Article Text

Abstract
Background Chimeric antigen receptor (CAR) T cells have shown considerable promise as a personalized cellular immunotherapy against B cell malignancies. However, the complex and lengthy manufacturing processes involved in generating CAR T cell products ex vivo result in substantial production time delays and high costs. Furthermore, ex vivo expansion of T cells promotes cell differentiation that reduces their in vivo replicative capacity and longevity.
Methods Here, to overcome these limitations, CAR-T cells are engineered directly in vivo by administering a lentivirus expressing a mutant Sindbis envelope, coupled with a bispecific antibody binder that redirects the virus to CD3+ human T cells.
Results This redirected lentiviral system offers exceptional specificity and efficiency; a single dose of the virus delivered to immunodeficient mice engrafted with human peripheral blood mononuclear cells generates CD19-specific CAR-T cells that markedly control the growth of an aggressive pre-established xenograft B cell tumor.
Conclusions These findings underscore in vivo engineering of CAR-T cells as a promising approach for personalized cancer immunotherapy.
- immunotherapy
- cell engineering
- receptors
- chimeric antigen
- antibody specificity
Data availability statement
Data are available on reasonable request. All data relevant to the study are included in the article or uploaded as supplementary information.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Background
Adoptive transfer of CD19-specific chimeric antigen receptor (CAR)-T cells has demonstrated considerable success for the treatment of B cell malignancies in patients with relapsed or refractory diseases,1 2 providing the basis for at least three cell therapies (Yescarta for non-Hodgkin’s lymphoma, Kymriah for acute lymphoblastic leukemia, and Tecartus for mantel cell lymphoma) approved by the US Food and Drug Administration (FDA) to date.3 However, the generation of CAR-T cell products in all instances involves time consuming and complex manufacturing processes that delay the immediate availability of these cellular therapies for patients with aggressive disease and also lead to exorbitant costs.4–8 Furthermore, activation, genetic manipulation, and ex vivo expansion of CAR-T cells inevitably leads to significant differentiation of T cells, which likely reduce their self-renewal capacity on adoptive transfer back into patients and consequently limiting the overall efficacy.9–15
Direct in vivo engineering of CAR-T cells, based on transducing T cells circulating in the peripheral blood with viral vectors, offers the potential to bypass the need for ex vivo manufacturing of patient-derived T cells entirely. Such viral vectors can serve as an off-the-shelf therapy immediately available to be infused in patients with aggressive disease, greatly expediting the therapy and markedly reducing the costs. Lentiviral (LV) vectors are already used to engineer CAR-T cells ex vivo in FDA-approved products, underscoring their ability to safely integrate the CAR transgene.3 16 17 Unfortunately, conventional LV vectors, such as those pseudotyped with VSV-G, exhibit extremely broad tropisms; the lack of cell target specificity prohibits their direct use in vivo to target T cells.18–20 Considerable efforts over the past two decades have led to engineering novel envelope glycoprotein pseudotypes, thereby establishing next-generation LVs retargeted to specific cell types via receptor binding.21 To date, the most successful strategies involve several common features: (1) ablation of wildtype cell surface receptor binding, (2) display of an additional targeting domain for binding a new cell type either covalently or non-covalently, and (3) preservation of membrane fusion activity for entry into cells.22
Paramyxovirus and alphavirus glycoproteins have received the most attention for retargeting LVs given their completely separate protein domains for receptor binding and membrane fusion. Buchholz and colleagues have pioneered much of the paramyxovirus glycoprotein engineering work using measles virus and Nipah virus as their preferred pseudotypes.22 Beginning with measles virus glycoproteins, the Buchholz team developed truncated versions of the hemagglutinin and fusion proteins that could effectively pseudotype LVs while codisplaying a covalently attached targeting ligand on the C-terminus of hemagglutinin.23 Specific mutations were also applied to reduce native measles virus receptor tropism, enabling an efficient LV system for transducing unstimulated T lymphocytes directly in vivo.24 25 More recently, Buchholz and colleagues applied a similar approach to Nipah virus glycoproteins, whereby a set of mutations was discovered to diminish native receptor binding and a specific site was established for adding surface exposed targeting ligands, such as single-chain antibodies or DARPins, for retargeting Nipah pseudotyped LVs to specific cell types.26 Using their Nipah LVs retargeted to CD3, CD4, and CD8, they have been the only group to date to generate functional CAR-T cells directly in vivo for cancer immunotherapy by viral vector delivery.22 27–31
