Article Text

Abstract
Background Defects in replication repair-associated DNA polymerases often manifest an ultra-high tumor mutational burden (TMB), which is associated with higher probabilities of response to immunotherapies. The functional and clinical implications of different polymerase variants remain unclear.
Methods Targeted next-generation sequencing using a 425-cancer gene panel, which covers all exonic regions of three polymerase genes (POLE, POLD1, and POLH), was conducted in a cohort of 12,266 patients across 16 different tumor types from January 2017 to January 2019. Prognostication of POL variant-positive patients was performed using a cohort of 4679 patients from the The Cancer Genome Atlas (TCGA) datasets.
Results The overall prevalence of somatic and germline polymerase variants was 4.2% (95% CI 3.8% to 4.5%) and 0.7% (95% CI 0.5% to 0.8%), respectively, with highest frequencies in endometrial, urinary, prostate, and colorectal cancers (CRCs). While most germline polymerase variants showed no clear functional consequences, we identified a candidate p.T466A affecting the exonuclease domain of POLE, which might be underlying the early onset in a case with childhood CRC. Low frequencies of known hot-spot somatic mutations in POLE were detected and were associated with younger age, the male sex, and microsatellite stability. In both the panel and TCGA cohorts, POLE drivers exhibited high frequencies of alterations in genes in the DNA damage and repair (DDR) pathways, including BRCA2, ATM, MSH6, and ATR. Variants of unknown significance (VUS) of different polymerase domains showed variable penetrance with those in the exonuclease domain of POLE and POLD1 displaying high TMB. VUS in POL genes exhibited an additive effect as carriers of multiple VUS had exponentially increased TMB and prolonged overall survival. Similar to cases with driver mutations, the TMB-high POL VUS samples showed DDR pathway involvement and polymerase hypermutation signatures. Combinatorial analysis of POL and DDR pathway status further supported the potential additive effects of POL VUS and DDR pathway genes and revealed distinct prognostic subclasses that were independent of cancer type and TMB.
Conclusions Our results demonstrate the pathogenicity and additive prognostic value of POL VUS and DDR pathway gene alterations and suggest that genetic testing may be warranted in patients with diverse solid tumors.
- biomarkers
- tumor
- genetic markers
- genome instability
- Immunotherapy
- translational medical research
Data availability statement
Data are available upon reasonable request. The raw sequencing data generated and/or analyzed during the current study are not publicly available due to privacy concerns and restrictions by the regulation of the Human Genetic Resources Administration of China as they contain germline information, but all other datasets analysed are available from the corresponding authors on reasonable request or on published The Cancer Genome Atlas databases.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Background
Replication and DNA repair mechanisms are intricately coordinated processes that are essential for genome integrity and proper functioning of the cell. Defective DNA replication and damage repair in cancer cells contribute to the accumulation of genomic alterations that drives tumor initiation and progression.1 2 At the same time, the reliance of the tumors on the residual repair capacity, as well as the increased neoantigen load, makes them particularly vulnerable to anticancer therapies, including chemotherapy, DNA repair inhibitors, and immunotherapy.3–5
While most tumors carry a relatively small number of somatic alterations, those that harbor mutations in DNA mismatch repair or DNA polymerases have been associated with an ‘ultra-mutator’ phenotype.6 The replication–repair-associated polymerases Pol ε, Pol δ and Pol η are key mediators of DNA replication and repair. Both Pol ε and Pol δ, the catalytic subunits of which are encoded by POLE and POLD1, respectively, carry a unique proofreading ability through their exonuclease domains to ensure DNA replicative fidelity.7 8 Both enzymes also participate in various DNA repair pathways, including nucleotide excision repair, base excision repair, and double-strand break repair.9 As a consequence, germline and somatic mutations in the exonuclease domain of POLE and POLD1 are associated with the initiation of multiple cancer types, including colorectal and endometrial cancers.10–14 Pol η belongs to a distinct class of error-prone DNA polymerases that are involved in translesion synthesis.15 It has been proposed that the error-prone DNA polymerases would replace the stalled replicative DNA polymerase at sites of DNA damage to efficiently bypass such lesions.16 17 Deficiency in POLH has been reported in genetic disorders, such as xeroderma pigmentosum18 and several cancers.19 20
While alterations in the polymerase genes might serve as a target for cancer therapies and a biomarker for patient outcome, it is important to extensively characterize the clinical and genomic correlates of these mutants and distinguish mere passenger alterations from true driving events. Using the massively parallel nature of next-generation sequencing (NGS), we analyzed data from germline and somatic POLE, POLD1, and POLH carriers (hereafter, POL+) in a set of 12,266 patients across 16 different cancer types using a targeted panel encompassing 425 cancer-relevant genes. A large cohort of POL+ patients from the The Cancer Genome Atlas (TCGA) database was used to validate some of our key findings.
Methods
Study cohorts
This study involved two independent cohorts of patients with diverse cancer types. One cohort (NGS cohort) of 12,266 patients from Zhejiang Cancer Hospital, National Cancer Center, Traditional Chinese Medical Hospital of Huzhou, The First Affiliated Hospital of Zhejiang University, and Sir Run Run Hospital (Nanjing Medical University) underwent targeted NGS using a 425-cancer gene panel (GeneseeqPrime) in a Clinical Laboratory Improvement Amendments (CLIA)-certified and College of American Pathologists (CAP)-accredited clinical testing laboratory (Nanjing Geneseeq Technology, Nanjing, China) from January 2017 to January 2019. An independent publicly available validation cohort (TCGA cohort) containing genomic profiling data by whole-exome sequencing of 4679 patients with cancer was selected from the TCGA database for mutational and survival analyses.
Targeted NGS
Targeted NGS was performed as previously described.21 In brief, genomic DNAs from tissue and whole blood control samples, or circulating tumor DNA (ctDNA) extracted from plasma samples were extracted and quantified by Qubit V.3.0 using the dsDNA HS Assay Kit (Thermo Fisher Scientific, USA). Library preparations were performed with KAPA Hyper Prep Kit (KAPA Biosystems, USA). Customized xGen lockdown probes (Integrated DNA Technologies) targeting 425 cancer-relevant genes were used for hybridization capture enrichment. Enriched libraries were on-beads PCR amplified, purified, and sequenced on the Illumina HiSeq4000 platform to a mean coverage depth of 948×, 3960×, and 267× for tumor, ctDNA, and blood control samples, respectively. No significant differences in tumor mutational burden (TMB) were observed between matched tissue and ctDNA samples (online supplemental figure S1A and B). In addition, there was a significant overlap in the detection of POL variants comparing the two sample types (online supplemental figure 1C). As tissue samples showed a trend towards higher TMB and enabled the detection of slightly more POL variants compared with ctDNA samples, tissue samples were included in the final analysis for patients with both tissue and plasma samples available. Of the 526 cases detected with POL variants, 403 were tissue samples and the remaining 123 were plasma samples.
