domains (physical, emotional, social, role, and cognitive), and symptoms (pain, fatigue, nausea/vomiting, constipation, diarrhea, insomnia, dyspnea, and appetite loss). For GHS/QoL, this scale includes two questions (‘how would you rate your overall health?’ and ‘how would you rate your overall QoL?’). Participants respond on a 4-point scale from ‘not at all’ to ‘very much’ for impact of each scale over the past week, with raw scores on all scales linearly converted to a 0–100 scale (higher scores reflect higher levels of functioning and higher levels of symptom burden). The questionnaire was administered on day 1 of each treatment cycle (treatment cycle defined as 8 weeks for groups 1 and 2 and 9 weeks for group 3). We analyzed longitudinal effects of cemiplimab on GHS/QoL, functioning status, and symptoms, including pain. Assessments per cycle or time were similar and thus are reported in cycles. Analyses of time to first clinically meaningful improvement for pain and time to first clinically meaningful deterioration for pain were also performed.
Safety assessments included treatment-emergent adverse events (TEAEs), laboratory tests, vital signs, and physical examinations. The severity of TEAEs was graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events (V.4.03).
Statistical analysis
Primary results for each group have previously been published,17–19 and clinical activity analyses presented here are intended to describe longer term outcomes. There is formal hypothesis testing for the October 2019 data cut (approximately 1-year additional follow-up). For groups 1 and 3, the null hypothesis was ORR of 15% and the alternative hypothesis was ORR not equal to 15%. For group 2, the null hypothesis was ORR of 25% and the alternative hypothesis was ORR not equal to 25%.
Clinical activity analyses were performed per intention-to-treat. Safety analyses were performed for all patients who received at least one dose of cemiplimab at data cut-off. QoL analyses were performed for patients included in the full analysis set with baseline and at least one post-baseline score for any QLQ-C30 functioning or symptom scale/item. Descriptive statistics were used to summarize QoL scores over time. Mixed-effects repeated measures models (SAS V.9.4) were used to estimate mean treatment effect (change from baseline while accounting for missing data) for all QLQ-C30 scales and items. The models used an AR(1) covariance structure. Covariates controlled in the model included dose group and baseline pain score. The study visit was considered as a random effect. Change of ≥10 points from baseline was considered clinically meaningful.25 Using this criterion, the number of patients experiencing a clinically meaningful change at cycle 6 and cycle 12 was evaluated, and time to the first clinically meaningful change was assessed. Data up to October 2019 were included in this analysis. Follow-up for patients in groups 1–3 was ongoing following the October 2019 data cut.
Results
Patients
Overall, 193 patients with advanced CSCC were eligible for inclusion in this analysis (online supplemental figure 1). Patient characteristics are provided in table 1 (N=193). Most patients were men (n=161, 83.4%), with median age of 72.0 (range: 38–96) years, and had a primary cancer site of head and neck (n=131, 67.9%). Median duration of follow-up was 15.7 months (range: 0.6–36.1) among all patients; with 18.5 months (range: 1.1–36.1) for group 1, 15.5 months (range: 0.8–35.6) for group 2, and 17.3 months (range: 0.6–26.3) for group 3. Median duration of exposure was 51.1 weeks (range: 2.0–109.3). Median number of doses was 18 (range: 1–48).
Supplemental material
Baseline characteristics
Clinical activity in the overall patient group
Overall, 89 of 193 patients had a response to therapy, for an ORR of 46.1% (95% CI: 38.9% to 53.4%); overall, 16.1% of patients achieved CR (table 2). ORR per ICR was 50.8% (95% CI: 37.5% to 64.1%) for group 1, 44.9% (95% CI: 33.6% to 56.6%) for group 2, and 42.9% (95% CI: 29.7% to 56.8%) for group 3. Per ICR, ORR was 48.4% in patients who had not received prior anticancer systemic therapy (n=128) and 41.5% among those who had (n=65). CR rates for this analysis were 20.3%, 12.8%, and 16.1% for groups 1, 2, and 3, respectively. CR rates over time (compared with primary analyses) are presented in figure 1. Among 31 complete responders, median time to CR was 11.2 months (IQR: 7.4–14.8). ORR per INV was 54.4% (95% CI: 47.1% to 61.6%) for all patients; 50.8% (95% CI: 37.5% to 64.1%) for group 1, 56.4% (95% CI: 44.7% to 67.6%) for group 2, and 55.4% (95% CI: 41.5% to 68.7%) for group 3.