Chen and Wang pioneered much of the early alphavirus pseudotype development for LVs using mutant and novel versions of Sindbis virus glycoproteins. Chen and colleagues developed novel Sindbis E2 glycoprotein domains, responsible for receptor binding, by applying site-specific mutations to ablate native receptor tropism and direct addition of adapter binding molecules, such as the ZZ domain of protein A, avidin, biotin-adaptor peptide, and integrin-targeting peptide.32–39 By mixing together LV and targeting ligand containing the adapter binding pair, such as Fc of antibody for ZZ domain, Sindbis-based LV could be redirected to a variety of receptors and cell types. A more recent extension of this strategy with Sindbis E2 was demonstrated using SpyTag and a disulfide bond-forming pair as adapter binding molecules for more permanent covalent incorporation of targeting ligand on viral surface.40 41 Wang and colleagues adopted a similar strategy with an HA tag in their E2 domain or direct coexpression of full IgG antibody molecules on viral surface while focusing much of their efforts on engineering enhanced Sindbis E1 fusogen variants.42–46 Although these various groups have demonstrated significant enhancement in transduction of immune and tumor cell types both in vitro and in vivo with their Sindbis pseudotyped LV systems, no one has yet to test the ability of a Sindbis LV system to transduce circulating primary T cells or generate CAR-T cells directly in vivo for cancer immunotherapy.
The current lengthy manufacturing processes to generate CAR-T cell products and increasing development of targeted gene delivery viral vectors motivated us to develop a LV-based gene transfer system with considerable specificity and efficiency for T cell targeting in vivo. To minimize transduction of non-target cells, we incorporated a mutated Sindbis pseudotyped lentiviral vector (SINV-LV), with mutations to the E2 glycoprotein that abrogate its native tropism to human cells (online supplemental figure S1A). To redirect the mutant SINV-LV to T cells, which lacks, any specific cell tropism or additional adapter molecule residues in the E2 receptor binding domain, we engineered a separate bispecific binder that can bind: (1) the mutant E2 glycoprotein on SINV-LV and (2) CD3, a ubiquitous coreceptor on all T cells (figure 1A). Our novel LV system thus comprises simply mixing SINV-LV with bispecific binders for non-covalent attachment and redirection to target T cells in vivo.
Supplemental material

Bispecific antibody binder enhances specificity and transduction efficiency of the mutant lentivirus. (A) Schematic of bispecific antibody in tandem Fab format (tFab) used for redirecting mutant Sindbis lentiviral vector (SINV-LV) to CD3+ T cells for targeted transduction. Orthogonal amino acid mutation sets are shown for constant and variable domains of each Fab to ensure correct pairing of heavy and light chains. (B) Binding affinity of control IgG (blue) and tFab (red) to human CD3ε analyzed by ELISA (n=2). (C) Binding affinity of tFab (red) to mutant Sindbis E2 glycoprotein analyzed by ELISA (n=2). (D) SINV-GFP transduction to CD3+ T cells is enhanced by addition of the tFab molecule in a concentration-dependent manner. At all tested concentrations, excess anti-CD3 IgG of the same clone blocked tFab-mediated SINV-GFP transduction, suggesting that transduction was specifically mediated via tFab binding CD3 in T cells. Data represent results of three independent experiments performed in triplicate (MOI=25) and analyzed using a two-way ANOVA with a post hoc Tukey’s test for multiple comparisons (****p<0.0001). (E) addition of tFab to mutated Sindbis pseudotyped lentiviral vector encoding green fluorescent protein (SINV-GFP) redirects the mutant lentiviral vector to CD3+ T cells in a mixed culture (CD3+ and CD3− cells together) demonstrating the specificity towards CD3+ T cells. (F) in mixed cultures of CD3+ (Sup-T1) and CD3− (BV-173) cells, SINV-GFP plus tFab demonstrated substantial selectivity towards CD3+ T cells as indicated by the increase in percentage of GFP+ cells. data represent results of three independent experiments performed in triplicate (MOI=25; (tFab)=30 nM) and analyzed using a two-way ANOVA with a post hoc Tukey’s test for multiple comparisons (****p<0.0001). ANOVA, analysis of variance; MOI, multiplicity of infection.