Supplemental material
Mutation calling for targeted NGS
Mutation calling for targeted NGS data was performed as previously described.21 In brief, paired-end reads were aligned to the reference human genome (hg19) using the Burrows-Wheeler Aligner,22 followed by PCR deduplication (Picard, http://broadinstitute.github.io/picard/) and local realignment around indels (GATK3).23 Somatic single-nucleotide variants (SNVs) and insertions/deletions were called using Mutect24 and Scalpel,25 respectively. All variants were further filtered: (1) <5 variant reads and <1% allele frequency; (2) >1% population frequency in the 1000G, ExAC, or gnomAD databases; and (3) through an internal database of recurrent sequencing errors.
TMB calculation
TMB of the GeneseeqPrime panel cohort was estimated by summing all SNVs and indels in the coding region of targeted genes, including synonymous alterations and excluding known driver mutations as previously described.26 TMB of the TCGA cohort was estimated by summing all SNVs and indels in the coding regions. The thresholds of high and ultra-high TMBs were determined based on a segmented linear regression analysis using the R package Segmented.27 For panel-based results, the cut-offs for high and ultra-high TMBs were 16 and 105 mutations (mut)/Mb, respectively. For the TCGA cohort, the cut-offs for high and ultra-high TMBs were 71 and 189 mutations, respectively.
Microsatellite instability (MSI) analysis
The stability of a total of 52 microsatellite sites covered by the GeneseeqPrime panel with a minimum of 15 bp repeats, including the classic MSI sites BAT-25, BAT-26, NR-21, NR-24, and MONO-27, was estimated and compiled into an overall MSI score. A site is considered qualified if it is covered by at least 101× depth of coverage. A sample is identified as MSI if more than 40% of the qualified sites with at least 100× coverage displayed instability. Analysis involving the comparisons of MSI, POL genes, and TMB status was limited to samples where MSI and TMB status were both determined.
Statistical analysis
Comparisons of proportion between groups were done using Fisher’s exact test. Trend analysis comparing TMB at increasing numbers of POL variants was performed using the Jonckheere-Terpstra test. For survival analyses, Kaplan-Meier curves were compared using the log-rank test, and HRs were calculated by Cox proportional hazards. Multivariate survival analysis was performed using the Cox regression model. A two-sided p value of less than 0.05 was considered significant for all tests unless indicated otherwise. All statistical analyses were done in R V.3.5.2.
Results
Patient characteristics and prevalence of POL family variants
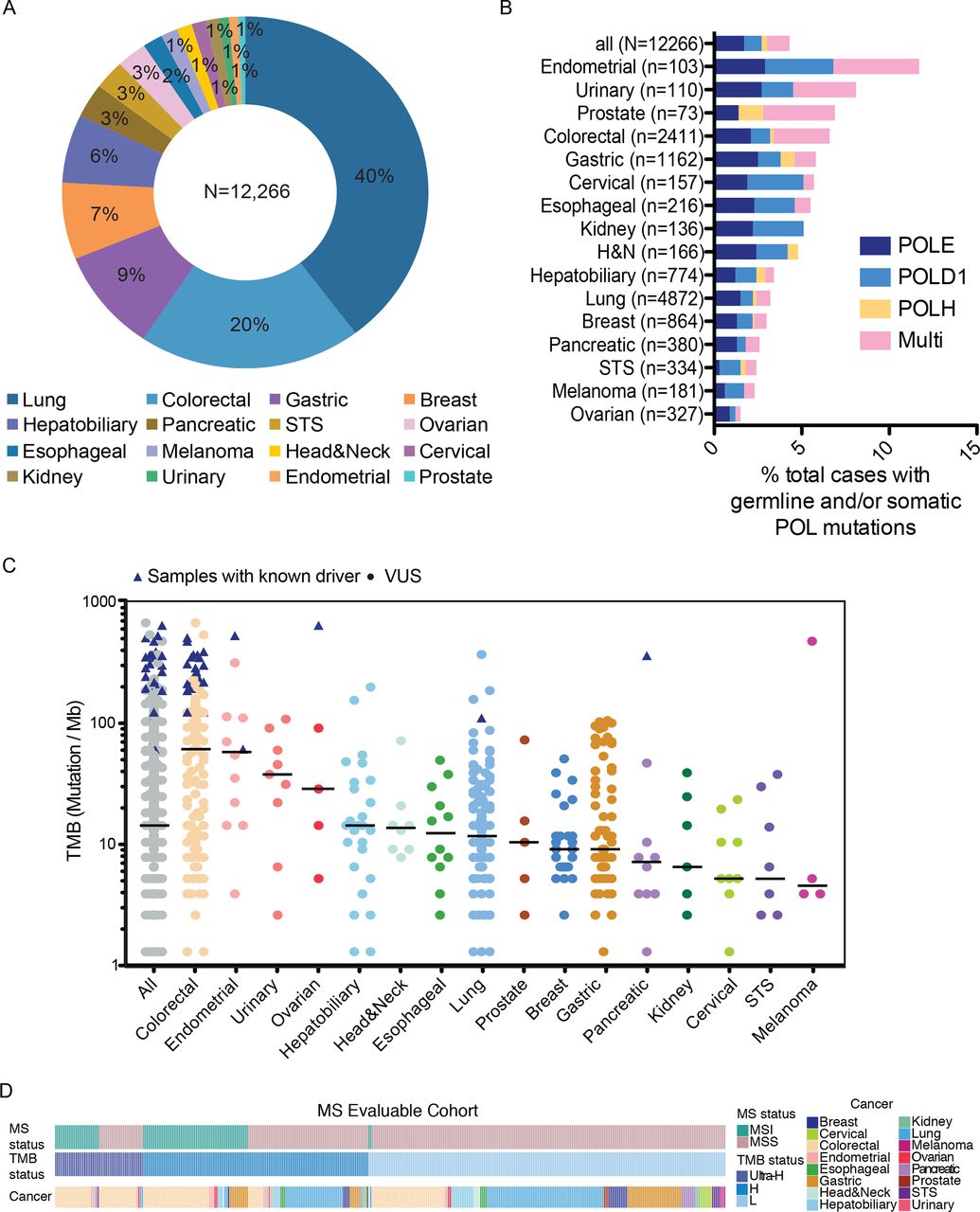
We analyzed sequencing data from a total of 12,266 patients with cancer with diverse solid tumors who underwent targeted NGS using a 425-gene panel (see the Methods section). The distribution of patients across 16 major cancer types is shown in figure 1A. We first analyzed the prevalence of somatic and germline variants in POLE, POLD1, and/or POLH, which is summarized in figure 1B and online supplemental table 1. Overall, a total of 526 (4.3%, 95% CI 3.9% to 4.7%) patients were detected with non-synonymous somatic alterations and/or rare germline variants in POLE, POLD1, and/or POLH, with a large portion (31%, 95% CI 27% to 35%) of patients carried more than one POL variant (figure 1B). Of these, 443 patients carried only somatic variants; 32 had only germline variants; and 51 patients with both somatic and germline variants. The frequencies of patients carrying somatic and/or germline POL variants were highest in endometrial cancers (12/103, 11.7%; 95% CI 6.2% to 19.5%), urinary cancers (9/110, 8.2%; 95% CI 3.8% to 15.0%), prostate cancers (5/73, 6.8%; 95% CI 2.3% to 15.3%) and colorectal cancers (CRCs) (160/2411, 6.6%; 95% CI 5.7% to 7.7%), which were also characterized by markedly higher proportions of patients with multiple POL variants than other cancer types. Overall, there appeared to be an enrichment of male patients in the POL variant-positive group compared with the entire cohort (POL+ vs POL−: 64%, 95% CI 60% to 68%, vs 54%, 95% CI 53% to 55%; p<0.001; χ2 test; online supplemental figure S2A,B). By controlling for cancer types, we found that only POL+ lung cancers were more frequently male compared with those with no alterations in the POL genes (74%, 95% CI 66% to 81%, vs 55%, 95% CI 54% to 57%; χ2 test, FDR adjusted p<0.001; online supplemental figure S2C), which we suspect might be related to higher rates of smoking in men.