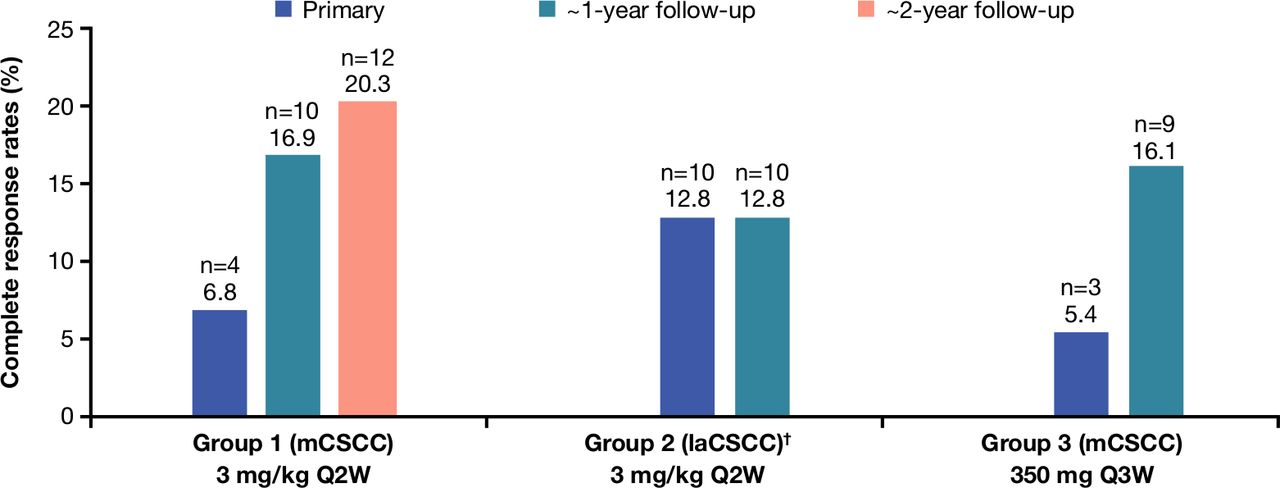
Complete response rates per independent central review. †At the time of the group 1 primary analysis, a prespecified group 2 interim analysis was performed. Among the 23 patients with laCSCC included in this prespecified interim analysis, there were no complete responses. laCSCC, locally advanced cutaneous squamous cell carcinoma; mCSCC, metastatic cutaneous squamous cell carcinoma; Q2W, every 2 weeks; Q3W, every 3 weeks.
Tumor response to cemiplimab per independent central review
Patients had deepening responses over time as evidenced by increasing CR rates compared with primary analyses17–19; the CR rate for group 1 increased from 6.8% in the primary analysis to 16.9% in the first follow-up analysis and to 20.3% at this subsequent follow-up analysis. For group 2, there were no CRs at the interim analysis, but the CR rate was 12.8% at the primary analysis and was unchanged at this follow-up analysis. For group 3, the CR rate increased from 5.4% at the primary analysis to 16.1% at this follow-up analysis. The median time to CR was 11.2 months.