Methods
Cell lines and primary cells
B cell lymphoma tumor cell lines (BV-173 and Daudi) and T cell lymphoma tumor cells (Sup-T1) were purchased from ATCC and cultured in RPMI-1640 medium (Gibco) supplemented with 10% HyClone FBS (GE Healthcare), penicillin (100 U/mL; Gibco), and streptomycin (100 U/mL; Gibco). All cells were maintained at 37 °C and 5% CO2 for growth. All cell lines are regularly tested for Mycoplasma, and the identity of each cell line was validated via flow cytometry for relevant surface markers and also monitored for morphological drift in culture. Cell lines were maintained in culture no longer than 30 days and then replaced with an earlier passage of cells thawed from cryopreservation. BV-173 cells were transduced with a gamma retroviral vector encoding the Firefly-Luciferase (FFluc) gene. Sup-T1 cells (CD3+TCR−) were engineered with the TCR specific for Tyrosinase368-376 (obtained from Frankel and colleagues)47 in order for Sup-T1 cells to express endogenous CD3 on the cell surface.48 49 Peripheral blood mononuclear cells (PBMCs) were isolated from fresh buffy coats (Gulf Coast Regional Blood Center) using Lymphoprep medium (Accurate Chemical and Scientific Corporation). PBMCs were then activated for 48 hours in bioreactors with soluble anti-CD3 (200 ng/mL; Miltenyi Biotec) and anti-CD28 (200 ng/mL; BD Biosciences) mAbs. Activated PBMCs were washed with PBS and allowed to rest at 37°C and 5% CO2 in growth culture medium for at least 24 hours prior to LV transduction or in vivo studies. Primary T cells were activated, cultured, and transduced in complete medium consisting of 45% Click’s Medium (Irvine Scientific), 45% RPMI-1640 (Gibco), 10% HyClone FBS (GE Healthcare), 2 mmol/L GlutaMax (Gibco), penicillin (100 U/mL; Gibco), and streptomycin (100 U/mL; Gibco) with 10 ng/mL IL-7 and 5 ng/mL IL-15 (PeproTech).
In vitro transduction assays
The CD3+ Sup-T1 tumor cell line was transduced with SINV-GFP in the presence of increasing concentrations of tFab to demonstrate BsAb-mediated enhanced transduction of target cells. Sup-T1 cells were seeded in sterile 96-well tissue culture treated plates (Corning Costar Cat # 3599) at 1×105 cells/well. SINV-GFP at a multiplicity of infection (MOI) of 25, based on qPCR titering, was premixed with various concentrations of tFab in serum-containing growth culture medium for 1 hour at room temperature to allow tFab to bind onto the surface of SINV-GFP particles before directly adding this transduction mixture to the plated cells. Each tFab concentration tested (1, 10, 30, and 50 nM) is reported as the final concentration of the tFab once diluted and added to cells for transduction in 96-well plates. To confirm enhanced transduction was dependent on the CD3 specificity of the tFab, excess IgG1α-CD3 (300 nM) was added to replicate sample wells at each tFab concentration to competitively block binding of CD3 as entry receptor for targeted transduction with SINV-GFP plus tFab. After 24 hours of transduction at 37 °C and 5% CO2, cells were washed twice with cold growth culture medium using low-speed plate centrifugation (300 × g) to remove residual antibody and LV prior to resuspension in fresh growth culture medium. Cells were allowed to grow and express GFP transgene for 72 hours at 37 °C and 5% CO2 prior to washing them into PBS and analyzing their GFP expression via flow cytometry using an Attune NxT flow cytometer with plate autosampler (Applied Biosystems).