Supplemental material

Prevalence of POL variants and mutational burden across diverse cancer types. (A) Distribution of cancer types in the entire study cohort. (B) Prevalence of germline and/or somatic POLE/POLD1/POLH variants across different cancer types. (C) TMB in each POL+ samples across different cancer types. TMB of samples with known POLE driver mutations are indicated by triangles. (D) Oncostrips showing microsatellite status, TMB status and cancer types in each individual patient with evaluable microsatellite stability. MS, microsatellite; MSS, microsatellite stable; TMB, tumor mutational burden; VUS, variants of unknown significance; STS, soft tissue sarcoma; H&N, head and neck cancer.
The median tumor mutational burden (mTMB) across all samples was 14.3 mut/Mb, with colorectal (61.3 mut/Mb), endometrial (58.0 mut/Mb), and urinary (37.8 mut/Mb) cancers having the highest mTMB (figure 1C). The thresholds for hypermutation and ultrahypermutation were 16 and 105 Mut/Mb, respectively, and were determined by segmented linear regression analysis (the Methods section). Of the samples from 409 POL variant-positive patients that qualified for MSI assessment, 90 (22.0%, 95% CI 18.1% to 26.3%) were classified as MSI-high, 25 of which (27.8%, 95% CI 18.9% to 38.2%) exhibited ultrahypermutation (figure 1D). In addition, 30.4% (97/319, 95% CI 25.4% to 35.8%) microsatellite stable (MSS) patients also had high (71/97, 73.2%; 95% CI63.2% to 81.7%) or ultra-high (26/97, 26.8%; 95% CI 18.3% to 36.8%) TMB. No significant association between TMB and age was observed (online supplemental figure S3).
Clinical and molecular characterizations of tumors with POL germline variants
Overall, 88 germline variants with unclear function in the polymerase genes (all non-synonymous variants with a<0.3% frequency in the general or the East Asian population based on gnomAD_exome entries, online supplemental table 1) were detected in the entire cohort. The distributions of POL germline variants across the three proteins are shown in online supplemental figure S4. Of these, 61 (69.3%, 95% CI 58.6% to 78.7%) were missense variants and the rest were splicing or truncating variants (online supplemental figure S5A), with a total of 28 variants occurred in the exonuclease domains of POLE or POLD1. There were four patients who carried more than one germline variants in the polymerase genes, whose clinical and mutational details are provided in online supplemental table 2). Of note, only one of these four patients (P325) displayed high TMB (144.74mut/Mb). The patient carried a p.N336S germline variant in the exonuclease domain of POLE, as well as a p.V124A variant in POLD1. While the p.N336S alteration might underlie the increase in mutational burden, the pathogenicity of the variant is unclear considering that another carrier of this particular variant had low TMB (6.52 mut/Mb).
Supplemental material
Notably in three cases, the patients subsequently acquired somatic ‘hot-spot’ mutations (hereafter, driver mutations) in POLE, which had been established to cause ultra-high TMB (online supplemental figure S5B). In one of these cases, the patient was diagnosed with hypermutated endometrial cancer (TMB=530.7 mut/Mb) at 54 years old and was found to carry a germline variant in POLH (p.K540R) and a concurrent p.V411L somatic mutation in POLE. Another case carried a germline variant in POLE (p.R494W) and a concurrent somatic driver mutation (p.A456P). The patient was diagnosed with CRC at the age of 49, with a TMB of 370.3 mut/Mb. In the third case, germline POLE variant (p.T466A) and the p.P436S somatic driver mutation were found in a child, who was diagnosed at 12 years old with hypermutated CRC (TMB=309 mut/Mb). This particular germline variant is located in the exonuclease domain of Pol ε, which is directly involved in DNA binding. All three germline variants, p.K540R in POLH, and p.R494W and p.T466A in POLE, were relatively rare with respective population frequencies of 0.003%, 0.0004%, and 0.001% based on gnomAD_exome database. Pathogenicity prediction algorithms suggested that the latter two variants might have severe functional impact on the protein (Sorting Intolerant From Tolerant [SIFT] score=0 and PolyPhen score=0.99 for both variants). Based on their associations with TMB, age of onset, population frequencies, and functional predictions, we suspected that the p.T466A germline variant in POLE was probably pathogenic.