DCR, durable DCR, median DOR, and Kaplan-Meier 12-month estimate of DOR per ICR are provided in table 2. With 1-year additional follow-up, median DOR was not reached (observed DOR range: 1.9–34.3 months). In responding patients, the estimated proportion of patients with ongoing response at 12 months was 87.8% (95% CI: 78.5% to 93.3%) (figure 2A). Estimated median PFS was 18.4 months (95% CI: 10.3 to 24.3) for all patients. The Kaplan-Meier estimated progression-free probability at 24 months was 44.2% (95% CI: 36.1% to 52.1%) (figure 2B). The Kaplan-Meier estimated probability of OS at 24 months was 73.3% (95% CI: 66.1% to 79.2%) (figure 2C). Median OS has not been reached. Percentage change in target lesions from baseline is shown in online supplemental figure 2, and swimmer plots are provided in online supplemental figure 3.

Kaplan-Meier curves for (A) DOR per ICR, (B) PFS per ICR and (C) OS. DOR, duration of response; ICR, independent central review; laCSCC, locally advanced cutaneous squamous cell carcinoma; mCSCC, metastatic cutaneous squamous cell carcinoma; OS, overall survival; PFS, progression-free survival; Q2W, every 2 weeks; Q3W, every 3 weeks.
Quality of life
Patient population and baseline QoL scores
Baseline scores for QLQ-C30 indicated generally moderate-to-high levels of functioning and moderate-to-low symptom burden (online supplemental table 1). The number of patients who completed QoL assessments at each cycle is provided in figure 3 and online supplemental table 1.

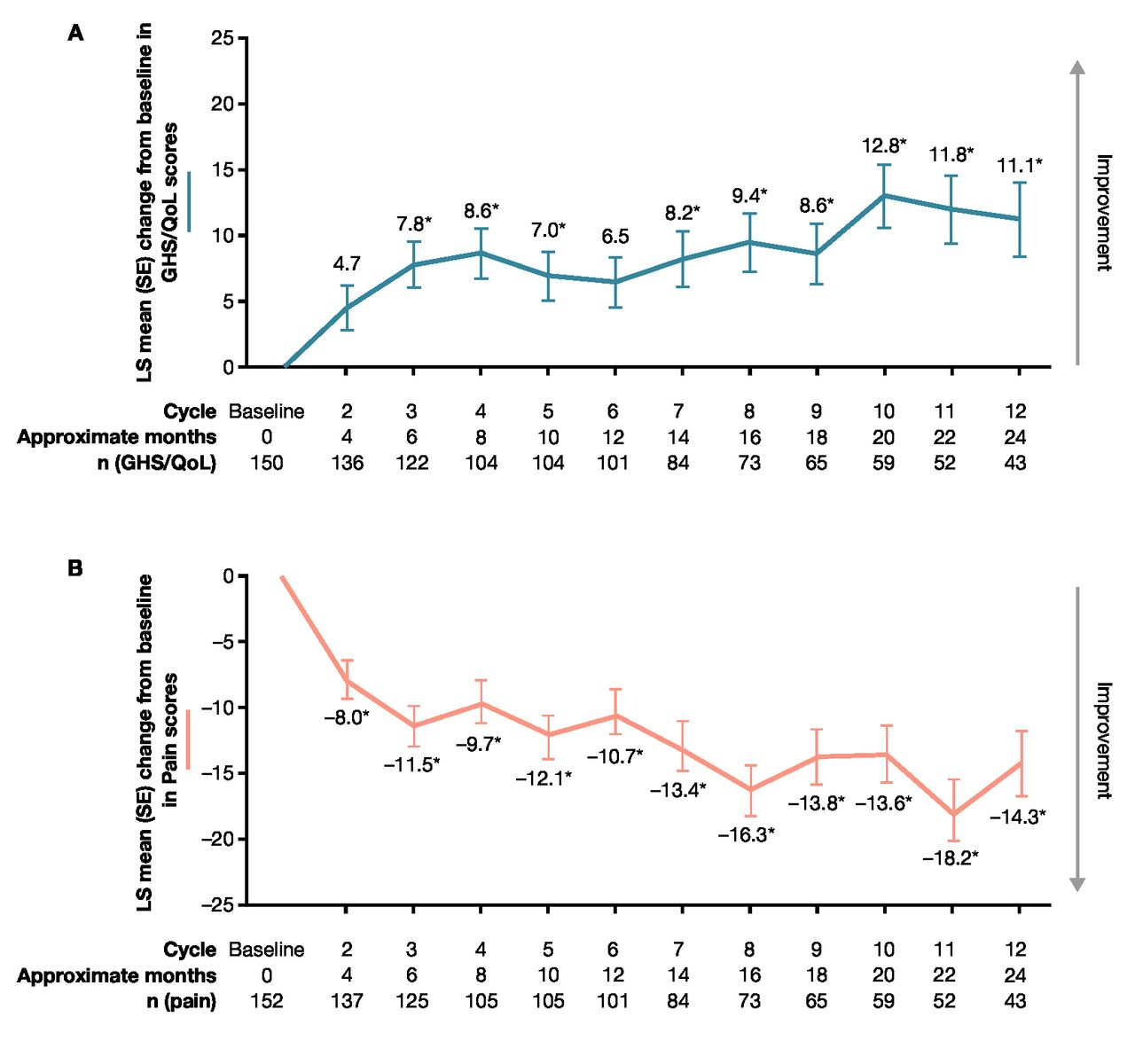
Change from baseline in (A) Global Health Status/quality of life (GHS/QoL) and (B) pain scores. *p<0.0001. An increase of ≥10 points from baseline is considered a clinically meaningful improvement, while a decrease of ≥10 points from baseline is considered a clinically meaningful deterioration. Data are shown for day 1 of each cycle. The questionnaire was administered on day 1 of each cemiplimab treatment cycle (treatment cycle defined as 8 weeks for groups 1 and 2 and 9 weeks for group 3). Equivalent months are shown. QoL, quality of life; LS, least squares.
Longitudinal QoL analysis