A similar transduction assay with CD3+ Sup-T1 and CD3− BV-173 tumor cell lines was established to demonstrate specificity and selectivity of SINV-GFP plus tFab transduction to CD3+ target cells. Sup-T1 and BV-173 cells were seeded together at a 1:1 ratio in each well of sterile 96-well tissue culture treated plates (Corning Costar Cat # 3599) at 1×105 total cells/well. SINV-GFP at a MOI of 25, based on qPCR titering, was premixed with 30 nM final concentration of tFab in serum-containing growth culture medium for 1 hour at room temperature before directly adding this transduction mixture to the plated co-culturing cells. A control transduction of SINV-GFP at MOI 25 without addition of tFab was also dosed to co-culturing cells. After 24 hours of transduction at 37 °C and 5% CO2, cells were washed twice with cold growth culture medium using low-speed plate centrifugation (300 × g) to remove residual antibody and LV prior to resuspension in fresh growth culture medium. Cells were allowed to grow and express GFP transgene for 72 hours at 37 °C and 5% CO2 prior to washing them into PBS for surface marker phenotype staining with anti-CD3 APC (BD Cat # 340440) and anti-CD19 PE (BD Cat # 340364). Phenotypic antibody staining was allowed to proceed for 30 min at 4°C followed by two PBS washes of samples to remove unbound antibodies. Washed cells were resuspended into PBS and analyzed for their GFP expression via flow cytometry using an Attune NxT flow cytometer with plate autosampler (Applied Biosystems).
Activated primary human PBMCs were transduced with SINV-CAR at a MOI of 10, based on qPCR for vector titer and total number of PBMCs, with and without addition of tFab to demonstrate functional CAR expression and subsequent cytotoxic activity of CAR-T cells in vitro. In brief, 2.5×105 activated PBMCs were transduced in 250 µL final volume per well of growth culture medium supplemented with IL-7 and IL-15 cytokines in 48-well tissue culture treated plates. SINV-CAR at a MOI of 10 (ie, 2.5×106 infectious units (IUs) based on qPCR titering) was premixed with 50 nM final concentration of tFab in serum-containing growth culture medium for 1 hour at room temperature before directly adding this transduction mixture to the plated PBMCs. SINV-CAR at MOI 10 was also dosed directly without addition of tFab for targeting along with other non-transduced control PBMC sample wells. After 6 hours of transduction at 37 °C and 5% CO2, PBMC samples were washed twice with cold growth culture medium to remove residual antibody and LV prior to resuspension in fresh growth culture medium and transfer to a new, sterile 24-well tissue culture treated plate for 84 hours of growth and CAR expression at 37 °C and 5% CO2. A portion of each sample well was collected and washed into PBS for phenotypic surface marker staining by a panel of antibodies and subsequent CAR expression analysis using an LSR Fortessa flow cytometer (BD Biosciences). The remaining PBMCs in each sample well were resuspended and counted by trypan blue dye exclusion for subsequent plating with CD19+ tumor B cells to demonstrate CAR functionality by a coculture cytotoxicity assay described in more details below.
In vitro coculture tumor cytotoxicity assay
Transduced and non-transduced control PBMCs (1.5×105 cells/well or 3×105 cells/well) were cocultured with tumor cell lines (BV-173 or Daudi, 1.5×105 cells/well in 24-well plates), in complete medium, in the absence of cytokines (E:T=1:1 or E:T=2:1). The effector-to-target (E:T) ratio was not normalized to CAR+ transduced T cells but was instead based on the total number of T cells in culture, including both transduced and non-transduced fractions taken together without separation. After 4–5 days of culture, cells were harvested and stained with CD3 (APC-H7, clone SK7 from BD Biosciences) and CD19 (FITC, clone SJ25C1 from BD Biosciences) monoclonal Abs to detect T cells and tumor cells, respectively. Residual tumor cells in culture were enumerated by flow cytometry. Culture supernatants were harvested after 24 or 48 hours of culture and IFN-γ and IL-2 measured using the DuoSet Human IFN-γ and DuoSet Human IL-2 ELISA kits respectively (R&D Systems). Data acquisition was performed on a Synergy2 microplate reader (BioTek) using the Gen5 software.