By contrast, 72% (60/83, 95% CI 61% to 82%) of cases with germline variants displayed relatively low TMB (online supplemental figure S5B). Of the MS-evaluable cohort, no significant difference was observed in the distribution of patients with and without germline POL variants according to their TMB status (Fisher’s exact test p=0.09) or MS status (Fisher’s exact test p=0.12, figure 1D and online supplemental figure S5C and D). Analysis of the MSS subgroup showed that cases with germline variants had higher TMB than those of the POL− cases (median TMB=5.74 mut/Mb vs 7.82 mut/Mb, Wilcoxon rank-sum test p=0.003), but lower TMB than those of cases with only somatic POL variants (7.82 vs 10.43, Wilcoxon rank-sum test, p=0.046; online supplemental figure S5E). No significant differences in TMB were observed between cases with germline and somatic variants across the major cancer types (CRC, p=0.20; gastric cancer, p=0.12; lung cancer, p=0.49; online supplemental figure S5F).
To further dissect the potential pathogenicity of the germline variants, we first compared the impact of different variant types on mutational burden. Missense variants were more likely to have a high mutational burden both overall (median TMB=11.1 vs 3.9 mut/Mb, Wilcoxon rank-sum test, p=0.0002) and when considering only patients with MSS tumors (median TMB=10.4 vs 3.9 mut/Mb, Wilcoxon rank-sum test, p=0.006; online supplemental figure S6). In addition, multiple disease-causing germline mutations have been previously identified,10 28–33 all of which are in the exonuclease domains of POLE (eg, p.L424V, p.N363K, p.Y458F, and p.A456P) and POLD1 (eg, p.S478N and p.P327L). Consequently, we surveyed for additional germline variants in the exonuclease domains of POLE and POLD1. As expected, germline variants in the exonuclease domains of POLE and POLD1 were associated with higher TMB compared with the rest of the germline variants (median TMB=13.0 mut/Mb vs 6.5 mut/Mb, Wilcoxon rank-sum test, p=0.04; online supplemental figure S6B), which was at least in part attributable to an enrichment of MSI in these variants (Fisher’s exact test, p=0.007; online supplemental figure S6C). Following correction using MSS, exonuclease domain variants only showed a trend towards increased TMB compared with others (median TMB=30.0 mut/Mb vs 7.8 mut/Mb, Wilcoxon rank-sum test, p=0.13; online supplemental figure S6B).
Subsequent analysis of the MSS subgroup revealed certain genetic features that correlated with TMB status. TP53 was the most frequently altered (29/48, 60%; 95% CI 45% to 74%) gene in this subgroup and displayed no significant difference between the tumor mutational burden-high (TMB-H) and tumor mutational burden-low (TMB-L) groups (Fisher’s exact test, p=0.52; online supplemental figure S6D). Within the major cancer types, patients with TMB-H CRC were enriched for ERBB4, KDR, MSH6, PDGFR, ARID1A, and PIK3R1 alterations (Fisher’s exact tests followed by multiple testing correction using the FDR adjustment method; online supplemental figure S6D). On the other hand, germline POL+ lung cancers showed increased association between high TMB and FLT1 alterations, and a slight trend towards reduced TMB in cases with oncogenic driver mutations in EGFR, ERBB2, and KRAS (FLT1, p=0.02; oncogenic driver mutations, p=0.28; Fisher’s exact tests). However, none of these genetic features in germline POL+ lung cancers were significant following multiple testing corrections by the FDR method (online supplemental figure S6D).
Clinical and molecular characterizations of tumors with somatic POL deleterious mutations
Somatic variants in the polymerase genes showed wide distribution across the entire protein coding regions (online supplemental figure S4). A total of 722 somatic variants in all three polymerase genes (POLE, 394; POLD1, 247; and POLH, 81) were detected in the cohort (n=494), with a median of one variant per patient (range 1–20). The most highly recurrent mutations in POLE were p.P286R and p.V411L, which are known ‘hot-spot’ deleterious mutations driving ultrahypermutation.6 In total, we detected known POLE deleterious mutations in 27 patients (figure 2A), with a total of 78 somatic variants across the three polymerase genes (median=3 variants per patient; range=1–6). The majority (22/27, 82%; 95% CI 62% to 94%) of these cases had CRCs, with the remaining cases being two endometrial cancers and one case each of pancreatic, lung and ovarian cancers. Consistently, all these cases exhibited ultrahypermutation (mTMB=243.8 mut/Mb, range: 61.4–644.2 mut/Mb), with the exception of one endometrial tumor carrying the p.A456P mutation (TMB=61.4 mut/Mb). In addition to POLE, we also detected previously reported POLD1 mutations6 in our cohort, including p. R311C, p.D316N, p.R409, and recurrent p.E245K and p.R689W variants (online supplemental table 3). However, these variants were inconsistently associated with high TMB and both of the p.R689W cases had MSI tumors. As a result, their driver status could not be established.
