Article Text

Abstract
Background Immune checkpoint inhibitors have revolutionized cancer treatment, but the benefits in refractory patients with esophageal cancer have been modest. Predictors of response as well as new targets for novel therapeutic combinations are needed. In this phase 2 clinical trial, we tested single-agent pembrolizumab in patients with advanced esophageal cancer, who received at least one prior line of therapy.
Methods Pembrolizumab 200 mg every 3 weeks was tested in 49 patients with refractory esophageal cancer: 39 with adenocarcinoma and 10 with esophageal squamous cell carcinoma. Major endpoints were radiological response by Immune-related Response Evaluation Criteria In Solid Tumors and survival. Tumor samples were evaluated for programmed cell death ligand 1 (PD-L1) expression, tumor mutational burden (TMB), and immune contexture by both NanoString mRNA expression analysis and flow cytometry. Peripheral blood mononuclear cells and a panel of circulating chemokines were also analyzed.
Results The overall response rate (ORR) was 8% (4 of 49 patients; 95% CI 2.3% to 19.6%). Median overall survival (OS) was 5.8 months (95% CI 4.0 to 9.5). ORR and OS were not associated with histology. For PD-L1-positive patients, ORR was 13.3% (95% CI 1.7% to 40.5%) and median OS was 7.9 months (95% CI 4.7 to 15.5). A trend toward improved OS was observed in seven patients with a TMB ≥10 mut/Mb (p=0.086). Tumors with a PD-L1 Combined Positive Score ≥1 showed enrichment of LAG3 (p=0.005) and IDO1 (p=0.04) gene expression. Baseline levels of circulating CXCL10, interleukin 2 (IL2) receptor α (IL2RA) and IL6 were associated with survival: CXCL10 favorably, (HR 0.37, p=0.002 (progression-free survival); HR 0.55, p=0.018 (OS)); IL2RA and IL6 unfavorably (HR 1.57, p=0.020 for IL6 (OS); HR 2.36, p=0.025 for IL2RA (OS)).
Conclusions Pembrolizumab monotherapy was modestly effective in refractory esophageal cancer. Circulating CXCL10 at baseline appeared to be a robust predictor of response. Other T cell exhaustion markers are upregulated in PD-L1-positive patients, suggesting that immunotherapy combinations such as anti-LAG3/programmed cell death protein 1 (PD-1) or anti-IDO1/PD-1 may be of promise in refractory esophageal cancer.
- immunotherapy
- biomarkers
- tumor
- cytokines
- gastrointestinal neoplasms
- tumor microenvironment
Data availability statement
Data are available on reasonable request via corresponding author.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Background
Esophageal cancer is the sixth-leading cause of cancer-related mortality worldwide.1 For patients with metastatic disease, chemotherapy has been the standard treatment, with median overall survival (OS) ranging from 9 to 12 months.2 Recently, immune checkpoint inhibitors have shown efficacy in this disease.3–8
Pembrolizumab is a humanized monoclonal antibody against programmed cell death protein 1 (PD-1).9 Binding of PD-1 ligands, programmed death ligand 1 (PD-L1) and 2 (PD-L2), to the PD-1 receptor on T cells, inhibits T cell proliferation and cytokine production. Pembrolizumab blocks the interaction between PD-1 and these ligands, promoting an antitumor immune response. A series of multinational phase III trials have demonstrated the activity of pembrolizumab and other anti-PD-1 antibodies in patients with advanced esophagogastric cancers.3–6 8 10 In these trials, the likelihood of response has been linked to the expression of PD-L1 on tumor and tumor-infiltrating immune cells. The identification of other biomarkers to differentiate responders and non-responders will be critical to selecting patients with the highest potential to benefit from immunotherapy and for development of future clinical trials.
Herein, we report the results of a phase II trial of pembrolizumab in patients with refractory esophageal cancer and of the exploratory tumor and immune profiling studies performed within samples obtained from this clinical study.
Materials and methods
Study design and population
This was an open-label, single-arm, multicenter phase II clinical trial of pembrolizumab in advanced, chemotherapy-refractory esophageal cancer (NCT02971956). Eligible patients were adults who had histologically-confirmed unresectable or metastatic esophageal squamous cell carcinoma (ESCC) or adenocarcinoma (EAC) of the esophagus or gastroesophageal junction (Siewert type 1), irrespective of PD-L1 expression. Patients must have received at least one prior therapy for their advanced disease. Participants were required to have measurable disease based on the Immune-related Response Evaluation Criteria In Solid Tumors (irRECIST).11
All eligible patients were required to have an Eastern Cooperative Oncology Group performance status of 0 or 1, and adequate organ function. Major exclusion criteria included prior therapy with a checkpoint inhibitor, diagnosis of immunodeficiency, systemic immunosuppressive therapy, central nervous system metastases, second malignancy, or autoimmune disease requiring systemic treatment. All patients provided written informed consent before study entry.
Treatment and assessments
Patients were treated with pembrolizumab 200 mg intravenously every 3 weeks. Treatment continued until disease progression, unacceptable adverse event(s) (AEs), or withdrawal of consent. Tumor imaging was performed at baseline and every 9 weeks. Response and progression were evaluated using the irRECIST criteria,11 as assessed by the independent Dana-Farber/Harvard Cancer Center Tumor Imaging Metrics Core. AEs were graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events, V.4.0.
Study outcomes
The primary endpoint of this study was overall response rate (ORR), defined as complete response or partial response (PR) by irRECIST, in patients with refractory EAC. Secondary endpoints were ORR, progression-free survival (PFS), OS, and duration of response (DOR) in patients with PD-L1-positive and -negative advanced EAC, as well as safety and tolerability in the overall cohort. Exploratory endpoints were ORR in patients with refractory ESCC and evaluation of the relationship between efficacy (response and survival) and potential predictive biomarkers. Unless otherwise specified, all response analyses were completed in patients with a PR or SD regardless of confirmed status, compared with patients with PD.