For GHS/QoL, improvements were observed from cycle 2, with statistically significant improvement from baseline observed at cycle 3 (least squares [LS] mean [SE] change 7.8 [1.6]; p<0.0001). Improvements in GHS/QoL had reached the clinically meaningful threshold (≥10-point change) by cycle 12 (LS mean [SE] change 11.1 [2.6]; p<0.0001) (figure 3). Among functioning scales, significant improvements were observed in emotional functioning and social functioning scales at cycle 3 and cycle 12 (online supplemental table 1). Physical functioning, role functioning, and cognitive functioning did not deteriorate and remained stable relative to baseline (online supplemental table 1). Regarding symptoms, significant improvements from baseline were also observed for symptoms of nausea/vomiting, pain, insomnia, appetite loss, and constipation by cycle 3 (online supplemental table 1), and as early as cycle 2 for pain (figure 3). These symptoms had all reached the clinically meaningful threshold (≥10-point change) by cycle 12 (online supplemental table 1). Across all functioning scales and symptom scales, the proportion of patients with clinically meaningful deterioration was generally low at both cycle 6 and cycle 12.
Early onset and durability of pain control
The Kaplan-Meier estimate of median time (95% CI) to first clinically meaningful improvement for pain was 2.1 (2.0 to 3.7) months overall. The Kaplan-Meier estimate of median time (95% CI) to first clinically meaningful deterioration for pain was 14.8 (9.2 to not evaluable [NE]) months overall. LS mean (SE) change from baseline in pain score was –11.5 (1.9) at cycle 3, and –14.3 (3.1) at cycle 12 (figure 3). LS mean change (SE) from baseline in pain score at first tumor response was –15.2 (1.5) in patients with objective response and –3.86 (2.1) in patients without objective response. The difference in LS mean change (95% CI) from baseline for pain score for responders versus non-responders was –11.3 (–16.3 to –6.3; p<0.0001). Among patients with objective response, the Kaplan-Meier estimate of median time (95% CI) to first clinically meaningful improvement for pain was 2.1 (1.9 to 2.1) months and the Kaplan-Meier estimate of median time (95% CI) to first clinically meaningful worsening for pain was 20.6 (9.2 to NE) months.