Supplemental material
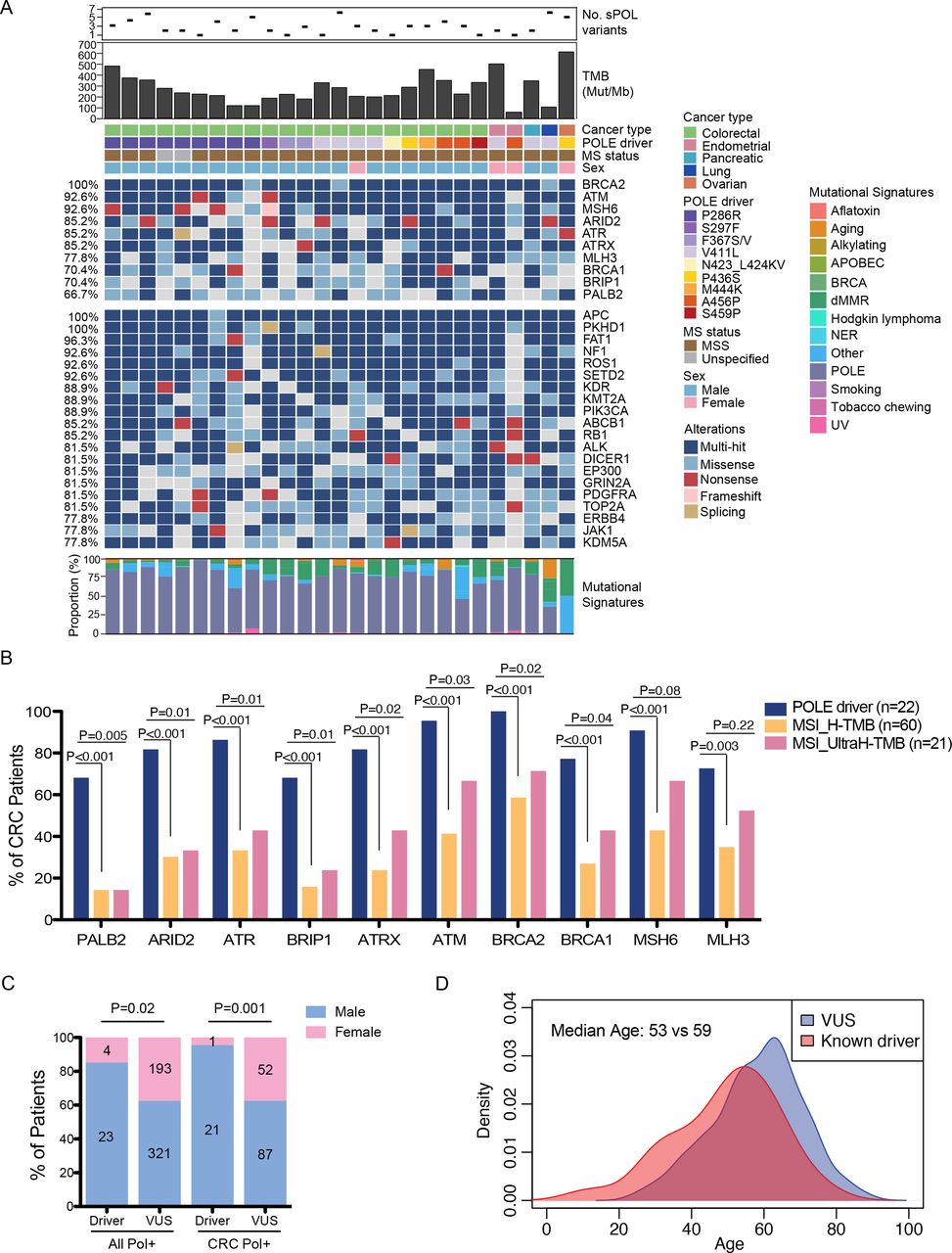
Mutational and clinical characteristics of POLE driver mutations. (A) Oncoplot illustrating top altered genes in samples carrying POLE driver mutations, including top 10 DDR pathway genes and the remaining top 20 most frequently altered genes. The number of somatic POL variants, TMB levels, the different driver mutations identified in the study cohort, as well as their associated cancer types, MS status and patient sex of each case are shown on top. Mutational signatures extracted from each sample are shown at the bottom. (B) Comparisons of the frequencies of DDR pathway alterations between POLE driver mutants and those with MSI-driven high or ultra-high TMB in CRC cases. (C) Comparisons of sex distributions between patients with known driver mutations and those with VUS in all or colorectal POL+ samples. (D) Comparisons of age distributions between patients with known driver mutations and those with VUS. CRC, colorectal cancer; DDR, DNA damage repair; MS, microsatellite; MSI, microsatellite instability; MSS, microsatellite stable; TMB, tumor mutational burden; VUS, variants of unknown significance.
Interestingly, DNA damage and repair (DDR) pathway genes were among the top altered genes in these driver-positive tumors, with BRCA2, ATM, and MSH6 alterations detected in over 90% of the cases (figure 2A). Although MSH6 and MLH3 were frequently mutated, all patients with evaluable MS status had MSS tumor. Similar mutational landscape was observed in the driver-positive samples of the TCGA colon cancer cohort (online supplemental figure S7A). To test if the enrichment of DDR pathway genes were specific to the pathogenic effects of POLE, rather than a high mutational load, we first examined the mutation rate (number of variants per gene) in each DDR pathway genes and in similarly sized non-DDR genes (±5% bp). Permutation analysis showed that ARID2, ATRX, BRCA1/2, MSH6 and MLH3 were significantly more prone to be mutated than similarly sized non-DDR genes in the driver-positive samples (p value<0.05; online supplemental table 4). Comparisons of variant classes revealed a higher frequency of nonsense mutations in DDR pathway genes compared with non-DDR genes in the driver-positive samples (Fisher’s exact test, p=0.03; online supplemental figure S7B). In addition, we compared the mutational profiles of CRC cases with POLE driver mutations and those with a high TMB due to MSI. Notably, alterations in all of the ten DDR pathway genes tested were markedly enriched in POLE drivers compared with MSI tumors (all had FDR adjusted P value<0.005; figure 2B). In addition, alterations in all DDR pathway genes, except for MLH3, remained associated with driver POLE in comparison with the ultra-hypermutated MSI tumors (ie, TMB >105 mut/Mb; figure 2B). Although the POLE driver-positive samples had higher TMB than hypermutated or ultra-hypermutated MSI tumors (online supplemental figure S7C), the probability of randomly selected sets of ten genes being significantly enriched in POLE driver samples was as low as 0.00006 (online supplemental table 5). Next, we mapped all silent and non-silent substitutions in each case to the 30 signatures of mutational processes34 from the Catalog of Somatic Mutation in Cancer (COSMIC) database described previously. As expected, these tumors showed predominantly POLE-associated signatures (figure 2A). Deficient mismatch repair (dMMR)-associated signatures were also detected at variable extent in each sample. One ovarian tumor with a deleterious p.P436S mutation in POLE and MSS status displayed predominantly dMMR- but no POLE-associated signatures. Comparisons of the clinical features of known drivers and variants of unknown significance (VUS) revealed strong associations of POL driver mutations with the male sex (figure 2C) and a younger age of disease diagnosis (median age=53 vs 59 years, Wilcoxon rank-sum test p=0.002; figure 2D).