Immunological studies
All patients provided a baseline tumor biopsy within 4 weeks prior to their first treatment cycle. EAC patients were offered another tumor biopsy at 8 weeks on treatment. Patients who initially responded to pembrolizumab and then developed resistance were asked to provide an end-of-treatment tumor biopsy. Pretreatment archival formalin-fixed paraffin embedded (FFPE) tissue was also analyzed, if available (online supplemental table S1).
Supplemental material
PD-L1 immunohistochemistry was performed on pretreatment FFPE tissue slides using a laboratory developed assay (QualTek Molecular Laboratories, Newtown, Pennsylvania, USA) with the anti-PD-L1 22C3 antibody (Merck & Co, Palo Alto, California, USA). PD-L1 expression was evaluated using the Combined Positive Score (CPS).12 13 The tumor mutational burden (TMB) was estimated and mismatch repair (MMR) deficiency was predicted from institutional targeted next-generation sequencing (Dana-Farber Cancer Institute OncoPanel), using DNA extracted from baseline specimens.14 15 For tumor flow cytometry, fresh baseline, on-treatment, and end-of-treatment biopsies were dissociated into single cell suspensions, stained with two 13-color antibody panels (online supplemental table S2), acquired on a BD Fortessa, and analyzed using FlowJo software (Treestar, Ashland, Oregon, USA). Immune cell subpopulations were not further annotated when the parent population consisted of less than 100 cells, unless specified otherwise. For peripheral blood mononuclear cells (PBMCs) flow cytometry, baseline samples were stained with three 10-color antibody panels (online supplemental table S3) and analyzed according to a previously published flow cytometry methodology.16 17 Approximately 100 000 stained PBMCs per patient were acquired on a BD LSRFortessa X-20, and analyzed using FlowJo software. Fluorescence Minus One controls were used to inform manual gating on each patient. The neutrophil-to-lymphocyte ratio (NLR) was calculated from the complete blood count and differential drawn on the first day of treatment. For gene expression profiling, RNA was extracted from FFPE slides and analyzed with the nCounter PanCancer Immune Profiling Panel (NanoString Technologies). Circulating cytokines and chemokines collected at the beginning of each treatment cycle were quantified by multiplex technology using the FLEXMAP 3D (Luminex Inc).
Statistical analyses
Associations between numerical and categorical variables were analyzed using Welch’s t-test, a Wilcoxon rank-sum test, or a Kruskal Wallis test (whichever appropriate) and associations between categorical and binary variables were analyzed using Fisher’s exact test or Pearson’s χ2 test (whichever appropriate) in R (V.4.0.3). Survival differences were analyzed using a Cox proportional-hazards regression model and logrank tests with the ‘survival’ package (V.3.2–7) in R. Patients lost to follow-up were censored at the time of last follow-up. DOR was calculated from time of first response to time of progression, while duration of stable disease was calculated from the start of treatment to time of progression. Heatmaps were generated by hierarchical agglomerative complete-linkage Euclidean distance clustering using the ComplexHeatmap package in R.18 Differential gene expression analysis was conducted using the DESeq2 V.1.28.1 package,19 and time series clustering using the TMixClust V.1.10.0 package in R.20 Gene Set Enrichment Analysis (GSEA) was performed using the method as described by Subramanian et al.21 All p values are two sided and considered statistically significant when p<0.05.
Results
Patient characteristics
Forty-nine patients received at least 1 cycle of treatment between February 2017 and October 2018 (table 1). The median age was 64 years (range 34–80). Thirty-nine patients (80%) had EAC; 10 patients (20%) had ESCC. Almost all patients (92%) had metastatic disease at enrollment. The majority of patients with prior treatment data available (51%) had received three or more lines of systemic therapy prior to pembrolizumab. Thirty-three patients (67%) had received prior radiation therapy. HER2 status was known for 38 out of 39 patients with EAC, of whom nine patients (23%) had HER2-positive tumors, all previously treated with trastuzumab-containing regimens. One patient had a known MMR-deficient tumor; however, MMR status was not routinely tested.
Baseline characteristics
Safety
All 49 evaluable patients were included in the safety analysis. Treatment-related AEs of any grade were reported in 38 patients (78%; online supplemental table S4); the most common were fatigue (27%), dyspnea (16%), rash (14%), anorexia (12%), arthralgia (10%), and pruritus (10%). Three patients (6%) discontinued therapy because of treatment-related AEs. Grade 3–4 treatment-related AEs occurred in six patients (12%), including two patients with grade 3 pneumonitis. One patient developed myasthenia gravis followed by respiratory failure and eventual cardiac arrest. No treatment-related deaths occurred.
Antitumor activity/treatment efficacy
Forty-three patients (88%) had at least one restaging scan; the remaining 6 patients had rapid clinical progression. PR was achieved in 8% (4/49; 95% CI 2.3 to 19.6%), stable disease (SD) in 22% (11/49), and progressive disease (PD) in 67% (33/49). Of 39 EAC patients, we observed a PR in three patients (8%, 95% CI 1.6 to 20.9%; two confirmed) and SD in eight patients (21%); three confirmed) for a disease control rate of 28% (figure 1A,B, online supplemental figure S1 and 2). PD was observed in 28 EAC patients (72%), including six with clinical progression before the first restaging scan. In the 10 patients with ESCC, one patient (10%) had a (confirmed) PR and three patients achieved SD (two confirmed). Among the four patients with a PR, the median DOR was 4.1 months (1.5–24.5 months). Median duration of SD was 6.4 months.
Supplemental material

Response to treatment by histology. (A, B) Show the radiological response by irRECIST of all 43 patients with advanced esophageal cancer treated with pembrolizumab that had at least one restaging scan, color coded by histology. The remaining six patients had progressed clinically before the first restaging scan at 9 weeks. (A) A spider plot showing the percentage change from baseline by time in weeks. (B) A waterfall plot showing the best overall percentage change from baseline per study subject. (C) Kaplan-Meier plots showing progression-free survival (PFS) and overall survival (OS) per histology. EAC, esophageal adenocarcinoma; ESCC, esophageal squamous cell carcinoma; irRECIST, Immune-related Response Evaluation Criteria In Solid Tumors.