Responder analysis for QoL assessment
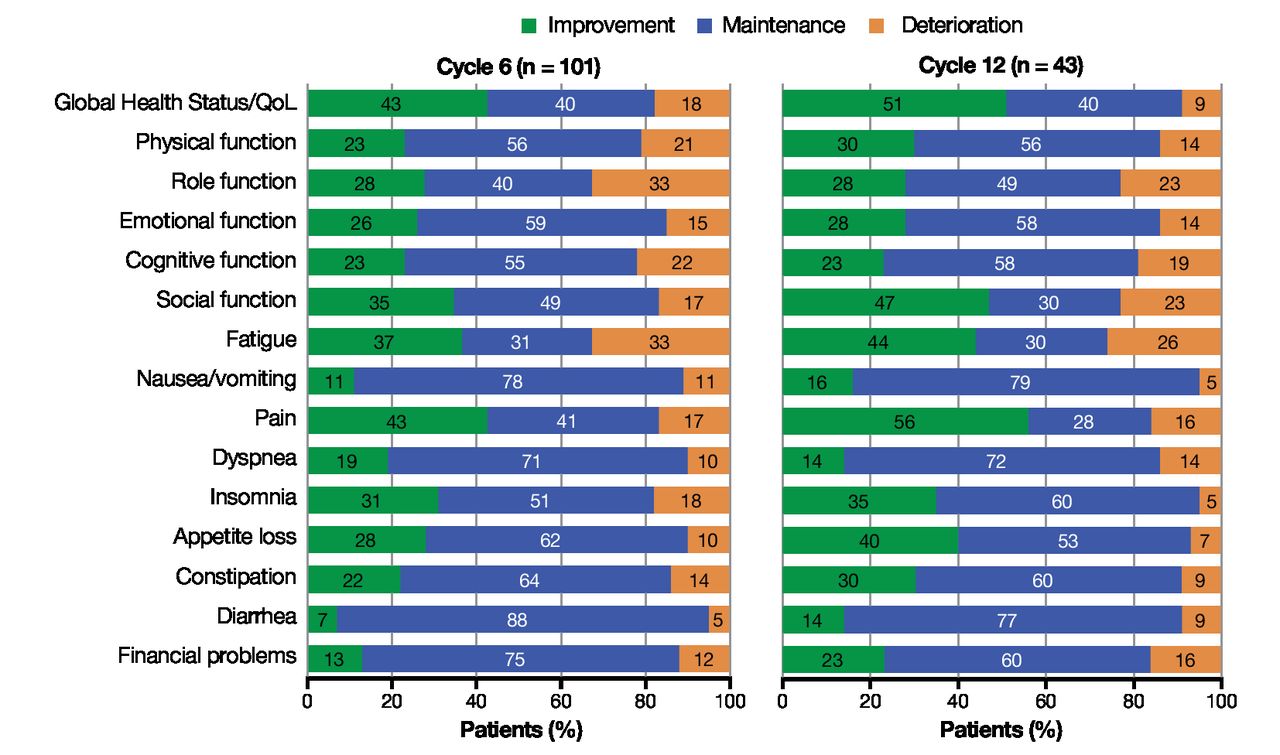
A substantial fraction of patients benefitted from treatment. Among all patients reporting clinically meaningful change (≥10-point change) by cycle 6, most patients experienced clinically meaningful improvements or stability in GHS/QoL (83%), functioning scales (68%–85%), and symptoms (68%–95%) (figure 4). Overall, 91% of responders experienced clinically meaningful improvement or stability in GHS/QoL scores at cycle 12, and most patients showed sustained improvement and stabilization across all functioning scales (77%–86%) and symptoms (74%–95%) by cycle 12 (figure 4). The proportions of patients with clinically meaningful deterioration in functioning scales were generally low at both evaluated time points (figure 4).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Proportion of responding patients reporting clinically meaningful change (≥10-point change) at cycle 6 and cycle 12. The questionnaire was administered on day 1 of each treatment cycle (treatment cycle defined as 8 weeks for groups 1 and 2 and 9 weeks for group 3). QoL, quality of life.