Tumor mouse model for testing efficacy of in vivo generated CAR-T cells
Female NSG mice (7–9 weeks of age, obtained from the University of North Carolina Animal Services Core) were used to establish our chronic myeloid leukemia xenograft tumor mouse model. Mice were irradiated at a low dose (100 rad) by a cesium irradiator on day −6 of the study prior to any cell engraftments. The following day (day −5), 5×105 FFLuc BV-173 tumor B cells were injected in 150 µL sterile PBS via intravenous tail vein. After allowing 5 days for tumor cell engraftment, 5×106 activated PBMCs were injected on day 0 in 150 µL sterile PBS via intravenous tail vein. Thirty min after infusing the PBMCs, mice were randomly separated into two different treatment groups: (1) SINV-CAR without tFab or (2) SINV-CAR with premixed tFab. In both groups, SINV-CAR was dosed at 2.5×107 IUs, based on qPCR, in 150 µL sterile PBS per mouse via intravenous tail vein injection. This dosage equated to 5×1010 viral particles per mouse, based on absolute particle counts of SINV-CAR using NanoSight NS500 (Malvern Panalytical) nanoparticle tracking analysis. tFab (5 µg/mouse) was premixed with SINV-CAR for 1 hour at room temperature in 150 µL sterile PBS prior to intravenous injections. B cell tumor growth was monitored weekly by bioluminescent imaging (BLI; total flux, photons/s) using an Ami HT optical imaging system (Spectral Instruments Imaging). Peripheral blood samples were taken weekly from mice via the submandibular route. Peripheral blood was subjected to red blood cell lysis followed by antibody staining and flow cytometry to assess number of human T cells (CD3+) and tumor B cells (CD19+) in circulation. Mice were sacrificed according to UNC guidelines for either tumor growth or occurrence of signs of discomfort, such as tumor-mediated paralysis. On sacrifice, peripheral blood was collected from cardiac puncture of the heart, and spleens were measured and weighed prior to smashing over cell strainers into single cell suspensions. Blood and spleen were subjected to red blood cell lysis, antibody staining, and flow cytometry using an LSR Fortessa flow cytometer (BD Biosciences) to detect and quantify CAR+ T cells and CD19+ tumor B cells in isolated tissues. Antibodies used for phenotypic staining of in vivo samples included CD3 (APC-H7, clone SK7), CD8 (Alexa Fluor 700, clone RPA-T8), CD45 (APC, clone 2D1) and CD19 (FITC, clone SJ25C1) along with CountBright absolute counting beads (Invitrogen). All flow cytometry data analysis was performed with FlowJo V.10 software.
Statistical analysis
All data are presented as mean±SD. GraphPad Prism V.8 software was used to generate graphs and perform statistical analyses. Either Student’s t-test or two-way analysis of variance (ANOVA) was used to determine statistically significant differences between treatment groups. Either a post hoc Tukey’s test or Bonferroni correction was performed to correct for multiple comparisons after two-way ANOVA. Survival analysis was performed using the Kaplan-Meier method with a log-rank test to determine statistical significance. All p values less than 0.05 were considered statistically significant.
Results
As proof of concept, we engineered bispecific binders in a tandem Fab format (tFab), comprised of two distinct Fab domains linked via a glycine-serine flexible linker and lacking the Fc antibody domain (figure 1A; online supplemental figure S1B). By applying different sets of orthogonal amino acid mutations50 to the two Fab domains (anti-CD3 and anti-E2), we were able to overcome traditional heavy/light chain mispairing51 and produce a pure population of bispecific tFab binders with properly paired Fabs by simple immobilized metal affinity chromatography purification (online supplemental figure S1C). We performed several immunoassays, including ELISAs (figure 1B,C), dot blots (online supplemental figure S1D), and immunogold labeling with transmission electron microscopy (online supplemental figure S1E), to characterize the specificity and affinity of the tFab binding to both human CD3ε and mutant Sindbis E2 glycoprotein. The tFab bound to both CD3ε and E2 at low nanomolar affinities (KD=19.7 nM and 4.7 nM, respectively) as assessed by ELISA, whereas control anti-CD3 IgG of the same Fab clone (IgG1α-CD3) bound only to CD3ε. Anti-CD3 IgG possessed higher binding affinity (KD=1.4 nM), which is likely a direct consequence of the dimeric nature of two Fabs per IgG molecule. Using different lentivirus pseudotypes including SINV, VSV-G, and measles virus in dot blot experiments, we confirmed tFab binds specifically to only SINV-LV.