At the time of analysis, 47 patients had passed away and 2 patients were lost to follow-up. Median OS for the EAC patients was 4.8 months (95% CI 3.2 to 9.5) and median PFS was 1.8 months (95% CI 1.8 to 2.0). For the entire population, the median OS was 5.8 months (95% CI 4.0 to 9.5) and the median PFS was 1.8 months (95% CI 1.8 to 2.0; figure 1C). The six patients with grade 3–4 treatment-related AEs showed a trend to improved PFS and OS (p=0.065 and p=0.17, respectively; logrank test). Although patients with HER2-positive EAC had inferior PFS (p=0.027), no significant associations between HER2 status and either OS or radiological response were noted. Prior radiotherapy or the number of prior lines of therapy were not associated with response or survival.
PD-L1 and response to pembrolizumab
Pretreatment PD-L1 CPS was available in 36 of 49 patients; PD-L1 CPS was determined from archival tissue in an additional 5 patients (online supplemental table S1). Fifteen of these 41 patients (37%) were PD-L1-positive (CPS≥1), of which 7 patients (17%) had a PD-L1 CPS≥10 (online supplemental figure 3A). The single MMR-deficient patient had PD-L1 CPS 1.
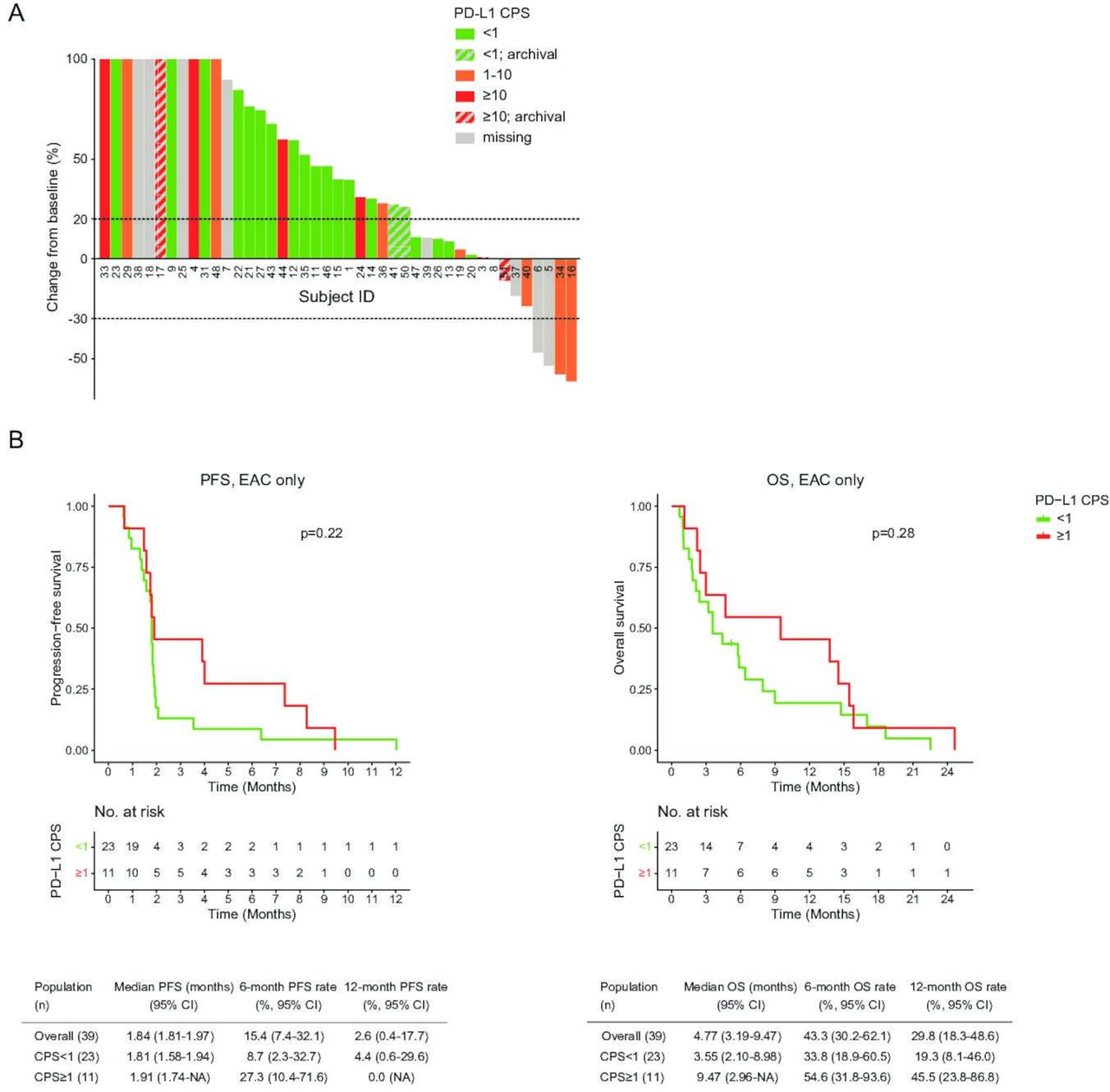
Eleven patients with PD-L1-positive EAC had a PR in 18% (2/11) and SD in 27% (3/11). In 23 PD-L1-negative EAC patients, no PR was seen and 17% (4/23) achieved SD (p=NS). Similar results were recorded for the entire study population (figure 2A, online supplemental figure S3B–D); the ORR seen in all PD-L1-positive patients was 13.3% (95% CI 1.7 to 40.5%).