Safety
In total, 192 (99.5%) patients experienced at least one TEAE of any grade regardless of attribution (online supplemental table 2). TEAEs leading to discontinuation were low (any grade: n=19, 9.8%; grade ≥3: n=14, 7.3%). The most common TEAEs (all grades) were fatigue (n=67, 34.7%), diarrhea (n=53, 27.5%), and nausea/vomiting (n=46, 23.8%). Overall, 94 (48.7%) patients experienced at least one grade ≥3 TEAE regardless of attribution. The most common grade ≥3 TEAEs were hypertension (n=9, 4.7%), anemia, and cellulitis (each n=8, 4.1%). No new TEAEs or treatment-related adverse events (TRAEs) resulting in death were reported for any group in this longer term follow-up, compared with previous reports.17–19 Grade ≥3 TRAEs were reported in 33 (17.1%) patients; the most common were pneumonitis (n=5, 2.6%), autoimmune hepatitis (n=3, 1.6%), anemia, colitis, and diarrhea (all n=2, 1.0%) (online supplemental table 3). In total, 57 patients (29.5%) experienced at least one sponsor-identified immune-related adverse event (irAE) of any grade, and 18 patients (9.3%) experienced at least one grade ≥3 irAE (online supplemental table 4). The most common grade ≥3 irAEs were pneumonitis (n=5, 2.6%), autoimmune hepatitis (n=2, 1.0%), and diarrhea (n=2, 1.0%).
Discussion
The pooled analysis presented here demonstrates durability of responses to cemiplimab in CSCC, increasing CR rates over time, and confirms the substantial antitumor activity of cemiplimab in patients with advanced CSCC established in previously published primary analyses.17–19 An estimated 87.8% of responders had not progressed at 12 months. With median DOR not reached after an additional year of follow-up, the present analysis reinforces the activity of cemiplimab in a patient population that previously had no widely accepted standard of care. Furthermore, DOR and OS are longer than has been previously described with other agents, prior to the approval of cemiplimab.26 Additionally, this analysis demonstrates that cemiplimab treatment is associated with improvement in GHS/QoL and pain scores.
Compared with primary analyses,17–19 patients had deepening responses over time, as evidenced by increasing CR rates. The median time to CR was 11.2 months, raising the possibility that prolonged cemiplimab treatment may be necessary for continued clinical activity in many patients with advanced CSCC.
In the KEYNOTE-629 Study (NCT03284424), patients (n=105; follow-up 9.5 months) with recurrent/mCSCC were treated with pembrolizumab 200 mg Q3W.27 ORR per ICR was 34.3% with an estimated DOR at 12 months of 65.6%. The overall CR rate during the follow-up period was 3.8%. It should be noted 86.7% of patients included in KEYNOTE-629 had previously received ≥1 line of systemic therapy, and this group achieved an ORR of 31.9%.27 Results for the laCSCC cohort in KEYNOTE-629 have not been published. Additionally, preliminary results from the CARSKIN trial of pembrolizumab (NCT02883556) reported an ORR of 38.5%, a CR rate of 5.1%, and a median DOR of 12.5 months in patients with no prior systemic therapy.28 In the present study, acknowledging that this may be in part due to longer follow-up, we report a higher ORR per ICR of 46.1%, an estimated DOR at 12 months of 87.8%, and a CR rate of 16.1%. However, given differences in trial designs, caution is required when comparing response rates across studies. There are no head-to-head trials assessing the comparative clinical activity of cemiplimab and pembrolizumab.