To evaluate the capacity of the SINV/tFab platform in targeting human T cells, we generated SINV-LV encoding an eGFP fluorescent reporter transgene (denoted as SINV-GFP), mixed with different amounts of tFab, and quantified the level of induced eGFP expression in a CD3+ human cell line (Sup-T1). We found a tFab dose-dependent transduction enhancement that saturated at ~50 nM concentration of tFab (figure 1D). Without addition of the tFab, the transduction efficiency of SINV-GFP alone was less than 1%, whereas 50 nM of tFab enabled transduction of >50% of the cells. The increased transduction was a direct consequence of the combination of SINV-GFP and tFab redirection, as demonstrated by competitive inhibition in the presence of excess amounts of anti-CD3 IgG1 (300 nM) (figure 1D). To further validate the specificity of viral targeting, we tested SINV-GFP/tFab in coculture experiments mixing CD3+ (Sup-T1) and CD3− (BV-173) cells. As expected, without addition of the tFab, SINV-GFP showed negligible transduction of either CD3+ or CD3− cells (figure 1E). In contrast, SINV-GFP/tFab showed a ~25 fold enhanced transduction of CD3+ versus CD3− cells (figure 1F). This is consistent with previous work from our group comparing the transduction efficiency of wildtype versus mutant SINV-GFP in the presence or absence of tFab against tumor cell lines in vitro, where we found mutations to the Sindbis glycoprotein significantly reduced non-specific, background transduction, while addition of tFab restored efficient transduction, presumably by enhancing binding and internalization via receptor-specific interactions.52
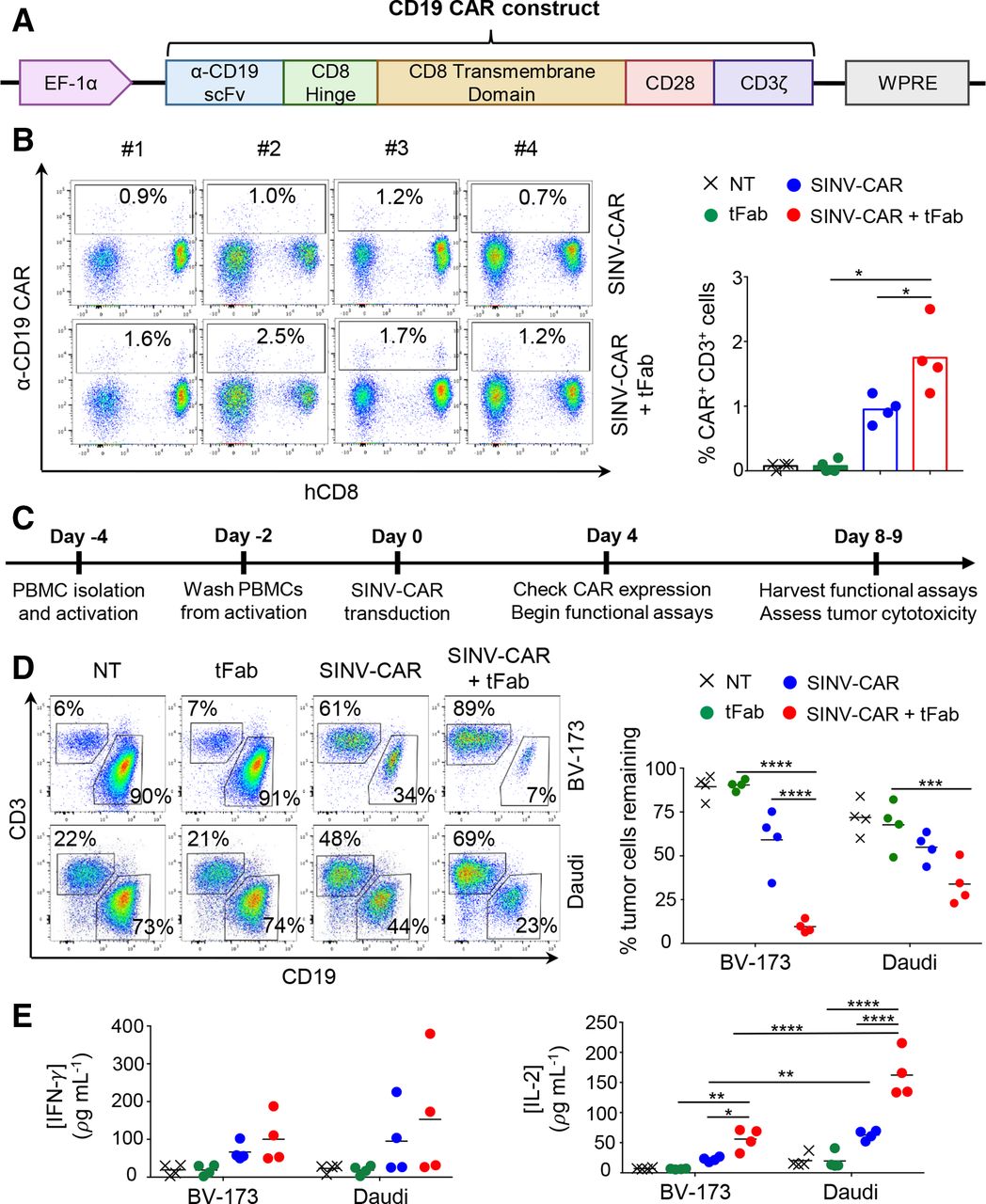
We next cloned a second-generation CD19-specific CAR encoding the CD28 costimulatory endodomain into our SINV-LV (SINV-CAR; figure 2A) and tested the transduction efficiency in primary human T cells. At relatively moderate multiplicities of infection (MOI=10) without spinoculation or transduction enhancers, the SINV-CAR/tFab yielded ~1.2%–2.5% CAR-T cells, including both CD4+ and CD8+ cells, which was ~2 fold higher fraction than the SINV-CAR alone (p=0.0437; figure 2B; online supplemental figure S2C). To determine if CAR-T cells were functionally active, we developed an in vitro coculture assay to measure CAR-T cell cytotoxicity and cytokine secretion in presence of CD19+ tumor cells (BV-173) (figure 2C). Even at very low effector-to-target (E:T) cell ratios (~1–5 CAR+ T cells per 100 tumor cells; see Methods section for description of calculations), CAR-T cells generated from SINV-CAR/tFab eliminated far more (up to ~6 fold) tumor cells within 4 days than CAR-T cells generated from SINV-CAR alone (figure 2D; online supplemental figure S2D). A similar trend was observed using another CD19+ tumor cell line (Daudi). The observed cytotoxic effect was consistent with the detection of IFN-γ and IL-2 in the culture medium collected within 24–48 hours of coculturing (figure 2E; online supplemental figure S2E). Although we noticed that the production of IFN-γ and IL-2 from CAR-T cells was higher when cocultured with the Daudi cell line, this is likely due to the expression of costimulatory markers (CD80 and CD86) from Daudi, which are not present in the BV-173 cell line (online supplemental figure S2F).

T cells transduced with SINV-CAR in combination with tFab express functional CD19.CAR and eliminate tumor B cells in vitro. (A) Schematic representation of the CD19.CAR cassette under the control of the EF-1α promoter and WPRE post-transcriptional regulatory molecule. (B) Flow cytometry plots (left) and summary (right) showing CAR expression in T cells transduced with SINV-CAR or SINV-CAR plus tFab. Non-transduced (NT) and tFab alone samples of T cells are provided as negative controls (n=4, mean shown). *P=0.0437 SINV-CAR plus tFab versus SINV-CAR; *p=0.0100 SINV-CAR plus tFab versus tFab with paired t-test. (C) Experimental schema for the transduction and subsequent coculturing of CAR-T cells with tumor B cells in vitro. (D) Representative flow plots (left panel) and summary (right panel) of the quantification of residual CD19+ tumor B cells (BV-173 and Daudi cell lines) remaining after coculturing with either NT, tFab, SINV-CAR, or SINV-CAR plus tFab treated T cells (E:T=2:1). All cells were collected after 4 or 5 days (BV-173 and Daudi, respectively) and stained with CD3 and CD19 mAbs to identify T cells and tumor cells, respectively, by flow cytometry (n=4, mean shown). ***P=0.0004, ****p<0.0001, two-way ANOVA. (E) Quantification of IFN (left panel) and IL-2 (right panel) cytokines in supernatant collected after 24 hours of coculturing NT, tFab, SINV-CAR, or SINV-CAR plus tFab treated T cells with tumor cell lines (E:T=2:1) (n=4, mean shown). *P=0.0393, **p<0.0087, ****p<0.0001, two-way ANOVA. ANOVA, analysis of variance; CAR, chimeric antigen receptor; tFab, tandem Fab format.