Response to treatment by PD-L1 Combined Positive Score (CPS). (A) A waterfall plot showing the best overall percentage change from baseline according to irRECIST per study subject, color coded by PD-L1 combined positive score (CPS). (B) Kaplan-Meier plots comparing progression-free survival (PFS) and overall survival (OS) of PD-L1-negative (CPS<1) and PD-L1-positive (CPS≥1) patients with esophageal adenocarcinoma (EAC). This includes PD-L1 CPS from archival tissue. irRECIST, Immune-related Response Evaluation Criteria In Solid Tumors; PD-L1, programmed death-ligand 1.
Median OS for PD-L1-positive EAC was 9.5 months (95% CI 3.0 to NA) vs 3.6 months (95% CI 2.1 to 9.0) for PD-L1-negative patients (p=NS). We also did not identify a significant difference in PFS between PD-L1-positive and -negative EAC patients (figure 2B). Similar results were recorded for the entire study population, with a median OS of 7.9 months (95% CI 4.7 to 15.5) in all PD-L1-positive patients (p=NS; online supplemental figure S4A). The 7 patients with CPS≥10 (4 EAC; 3 ESCC) did not have statistically superior survival (online supplemental figure S4B).
Tumor mutational burden
TMB could be estimated for 27 patients (21 EAC; 6 ESCC). Seven patients (25.9%) had a TMB of 10 mut/Mb or higher (5 EAC; 2 ESCC). A TMB of 10 mut/Mb or higher showed a trend toward improved OS (p=0.086, logrank test). There were no associations between TMB and radiological response or PFS.
Immune infiltrate (flow cytometry) and response to pembrolizumab
To characterize the immune response, we first analyzed the tumor immune microenvironment by flow cytometry on fresh pretreatment biopsies of 42 patients (37 EAC; 5 ESCC). After exclusion of cases with <100 viable CD45+ single cells, 20 samples were taken from the primary tumor, 10 from a liver metastasis, and 7 from another location (figure 3A). Median cell viability was 57.9% (IQR 36.9%–77.2%), resulting in a median of 1766 live single cells per patient (IQR 857–8352). Based on biopsy size, 25 samples were profiled with a T cell and a myeloid cell panel and 12 samples were profiled with only the T cell panel.

Flow cytometric analysis of tumor biopsies at baseline. (A) Overview of major cell populations as a percentage of CD45+ cells, ordered by best overall radiological response (%). (B) Comparison of major cell populations as a percentage of CD45+ cells by biopsy site. (C) Comparison of CD4 T cell exhaustion and activation markers by best overall radiological response. (D) Comparison of major cell populations relative to CD45+ cells by best overall radiological response. (E) Progression-free survival (PFS) and overall survival (OS) Cox proportional hazards survival analysis results; corrected for histology and biopsy location. BOR, best overall response; CPS, Combined Positive Score; MMR, mismatch repair; EAC, esophageal adenocarcinoma; ESCC, esophageal squamous cell carcinoma; PD, progressive disease; PR, partial response; SD, stable disease; gMDSC, granulocytic myeloid-derived suppressor cell.
The immune infiltrate differed profoundly by biopsy location: Granulocytes were more abundant in tumor biopsies from the esophagus compared with liver metastases and other locations (p=0.013, Kruskal Wallis test), whereas natural killer (NK) cells were more abundant in biopsies from liver metastases (p=0.028, Kruskal Wallis test; figure 3B). There were no significant differences in major immune cell populations (T cells, B cells, myeloid cells, monocytes, granulocytes, NK cells, and NK T cells) between EAC and ESCC patients. Surprisingly, the primary tumor of the MMR-deficient patient contained a relatively low proportion of CD3 T cells (3.5%) compared with the median of the other samples (20%).
We analyzed if the pre-existing immune infiltrate could predict radiological response to pembrolizumab, comparing patients with clinical benefit (either PR or SD) to those without (PD), correcting for biopsy location and histology by linear regression (online supplemental table S5). There was no difference in the proportion of CD4+ or CD8+ T cells (relative to CD3+ T cells) or CD8:CD4 ratio between patients with and without clinical benefit. Patients with clinical benefit had a significantly smaller proportion of PD-1+ CD4+ T cells than those without (median 22.7% vs 43.6% of total CD4+ T cells, p=0.035 (linear regression, adjusted for biopsy location); figure 3C). As for other cell types, the proportion of granulocytes was significantly larger in patients with clinical benefit than those without (median 81.9% vs 31.8% of CD45+, p=0.0014 (linear regression, adjusted for biopsy location and histology)), whereas B cells were proportionally less abundant in patients with clinical benefit than those without (median 0.16% vs 1.27% of CD45+, p=0.027 (linear regression, adjusted for biopsy location and histology); figure 3D).
Several significant associations between immune cell populations at baseline and survival were found, corrected for biopsy location and histology by multivariate Cox proportional hazards regression (figure 3E). Expression of PD-1 and TIM3 on CD4+ T cells was associated with early progression, with a HR of 1.028 for PD-1 (p=0.039), an HR of 1.039 for TIM3 (p=0.047), and an HR of 1.075 for combined PD-1/TIM3 expression (p=0.024). Consistent with the association between a higher CD15+ granulocyte proportion and a favorable radiological response, an association between granulocyte presence and a decreased risk of early progression (HR 0.976, p=0.031) was found. Moreover, the CD14+ monocyte proportion was associated with early death (HR 1.051, p=0.016). We also explored whether the baseline immune infiltrates of PD-L1-positive (CPS≥1) patients differed from PD-L1-negative patients but did not find any significant associations.
Four EAC patients had a biopsy on day 56 (week 8) and 1 patient had a biopsy at the end of treatment (EOT); these samples were examined by flow cytometry. Two patients had PD as best overall response (BOR), two had SD, and the EOT biopsy was from the MMR-deficient patient who achieved a PR (online supplemental figure S5A). Irrespective of response, we observed an increase in tumor-infiltrating T cells and NK T cells and a decrease in myeloid cells with treatment. B cells and NK cells tended to decrease in patients without clinical benefit (PD) and increase in patients with clinical benefit (PR+SD; online supplemental figure S5B,C).