Pain is a key issue for patients with advanced CSCC, especially those with unresectable disease.15 The mean (SD) baseline pain score of patients with advanced CSCC receiving cemiplimab of 29.8 (30.4) was significantly worse than reported by patients with advanced head and neck cancer (24.9 [26.3]; p<0.05; n=1722) and the general population (20.9 [27.6]; p<0.0001; n=7802).29 Here, the clinical responses observed correlated well with pain improvement, which positively impacted patient QoL. Cemiplimab resulted in pain reduction by cycle 2, with clinically meaningful reduction (≥10 points) from cycle 3, maintained through to cycle 12. The Kaplan-Meier estimate of median time (95% CI) to first clinically meaningful improvement for pain was 2.1 months overall, and this was sustained to 14.8 months, demonstrating durability of pain control with cemiplimab. GHS/QoL improvement was observed as early as cycle 3, with clinically meaningful improvement seen by cycle 12. By cycle 6, most patients experienced clinically meaningful improvement or stability in GHS/QoL and functioning status, while maintaining low symptom burden.
Eighteen patients (9.3%) experienced at least one grade ≥3 irAE, with a low treatment discontinuation rate of 9.8%. As previously reported,19 the safety profile for cemiplimab continues to be consistent with that previously reported for other anti-PD-1/PD-ligand 1 agents.30 There were no new safety signals or types of toxicities compared with previous analyses.
This analysis confirms the substantial clinical activity of cemiplimab, including new findings of improved CR rates over time compared with primary analyses, and an impressive and increasing DOR based on Kaplan-Meier estimate at key landmarks in patients with advanced CSCC. Additionally, cemiplimab treatment resulted in a clinically meaningful reduction in pain as early as cycle 2, maintained to cycle 12. Further, clinical response to cemiplimab was associated with a reduction in pain. Most patients experienced clinically meaningful improvements or maintenance in GHS/QoL, functioning, and symptoms. These results provide further support for cemiplimab as an agent with favorable data to support its use for the treatment of advanced CSCC.
Supplemental material
Supplemental material
Supplemental material
Supplemental material
Supplemental material
Data availability statement
Data are available upon reasonable request. Qualified researchers may request access to study documents (including the clinical study report, study protocol with any amendments, blank case report form, statistical analysis plan) that support the methods and findings reported in this manuscript. Individual anonymized participant data will be considered for sharing once the indication has been approved by a regulatory body, if there is legal authority to share the data and there is not a reasonable likelihood of participant re-identification. Submit requests to https://vivli.org/.
Ethics statements
Ethics approval
This study was conducted in accordance with the principles of the Declaration of Helsinki and the International Conference on Harmonization Good Clinical Practice guidelines. The study protocol and all amendments were approved by the institutional review board/ethics committee at each participating study site. All patients provided written informed consent.
Acknowledgments
The authors would like to thank the patients, their families, all other study investigators, and all investigational site members involved in this study. Medical writing support, which was provided by Kate Carolan, of Prime (Knutsford, UK) according to Good Publication Practice guidelines, was funded by Regeneron Pharmaceuticals and Sanofi. Responsibility for all opinions, conclusions, and data interpretation lies with the authors.
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Twitter @squamous cell
Presented at Data from this manuscript have previously been presented at ASCO 2020: Rischin D et al. J Clin Oncol. 38, no. 15_suppl (May 20, 2020) 10018-10018 and Migden MR et al. J Clin Oncol. 38, no. 15_suppl (May 20, 2020) 10033-10033.
Contributors DR, CDS, AG, AH, ES, JB, S-YY, SL, ZC, EO, C-IC, VM, MS, IL, MGF, and MRM conceived and designed the study. DR, NIK, CDS, AG, ALSC, KDL, AML, LH-A, BGMH, DS, AH, and MRM recruited patients and collected the data. S-YY and SL did the data analysis. EO worked on safety analysis strategy and contributed to safety data analysis. All authors contributed to data interpretation, as well as critical review, revision, and approval of the report.
Funding This work was funded by Regeneron Pharmaceuticals and Sanofi.