Supplemental material
Supplemental material
Potential clinical impact of somatic VUS
Apart from the POLE driver mutations, we have detected a large number of somatic POL variants of which the functional and clinical significance remained elusive. Thus, we sought to investigate further into the pathogenesis of these VUS. As all known POLE driver mutations occur in the exonuclease domain, we first evaluated the effect of VUS on TMB across different protein domains comparing the MSS patients with only one VUS to eliminate the potential confounding effects of MSI and multiple VUS. Not surprisingly, patients carrying VUS in the exonuclease domain of POLE were more likely to have increased TMB compared with those with VUS that occur in the other domains of the protein (median TMB=16.3 vs 7.8 Mut/Mb, Wilcoxon rank-sum test p=0.04; online supplemental figure S8A). Similarly, VUS in the exonuclease domain of POLD1 were also associated with higher TMB compared with VUS in other domains (median TMB=10.4 vs 7.2 mut/Mb, Wilcoxon rank-sum test p=0.02; online supplemental figure S8B). On the other hand, no clear association with high TMB was observed across various protein domains of POLH (online supplemental figure S8C). However, the functional significance of different POLH domains requires further investigation as only a small number of VUS were identified.
Given the large number of patients with multiple POL variants, we next tested the hypothesis that VUS of variable penetrance have additive effects on TMB. Indeed, we observed an exponential increase in TMB in samples carrying a higher number of POL variants across the entire study cohort (Jonckheere-Terpstra test p<0.001, figure 3A) or patients with MSS tumors (Jonckheere-Terpstra test p<0.001, online supplemental figure S9A). As tumors with multiple variants in a given gene are also likely to have higher TMB, we further assessed the association of TMB with the total number of POL genes affected. While the number of variants per polymerase gene was quite comparable across different samples, alterations that affected more POL genes again highly correlated with increases in TMB in all of the POL driver-negative cases (Jonckheere-Terpstra test p<0.001, online supplemental figure S9 B and C), as well as the MSS POL driver-negative cases (Jonckheere-Terpstra test p=0.004, online supplemental figure S9D and E). In addition, to examine the association of TMB with multiple variants in genes other than POL, we selected a number of genes with similar or larger sizes. However, the increasing number of variants in these genes did not lead to corresponding increases in TMB in the POL− samples (online supplemental figure S10A–E). Permutation analysis using 500 random sets of genes with a size similar to that of POL genes (size ±5%) showed that POL genes had a significantly higher mutation rate than 99.8% (499/500) of the random gene sets. In addition, the background mutation rate (variant per gene/gene set) was significantly lower than that of POL genes in patients with TMB-H (permutation test p<0.001, online supplemental figure S10F). Notably, there were three cases with MSS status and no clear POLE drivers that also displayed ultra-hypermutation, all of which carried multiple variants in at least two of the POL genes assessed. For example, a 47-year-old man with CRC carried a total of two germline variants in POLE and POLD1, and a total of five somatic variants in POLE and POLH (figure 3B). There were two hits in the exonuclease domain of POLE, including one germline p.N336S variant and one somatic p.T278K variant. However, no strong evidence exists supporting the pathogenicity of the p.N336S and p.T278K variants. Specifically, we identified two additional patients with MSS carrying the p.N336S variants (one germline and one somatic). Both were patients with lung cancer with only one somatic POL variant, yet neither exhibited ultra-high TMB characteristic of pathogenic POLE (one germline p.N336S: TMB=6.52, one somatic p.N336S: TMB=29.99). No other patient with MSS with p.T278K was identified in our cohort. Even in the absence of clear driver mutations, the tumor displayed a strong POLE-associated mutational signature. We suspect that the ultra-high TMB in this case (145 mut/Mb) is a result of the collective effect of multiple POL variants. However, we could not rule out the possibility that higher rates of POL VUS were merely a consequence of high TMB. Future functional or biochemical validations would be necessary to establish the causal relationship between multiple POL VUS and TMB.

Functional characterization of POL VUS. (A) Changes in TMB levels comparing all samples with increased number of POL variants (Jonckheere-Terpstra test p<0.001). (B) One representative case of ultrahypermutator carrying multiple germline and somatic variants in all three polymerase genes and no known drivers. (C) Kaplan-Meier estimates of overall survival in the POL+ TCGA cohort comparing patients with different numbers of POL genes affected. (D) Increased frequencies of driver mutations in patients with more POL genes affected. (E) Kaplan-Meier estimates of overall survival in the POL+ driver-negative TCGA cohort comparing patients with different numbers of POL genes affected. MSS, microsatellite stable; TCGA, The Cancer Genome Atlas; TMB, tumor mutational burden; VUS, variants of unknown significance.
Using a cohort of TCGA patients with variants in the POLE, POLD1, and/or POLH genes, we examined the clinical significance of multiple POL variants on patient prognosis. We found that patients with an increased number of altered POL genes affected had better chance of prolonged overall survival (log-rank p=0.06, figure 3C). However, driver mutations were detected in a higher frequency in samples with multiple POL genes affected (figure 3D). To rule out the influence of driver mutations on patient survival, we compared POL+ samples with VUS only. Similarly, we found that patients with VUS in all three POL genes had a longer overall survival than those with one or two POL genes affected (5-year survival rate of patients with three vs two vs one POL gene affected: 100% vs 65% vs 62%, figure 3E), which might suggest loss of functional compensation as a consequence of simultaneous perturbation of multiple POL genes. A similar trend towards increased survival persisted following correction for cancer types and TMB (online supplemental table 6).
Supplemental material
Mutational characterization of POL VUS
Given the potential additive value of POL VUS in driving similar functional and clinical consequences to those of driver mutations, we sought to compare the mutational spectra and mutational signatures of POL+ patients displaying high TMB as a result of driver mutation, VUS, or MSI, as well as those of patients with TMB-L. The comparisons were performed in patients with CRC, where the majority of POLE driver mutations were found. Similar to known POLE driver mutations, comutations in the DDR pathway genes were also relatively common in patients with TMB-H MSS with POL VUS, which were nearly absent in the TMB-L tumors (figure 4A). By contrast, the mutational frequencies of the top CRC genes, including TP53, APC, KRAS, and FBXW7, among others, were highly comparable between POL VUS carriers with different TMB status (figure 4A). Extraction of the mutational signatures revealed largely distinct patterns associated with the four different groups (figure 4B,C). In particular, POLE drivers and, to a lesser extent, the TMB-H VUS tumors were associated with pronounced increases in C>A and T>G single-nucleotide substitutions compared with the MSI or TMB-L tumors (figure 4B). The increased T>G single-nucleotide substitutions were highly consistent with the COSMIC polymerase hypermutation signatures V.3.2 (March 2021). Notably, in the POLE driver-positive samples, the T>G substitutions were more prominent in DDR pathway genes than non-DDR genes (Fisher’s exact test, p<0.0001; figure 4C), suggesting that DDR pathway changes might be early events in POL pathogenesis. More detailed examination of the mutational frequencies of all 96 possible trinucleotide contexts revealed notable enrichments of the G(C>A)G and the T(T>G)T signatures in POLE drivers (figure 4D). While the TMB-H VUS tumors also displayed high levels of C>A and T>G substitutions, only an enrichment of T(T>G)T signature was observed compared with the MSI or TMB-L tumors. The G(C>A)G signature was otherwise absent from all of these three driver-negative groups.