Circulating immune cells and response to pembrolizumab
Baseline PBMCs of 47 patients were analyzed by flow cytometry, using antibodies to annotate major immune populations, immune checkpoint expression, and T cell activation and memory status. Patients with clinical benefit (PR+SD) had less circulating naïve CD8 T cells (p=0.026, Wilcoxon rank-sum test), less activated CD69+ CD4 T cells (p=0.014, t-test), and a trend toward less Tregs (p=0.087, t-test) than those without. A relatively high number of circulating Tregs was associated with shortened OS (HR 1.3, p=0.018), whereas circulating CD8+ NK T cells were associated with extended OS (HR 0.97, p=0.0036). There were no significant differences in PD-1, TIM3, and LAG3 on CD4 or CD8 T cells between patients with and without clinical benefit (online supplemental figure S6).
Despite there being no association between clinical response and prior radiotherapy or the number of prior lines of therapy, there were differences in PBMCs (online supplemental figure S7 and 8). Notably, patients who received three or more lines of prior systemic therapy had more circulating CD8 T cells, and less CD4 T cells, which expressed more activation marker HLA-DR. Patients who received prior radiotherapy had less circulating CD3 T cells, more Tregs, and more PD-1 positive CD4 T cells. Apart from PBMCs, we also evaluated the neutrophil-to-lymphocyte ratio (NLR) at baseline. An increased NLR was associated with inferior survival (PFS: HR=1.15 (95% CI 1.05 to 1.25), p=0.0015; OS: HR=1.16 (95% CI 1.07 to 1.25), p=0.00048; Cox regression, corrected for histology; online supplemental figure S9).
Sequential cytokine/chemokine analyses and response to pembrolizumab
Further immune profiling was performed using multiplex cytokine/chemokine quantification on plasma in 45 of 49 patients, with 123 sequentially collected and analyzed samples in total (online supplemental table S6). Calculable results were obtained for the following 12 cytokines and chemokines: interleukin 1 (IL1) receptor antagonist (IL1RN), IL2 receptor α (IL2RA), IL6, IL7, IL10, CCL2, CXCL2, CXCL5, CXCL6, CXCL8 (IL8), CXCL10, and CSF2 (granulocyte-macrophage colony-stimulating factor, GM-CSF).
We first queried associations between baseline cytokine/chemokine levels and response in our entire population (adjusted for histology by multiple regression analysis). We found no significant differences in baseline cytokine levels between patients with clinical benefit (PR+SD) and those without, although there was a trend toward a higher CXCL10 concentration with clinical benefit (median of 107.5 pg/mL vs 67.9 pg/mL, p=0.055; adjusted for histology by linear regression on log-transformed data; figure 4A). When applying a stricter definition of clinical benefit, i.e. confirmed SDs or any PR, CXCL10 was significantly higher among those with clinical benefit (p=0.017). IL2RA was significantly lower in patients with a confirmed SD or any PR (p=0.040). CXCL10 concentration at baseline was also associated with a decreased risk of both early progression and death (HR 0.37/p=0.0022 for PFS, HR 0.55/p=0.018 for OS; Cox regression on log-transformed data, adjusted for histology; figure 4B). Levels of IL6 and IL2RA at baseline, in contrast, were associated with an increased risk of early death (HR 1.57/p=0.020 for IL6; HR 2.36/p=0.025 for IL2RA), and levels of IL1RN showed a trend toward an increased risk of early progression (HR 2.01, p=0.051). We confirmed these findings with logrank analyses (not adjusted for histology) by splitting the cohort into three groups, by concentration, for CXCL10 (PFS p=0.0038, OS p=0.0034) and IL2RA (OS p=0.021; figure 4C).
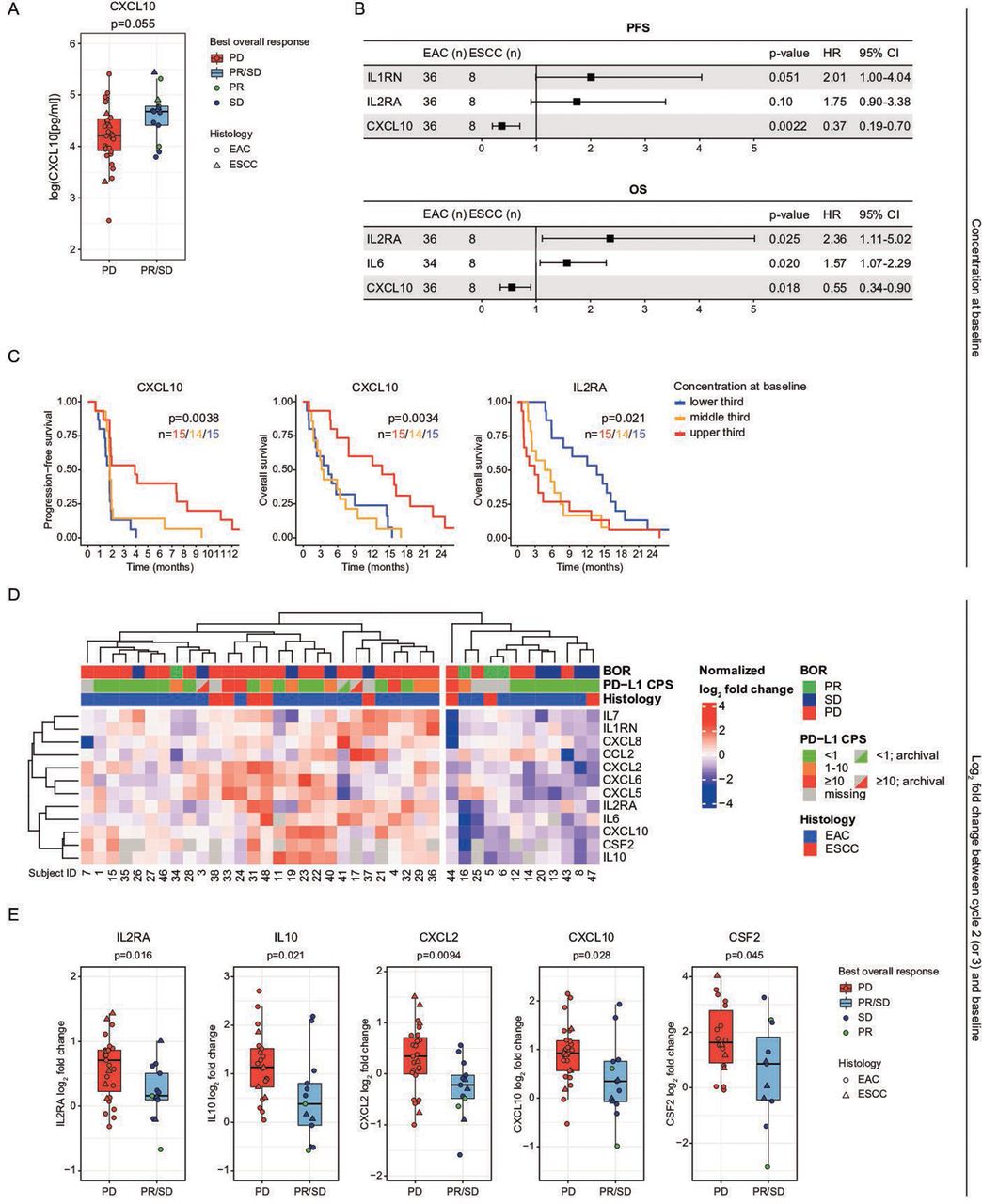