Competing interests DR—institutional research grant and funding from Regeneron Pharmaceuticals, Roche, Merck Sharp & Dohme, Bristol-Myers Squibb, GlaxoSmithKline, and Sanofi; uncompensated scientific committee and advisory board from Merck Sharp & Dohme, Regeneron Pharmaceuticals, Sanofi, GlaxoSmithKline, and Bristol-Myers Squibb; and travel and accommodation from Merck Sharp & Dohme and GlaxoSmithKline. NIK—grants from Regeneron Pharmaceuticals; grants and advisory board fees from Bristol-Myers Squibb and HUYA Bioscience International; advisory board fees from EMD Serono, Regeneron Pharmaceuticals, Genentech, AstraZeneca (data safety monitoring committee), Incyte (data safety monitoring committee), Iovance, Merck, ARRAY Biopharma, Jounce Therapeutics, and Immunocore; grants from Merck, Novartis, GlaxoSmithKline, Celgene, and Amgen; honorarium from Sanofi; and common stock ownership of Bellicum Pharmaceuticals, Mazor Robotics, Amarin, and Transenetrix. CDS—steering committee member for Castle Biosciences; steering committee member and consultant for Regeneron Pharmaceuticals; consultant for Sanofi; received research funding from Castle Biosciences, Regeneron Pharmaceuticals, Novartis, Genentech, and Merck; and is a chair for the National Comprehensive Cancer Network. AG—personal fees and non-financial support (advisory board and travel support) from Bristol-Myers Squibb and Sun Pharma; personal fees (advisory board) from Merck KGaA, Eisai, and Pfizer; non-financial (travel) support from Astellas; and clinical trial unit support from PPD Australia. ALSC—consulting and advisory roles at Regeneron Pharmaceuticals and Merck; and research funding from Regeneron Pharmaceuticals, Novartis, Galderma, Jounce Therapeutics, and Merck. KDL—grants and personal fees from Regeneron Pharmaceuticals during the conduct of the study. AML—uncompensated advisory board from Merck Sharp & Dohme and Bristol-Myers Squibb with travel and accommodation expenses; and uncompensated consultancy for Eisai. LH-A—performed consulting and advisory roles at Massive Bio; speakers’ bureau roles at Sanofi and Regeneron Pharmaceuticals; received travel, accommodations, and expenses from Regeneron Pharmaceuticals, Sanofi, and Bristol-Myers Squibb; and research funding from Bristol-Myers Squibb, Regeneron Pharmaceuticals, Immunocore, Merck Sharp & Dohme, Polynoma, Corvus Pharmaceuticals, Roche, Merck Serono, Amgen, MedImmune, and Takeda. BGMH—consulting or advisory roles at Merck Sharp & Dohme, Bristol Myers Squibb, AstraZeneca, Pfizer, Roche, Eisai, and Merck; and institutional research funding from Amgen. DS—institutional patients’ fees from Regeneron Pharmaceuticals; advisory board honorarium fees from Amgen and Leo Pharma; speaker fee from Boehringer Ingelheim; advisory board, speaker honorarium, and patients’ fees from Roche, Novartis, Bristol-Myers Squibb, and Merck-EMD; advisory board and speaker honorarium fees from Incyte and Pierre Fabre; advisory board honorarium and patients’ fees from MSD; steering committee honorarium fees from 4SC; advisory board fees from AstraZeneca, Pfizer, and Array; and advisory board and patients’ fees from Philogen. AH—institutional grants, speaker’s honoraria, and consultancy fees from Amgen, Bristol-Myers Squibb, Merck Sharp & Dohme/Merck, Pierre Fabre, Provectus, Roche, and Novartis; institutional grants and consultancy fees from Merck Serono, Philogen, and Regeneron Pharmaceuticals; and consultancy fees from OncoSec. ES, JB, S-YY, SL, ZC, EO, C-IC, VM, IL, MGF—employees of and shareholders in Regeneron Pharmaceuticals. MS—employee and shareholder in Sanofi. MRM—honoraria and travel expenses from Regeneron Pharmaceuticals, Sanofi, Novartis, Genentech, Eli Lilly, and Sun Pharma; and institutional research funding from Regeneron Pharmaceuticals, Novartis, Genentech, and Eli Lilly.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.