Molecular signatures of different types of POL mutations. (A) Comparisons of mutational frequencies of top CRC genes and DDR pathway genes between POL VUS carriers with different TMB status. (B) Single-nucleotide substitution signatures comparing different subcohorts of patients with POL+ CRC. (C) Comparisons of single-nucleotide substitution signatures between the DDR pathway and non-DDR genes in pole driver-positive samples. (D) Trinucleotide context frequencies comparing different subcohorts of patients with POL+ CRC. CRC, colorectal cancer; DDR, DNA damage and repair; MSS, microsatellite stable; TMB, tumor mutational burden; TMB-H, tumor mutational burden-high; TMB-L, tumor mutational burden-low; VUS, variants of unknown significance.
Prognostic significance of POL and DDR pathway gene alterations
Our results revealed a strong association between DDR pathway gene mutations and the penetrance of POL variants. Finally, in a cohort of 4679 TCGA patients that encompasses 11 cancer types, including breast, gastric, and endometrial cancers and CRCs, we assessed the effects of DDR pathway gene alterations on top of POL variants on patient survival. The DDR pathway genes analyzed are shown in figure 4A. Variants in POLE/POLD1/POLH represented a strong prognostic factor (figure 5A), with patients having driver mutations showing increased survival probability than the VUS carriers (figure 5B). Remarkably, loss of function mutations (nonsense or frameshift) in DDR pathway genes were associated with further improved overall survival regardless of the POL variant status (driver, VUS or POL−; figure 5A,B). POL and DDR pathway alterations remained independent predictors in multivariate Cox regression analysis adjusting for cancer types (p<0.0001 and p=0.01, respectively; online supplemental table 7).
Supplemental material

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Prognostic subclasses of POL and DDR pathway gene alterations. (A,B) Kaplan-Meier estimates of overall survival in the TCGA cohort showing distinct prognostic subclasses defined by POL and DDR pathway gene mutational status. (C) Kaplan-Meier estimates of overall survival in the TCGA cohort comparing patients with varying number of defective DDR pathway genes. (D) Kaplan-Meier estimates of overall survival in the TCGA cohort comparing patients with different types of DDR pathway gene alterations (DDR_loss, missense, DDR+ patients with both loss of function and otherwise missense mutations; DDR_loss only, DDR+ patients with only loss of function mutations; DDR_missense only, DDR+ patients with only missense mutations that are not nonsense mutations; DDR−, patients with no DDR alterations). DDR, DNA damage repair; TCGA, The Cancer Genome Atlas.
Taking into consideration that both POL variants and loss of function in DDR pathway genes might be correlated with increased TMB, we assessed the potential interaction of these variables on patient survival in the subset of patients with evaluable TMB. While patients with ultra-high TMB were associated with prolonged overall survival, those with high-TMB initially did poorly compared with those with low-TMB and only began to manifest its survival benefit around 11 years following disease diagnosis at the crossover of the two survival curves (ultra-high vs high and low TMBs, HR=0.82, 95% CI=0.66 to 1.02; p=0.07; online supplemental figure S11). Multivariate Cox regression analysis incorporating TMB, DDR and POL gene mutational status, as well as cancer type, showed POL variants, and, to a lesser extent, loss of function in DDR pathway genes as strong predictors of patient outcome independent of high-TMB or ultra-TMB status (online supplemental table 7).
We hypothesized that multiple hits on the DDR pathway genes would have similar additive effects on patient survival. As expected, accumulation of defective genes in the DDR pathway was associated with increasing probability of survival (log-rank p<0.0001, figure 5C). This association remained significant following correction for cancer type and different TMB status (online supplemental table 7). To verify that DDR pathway genes are functionally important for prolonged survival in patients with cancer, we also tested the effects of different types of DDR pathway gene alterations on patient survival. Indeed, patients with only loss of function mutations in DDR pathway genes showed markedly increased overall survival compared with those with otherwise missense mutations (median overall survival=20.4 years vs 7.7 years, HR=1.42, 95%CI=0.98 to 2.06, log-rank p=0.06; figure 5D and online supplemental figure S12A). Notably, this survival difference could not be simply explained by the increase of mutational burden with defective DDR as loss of function and otherwise missense mutations exhibited similar proportions of POL or DDR pathway genes mutated (online supplemental figure S12B). Similarly, no difference in TMB was observed between samples with these two types of DDR alterations using the TMB-evaluable subset (Wilcoxon rank-sum test, p=0.38; online supplemental figure S12C).
Discussion
In this study, we described the prevalence of somatic and germline POLE/POLD1/POLH variants in a cohort of 12,266 patients across diverse cancer types and conducted comprehensive investigations into mutational and functional consequences associated with POL alterations. In addition to the high prevalence of POL variants in endometrial (11.7%) and colorectal (6.6%) cancers,11 12 35 we also observed relatively high frequencies of POL variants in previously under-reported urinary (8.2%) and prostate (6.8%) cancers. Although the functional significance of POL VUS can be difficult to interpret at the individual level, those in the exonuclease domain of POLE and POLD1 are more likely to be associated with high TMB. In addition, multiple POL VUS might potentially act in concert to drive the hypermutation or even the ultra-hypermutation phenotype. Mutational analyses of the POL+ samples uncovered a strong linkage between POL and DDR pathway alterations, which showed cumulative and independent effects on TMB and patient survival across a wide range of cancers in both our cohort and the TCGA cohorts.