Analyses of a panel of circulating chemokines (Luminex). (A) Boxplot showing a trend toward a higher circulating CXCL10 concentration at baseline in patients with a best overall radiological response of stable disease (SD) or a partial response (PR). (B) Progression-free survival (PFS) and overall survival (OS) Cox proportional hazards analysis of circulating chemokine concentrations at baseline; corrected for histology. Chemokine concentrations were log-transformed to obtain a normal distribution. Only associations with a p<0.1 are shown. (C) Kaplan-Meier plots comparing circulating chemokine concentrations at baseline of several chemokines shown in B. (D) Heatmap of log2 fold changes in circulating chemokine concentrations at treatment cycle 2 (cycle 3 if cycle 2 was missing) relative to baseline. (E) Boxplots showing larger increases in several circulating chemokines at cycle 2 (or 3) relative to baseline in patients with progressive disease than in patients with a partial response or stable disease as best overall radiological response. (F) PFS and OS Cox proportional hazards analysis of log2 fold changes in circulating chemokine concentrations at cycle 2 (or 3) relative to baseline, corrected for histology. Only associations with a p<0.1 are shown. (G) Kaplan-Meier plots comparing circulating chemokine concentrations at cycle 2 (or 3) relative to baseline of the chemokines shown in F. BOR, best overall response; CPS, Combined Positive Score; EAC, esophageal adenocarcinoma; ESCC, esophageal squamous cell carcinoma; IL2RA, IL2 receptor α; PD-L1, programmed death-ligand 1.
We then analyzed dynamic changes in cytokine/chemokine profiles during treatment. A clustering analysis to evaluate patterns in changes in cytokines and chemokines during treatment (start of treatment cycle 2 (or 3) relative to baseline), showed that patients with clinical benefit (PR+SD) were enriched in patients with overall decreases in cytokines/chemokines (7 of 12 patients (58.3%)) compared with a those with overall increases (6 of 28 patients (21.4%), p=0.032, Fisher’s exact test; figure 4D). For IL2RA, IL10, CXCL2, CXCL10, and CSF2, a significant decrease was observed at cycle 2 (or 3) relative to baseline in patients with clinical benefit (linear regression, adjusted for histology; figure 4E). A significant decrease was also seen for CXCL8 (p=0.0081; Wilcoxon rank-sum test, not adjusted for histology). Cox survival analyses, adjusted for histology, confirmed that an increase in IL2RA was a poor prognostic indicator; the log2 fold change of IL2RA was associated both with an increased risk of early progression (HR 2.85, p=0.010) and early death (HR 2.17, p=0.046). An increase in CXCL2 trended toward both an increased risk of early progression (HR 1.77, p=0.062) and early death (HR 1.76, p=0.071; figure 4F). These findings were confirmed with logrank tests (not adjusted for histology) for IL2RA (PFS, p=0.018) and CXCL2 (OS p=0.016; figure 4G).
We evaluated chemokine data relative to circulating immune cell composition from flow cytometry. Increasing baseline CXCL10, which was associated with superior survival, correlated significantly with decreasing circulating activated CD69+ and CD4 and CD8 T cells (p=0.00043, p=0.0032), central memory CD4 T cells (p=0.033), and naive CD8 T cells (p=0.0015; online supplemental table S7). Increasing baseline IL6, which was associated with inferior OS, correlated significantly with increasing TIM3+ CD4 and CD8 T cells (p=0.0072, p=0.036), increasing PD-L1+ CD8 T cells (p=0.032), and increasing CD14+ HLA-DR− monocytes (p=0.042). Increasing baseline IL2RA, also associated with inferior OS, was positively correlated with CD71+ CD8 T cells (p=0.032).
Gene expression profiling (NanoString) and response to pembrolizumab
NanoString results were obtained in 21 patients at baseline (19 EAC; 2 ESSC), of whom 4 had clinical benefit (2 PR, 2 SD) and 17 did not have clinical benefit; 12 were PD-L1-negative, 8 PD-L1-positive, and 1 patient had a missing PD-L1 status. Biopsies originated from primary tumors in 12 cases, from liver metastases in 5 cases, and other metastases in 4 cases (online supplemental figure S10A).
Individual differential gene expression analysis of baseline biopsies, controlled for histology and biopsy location, showed that gene expression of NLRC5 (log2 fold change=−1.60, adjusted p=0.032) and SSX1 (log2 fold change=−3.85, adjusted p=0.0031) were enriched in patients with clinical benefit (PR+SD) compared with those without, whereas expression of LBP (log2 fold change=5.99, adjusted p=0.011) and AMBP (log2 fold change=7.19, adjusted p=0.0027) were enriched in patients without clinical benefit compared with those with clinical benefit (online supplemental figure S10A and table S8).
Patients with PD-L1-positive tumors (CPS≥1) had significantly higher gene expression of LAG3 (log2 fold change=2.17, adjusted p=0.0049), indoleamine 2,3-dioxygenase 1 (IDO1; log2 fold change=2.22, adjusted p=0.041), CCL7 (log2 fold change=2.08, adjusted p=0.041), and SPP1 (log2 fold change=2.90, adjusted p=0.041; figure 5, online supplemental table S9). GSEA showed downregulation of genes related to cytotoxicity at baseline (unadjusted p=0.008; FDR q value=0.023) in patients without clinical benefit (PD) compared with those with clinical benefit (PR+SD; online supplemental figure S10C and table S10.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
NanoString gene expression analysis of tumor biopsies at baseline. Volcano plot of the differential gene expression analysis comparing PD-L1-negative (CPS<1, n=12) and PD-L1-positive (CPS≥1, n=8) patients, showing enrichment of genes associated withT cell suppression (LAG3, IDO1) in PD-L1-positive patients. CPS, Combined Positive Score; PD-L1, programmed death-ligand 1.
In addition, we had RNA from nine archival biopsies and three on-treatment biopsies of EAC patients taken 56 days after starting anti-PD-1 therapy. Unsupervised time series clustering was used to identify clusters of genes that exhibited a similar trend in expression over time. We first compared baseline and on-treatment biopsies. Among the genes generally increasing during treatment were two associated with TH1 CD4 T cell differentiation (STAT1, SPP1) and two with MHC class II antigen presentation (HLA-DPB1, HLA-DRA). By contrast, apoptosis-inducer TRAIL (TNFSF10) was generally decreasing (online supplemental figure S11A). We also compared expression profiles from available baseline biopsies with archival biopsies taken at time of original cancer diagnosis. This comparison showed 18 out of 20 immunity-related genes generally decreasing by the time that PD-1 therapy was initiated, among which were several associated with antigen presentation (HLA-C, CD74) and the IL2 receptor ɣ chain (IL2RG) which is constitutively expressed on T cells. In contrast, vascular endothelial growth factor A (VEGFA) was among the genes generally increasing prior to the start of trial (online supplemental figure S11B).
Discussion
In this phase II trial, pembrolizumab demonstrated modest efficacy in unselected, chemotherapy-refractory patients with esophageal cancer, yielding an ORR of 8% in 4 of 49 patients (95% CI 2.3 to 19.6%). This outcome is consistent with anti-PD-1 checkpoint inhibitor monotherapy in similar cohorts with advanced esophageal or gastric adenocarcinoma.3 13
In the last year, multiple trials of immunotherapy have demonstrated a survival benefit in front-line esophagogastric cancer.5 7 8 Although these trials expand the use of checkpoint inhibitors, their inclusion of chemotherapy confounds our ability to identify patients who are more likely to benefit from immunotherapy. While checkpoint inhibitors have shown more modest activity in refractory disease, monotherapy studies provide opportunities to define factors that modulate efficacy. In KEYNOTE-059, increased response to pembrolizumab in advanced gastric cancers was seen in PD-L1-positive tumors (CPS≥1) when compared with PD-L1-negative tumors.13 KEYNOTE-180 reported a response rate of 9.9% in esophageal cancer patients; however, responses were not enriched in the 47.9% of patients with PD-L1 CPS≥10, nor was there a difference in response by histology.3 In KEYNOTE-181, second-line pembrolizumab was associated with significant improvements in OS compared with chemotherapy for ESCC and for tumors with PD-L1 CPS≥10, irrespective of histology.4 Similar results were achieved for nivolumab in the ATTRACTION-2 and −3 studies.6 10
TMB has also been shown to correlate with response, for example in KEYNOTE-158.22 In that trial, second-line pembrolizumab in patients with advanced TMB-high solid tumors (≥10 mut/Mb by FoundationOne CDx assay) achieved an ORR of 29%. However, only 9.8% of esophageal cancers exceed this cut-off.23 Pembrolizumab is associated with response and survival benefit in MMR-deficient solid tumors, too.24–27 While these results show that histology, PD-L1 status, and TMB can assist in clinical decision making for esophagogastric cancers, they also demonstrate the need for more precise biomarkers.
There are several potential factors for the modest clinical results of our trial. First, the use of immunosuppressive therapies preceding pembrolizumab may reduce benefit.28 Second, pembrolizumab may be more effective in Asian than non-Asian populations. For example, Asian advanced gastric cancer patients appeared to have an enhanced survival benefit from first-line pembrolizumab versus chemotherapy compared with non-Asian patients in KEYNOTE-062.29 30 It has been shown that gastric adenocarcinomas from Asian and non-Asian patients exhibit a distinct tumor immunity signature related to T cell function.31 Third, the combination of immune checkpoint inhibitors with chemotherapy may enhance efficacy. Indeed, studies in murine gastric cancer models showed enhanced efficacy by adding chemotherapy to anti-PD-1 therapy, possibly due to the observed reduced myeloid-derived suppressor cell (MDSC) infiltration.32
To identify novel candidate biomarkers, we analyzed biopsies at several time points (archival, at baseline, on-treatment, and by the EOT), as well as circulating cytokines (at baseline and during treatment). However, analyses were hampered by the low number of responders. Despite clear trends toward improved responses and survival with PD-L1-positivity in baseline biopsies, these results were not statistically significant. Apart from our lack of power, reasons for this discrepancy with previous reports could be the difference in antibody, method, and thresholds,33 as well as intracase-heterogeneity, intraobserver and interobserver variability, intersite variability and limited robustness of the test.