The classification of VUS has remained a major challenge in human genetics, particularly in the era of large-scale genomic sequencing. The large size of POL genes and the non-redundant nature of most POL VUS make it increasingly difficult to accurately interpret the pathogenicity of each variant. Functional classification of germline variants is particularly challenging in the absence of medical and genetic information from the family members for validation. Previous studies have identified likely pathogenic germline mutations in the exonuclease domains of POLE (eg, L424V, N363K, Y458F, and A456P) and POLD1 (eg, S478N and P327L) that predispose to colorectal, endometrial and brain cancers.10 28–33 None of these reported germline mutations were detected in our cohort. However, we showed that germline variants in the exonuclease domains of POLE and POLD1 were associated with higher TMB than variants in other parts of the proteins. In addition, we identified a p.T466A germline variant in the exonuclease domain of POLE that was present in an early-onset CRC case, which is probably pathogenic. The rare incidence of deleterious germline mutations in the polymerase genes might be due to the detrimental effect of high mutational burden, and consequently, they were selected out early on in the process of normal development.
Similar to germline variants, the majority of somatic variants in POL genes were not associated with ultra-high TMB. Consistent with previous reports,6 established somatic driver mutations in our cohort all occur in the exonuclease domain of POLE, the disruption of which has been associated with a decreased DNA replication fidelity and an increased DNA excision rate.6 Indeed, we found that VUS in the exonuclease domains of POLE and POLD1 were also associated with high TMB. A number of POLD1 driver mutations have also been reported,6 10 yet we found that such mutations were inconsistently associated with ultra-hypermutation. This discrepancy likely reflects potential sampling biases that might be accounted for by the presence of additional POL/DDR/mismatch repair (MMR) pathway gene mutations or exposure to environmental mutagens, given the small sample size carrying these variants.
The most intriguing finding of our study is the co-occurrence of multiple somatic POL variants and DDR pathway alterations in a large proportion of samples. Driven by the hypothesis that disruption of multiple genes in compensatory pathways promotes disease pathogenesis, we found increased TMB and patient survival with the presence of multiple POL VUS or with comutations in DDR pathway genes. Their functional and clinical impact is likely explained by the combined effect of the reduced fidelity of the polymerases and/or the accumulation of DDR pathway alterations. Notably, a subset of patients with multiple VUS in POL also exhibited the polymerase hypermutation signatures in the absence of known driver mutations (figures 3B and 4B,D), further supporting that POL VUS might be of functional consequences particularly when there were multiple hits in the polymerase genes. Future functional studies are needed to verify the cause-and-effect relationship between POL VUS and TMB, as well as the evolutionary dynamics of co-occurring DDR pathway mutations.
It has been shown that POLE/POLD1 variants, including both drivers and VUS, are predictive of favorable immunotherapy outcome.35 However, to what extent POL variants of variable penetrance exert their influence on patient survival has been unclear. Based on the mutational statuses of POL and DDR pathway genes, we made several important findings that clearly elucidate the functional and clinical relevance of testing for POL variants and unveiled distinct subclasses of patients with varying probability of survival. First, we showed that the pathogenicity of POL variants is positively correlated with an increased survival outcome. In particular, patients with POL driver mutations or those with multiple POL genes affected experienced highly prolonged overall survival. As hypermutation in POL+ patients was often accompanied by the simultaneous loss of the DDR pathway genes, we found that incorporation of DDR deficiency status allowed for further stratification of patients and DDR deficiency was associated with added survival benefit irrespective of POL mutational status. Interestingly, even for patients harboring POL VUS with minimal functional consequences at the level of TMB or those without the acquisition of additional DDR pathway alterations, they still had higher survival probability than the POL− and DDR-negative population. Furthermore, the prognostic values of POL and DDR pathway alterations were independent of TMB. Finally, our findings on POL genes and their associated DDR pathway mutations are highly robust and generalizable across a multitude of cancer types. Thus, while it is generally accepted that most VUS are merely passenger mutations with no functional consequences, the presence of POL VUS regardless of their impact on TMB may serve as a biomarker for overall survival, as well as immunotherapy outcome.
To our knowledge, our study represents the largest set of patients screened for POLE/POLD1/POLH germline and somatic variants with characterization of the mutational landscape and functional relevance of driver mutations and VUS. Our extensive characterization of POL variants revealed the accumulation of additional DDR pathway alterations as one potential underlying pathogenic mechanism of their hypermutation phenotype. In addition to POL drivers, POL VUS in combination with DDR pathway deficiencies may act in a dose-dependent manner to impact patient survival. Combinatorial assessment of POL and DDR pathway gene alterations allows for optimal stratification of patients based on their clinical outcome and may also be predictive of benefit from a wide range of cancer therapies, including chemotherapy, immunotherapies, and poly (ADP-ribose) polymerase (PARP) inhibitor treatments.
Data availability statement
Data are available upon reasonable request. The raw sequencing data generated and/or analyzed during the current study are not publicly available due to privacy concerns and restrictions by the regulation of the Human Genetic Resources Administration of China as they contain germline information, but all other datasets analysed are available from the corresponding authors on reasonable request or on published The Cancer Genome Atlas databases.
Ethics statements
Patient consent for publication
Ethics approval
The study was conducted in accordance with Declaration of Helsinki. All data analyzed were collected as part of routine diagnosis and treatment. Informed written consent was obtained from each subject or each subject’s guardian.
Acknowledgments
We thank all the patients and family members who gave their consent on presenting the data in this study, as well as the investigators and research staff at all hospitals and research sites involved.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
JY and LY contributed equally.
Correction notice This article has been corrected since it was first published. Dr Lingjun Zhu has now been added as a corresponding author, and ‘National Cancer Center/National Clinical Research Center for Cancer/Cancer Hospital’ has been added to Dr. Lin Yang's affiliation.
Contributors JY, LY, XW, YS, LZ, and XCheng conceived and supervised the study. JY, LY, JCY, JP, YW, and HB designed the experiments. JCY, GX, MX, XChen, JP, YW, and HB acquired and analyzed the data for this work. All authors participated in data interpretation. JY, LY, and JCY drafted the manuscript. JY, LY, JCY, XChen, HB, XW, YS, LZ, and XCheng provided critical revision of the manuscript for important intellectual content.
Funding This work was supported by in part by a grant from Jiangsu Provincial Medical Talent awarded to LZ (ZDRCA2016089).
Competing interests JCY, XChen, JP, YW, HB, XW, and SY are employees or shareholders of Nanjing Geneseeq Technology Inc. All remaining authors have declared no conflicts of interest.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.