We also evaluated another common predictor of immunotherapy response, TMB. No significant associations with radiological response or survival were identified, although a TMB ≥10 mut/Mb did show a trend toward improved OS. With 25.9% of patients having a TMB ≥10 mut/Mb, this proportion was significantly higher than the 9.8% described previously.23 However, our results are derived from an institutional sequencing assay (OncoPanel) rather than the FoundationOne CDx test.34 Both OncoPanel and FoundationOne CDx estimate TMB by somatic-only targeted sequencing of a panel of genes, but they differ in multiple aspects, such as specific gene content, overall panel size, sensitivity, and variant filtration strategy; potentially leading to inconsistencies.
We next evaluated other predictors of response. One possible contributing factor to the scarcity in observed responses could be a terminally exhausted tumor-immune microenvironment, especially considering the multiple lines of chemo(radio)therapy preceding pembrolizumab treatment. Although we did not find a clear association between exhaustion and non-response, we did find associations between PD-L1-positivity by IHC and the expression of genes associated with T cell suppression, such as LAG3 and IDO1. These results raise the potential for combined LAG3/PD-1 or IDO1/PD-1 inhibition in this population. Combined LAG3/PD-1 blockade has been shown to enhance the anti-tumor immune response in both preclinical and clinical studies.35 36 Beyond these targets, our time series clustering analysis revealed decreasing expression of genes related to antigen presentation and T cells combined with increasing expression of VEGFA during pembrolizumab treatment, which could make a case for earlier immunotherapy or immunotherapy combined with anti-VEGF therapy.
The flow cytometry analysis was unexpectedly impacted by biopsy location, which acted as a third variable. After correction for this confounder, a granulocytic tumor infiltrate at baseline was consistently associated with a superior outcome, and CD4 T cell PD-1 expression with inferior outcome. Intriguingly, despite these associations from the tumor biopsies, our analysis of the ratio of circulating NLR found a high NLR to be associated with poor responses, consistent with prior data.37 38 PD-L1+ neutrophils have been associated with disease progression in gastric cancer, raising questions about how PD-1 blockade impacts neutrophils and their interaction with other immune populations within the tumor microenvironment.39
Notable results came from the analysis of circulating cytokines. At baseline, patients with increased plasma levels of CXCL10 had a significantly decreased risk of progression and death and trended toward superior radiological response. CXCL10, secreted by macrophages in response to IFN-γ, is known to attract TH1 cells, cytotoxic T cells, and NK (T) cells.40 An association between intratumoral CXCL10 and an increase in response to dual PD-1/CTLA-4 blockade has been described in melanoma, with CXCL10 correlating with CD8 T cell infiltration.41 Importantly, efficacy was dependent on CXCL10 receptor CXCR3, as CXCR3 neutralization abrogated CD8 T cell infiltration and efficacy of immune checkpoint blockade.41
Plasma IL2RA, IL1RN, and IL6 concentrations at baseline were associated with short survival. The α chain of the IL2 receptor (IL2RA), also known as CD25, is upregulated after activation of naïve T cells and constitutively expressed on Tregs, forming, together with the β and γ chain, a high-affinity receptor for IL2.42 Cleaved, circulating IL2RA correlates with T cell IL2RA expression and has been associated with a poor prognosis in several tumor types including ESCC.43 Moreover, soluble IL2RA is associated with suppression of effector T cells,42 and inhibited response to CTLA4 blockade in melanoma patients, presumably through the parallel proportional increase in Tregs.44 IL-6 has been shown to promote epithelial-to-mesenchymal transition, cancer-associated fibroblast activation, and MDSC proliferation.45–47 Similar to our result, a high circulating IL-6 concentration has been associated with resistance to immune checkpoint inhibition before.48
Conclusions
In conclusion, although efficacy of pembrolizumab in relapsed/refractory esophageal cancer was limited, our correlative studies raise key hypotheses regarding candidate biomarkers such as CXCL10 to guide therapy. Moreover, PD-L1 expression was associated with a terminally exhausted tumor-immune microenvironment, suggesting the greater potential efficacy of anti-PD-1 antibodies in earlier lines of therapy in esophageal cancer.
Data availability statement
Data are available on reasonable request via corresponding author.
Ethics statements
Patient consent for publication
Ethics approval
This study was approved by the Harvard Cancer Center Institutional Review Board and was carried out in accordance with the Declaration of Helsinki.
Acknowledgments
The authors would like to acknowledge the DFCI Oncology Data Retrieval System (OncDRS) for the aggregation, management, and delivery of the clinical and operational research data used in this project. The content is solely the responsibility of the authors.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Twitter @labrat3
LKdK and AKP contributed equally.
PCE and AJB contributed equally.
Contributors Data collection: JA, MU, NR, FGA, NJM, JMC, DAR, JWC, BF, LKB, MEC, AJR, MGJ, PHL, MJN, and MS.Study conception and design: AJB and PCE. Analysis and interpretation of results: LKdK, AKP, SD, HZ, PCE, and AJB. Draft manuscript preparation: LKdK, AKP, SD, PCE, and AJB.
Funding This trial and correlative research were supported by Merck.
Competing interests AJB received funding support from Bayer, Merck and Novartis. JMC received research funding to his institution from Abbvie, Merus, Roche, and Bristol Myers Squibb, received research funding from Merck, Astrazeneca, Esperas Pharma, and Tesaro, received consulting fees from Bristol Myers Squibb, and received travel funding from Bristol Myers Squibb. PCE has consulted for and has received honoraria from ALX Oncology, Arcus Bioscience, Astellas, Astra-Zeneca, Blueprint Medicines, Bristol-Myers Squibb, Celgene, Daiichi-Sankyo, Five Prime, Ideaya, Istari, Legend, Lilly, Loxo, Merck, Ono, Taiho, Takeda, Turning Point Therapeutics, Xencor and Zymeworks.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.