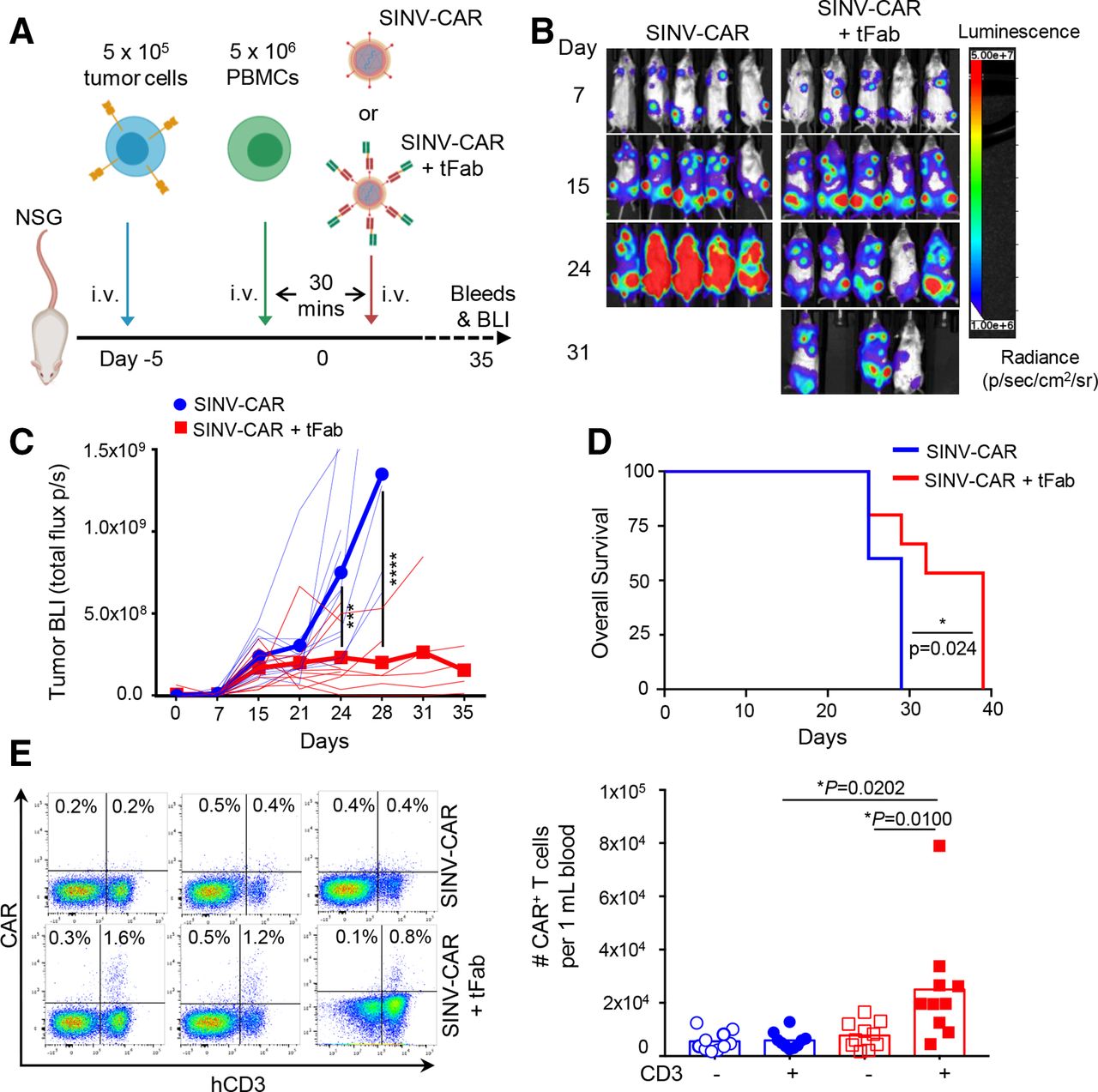
Based on these findings, we next evaluated the efficacy of the SINV-CAR/tFab vector system in a xenograft mouse model (figure 3A). CD19+ BV-173 cells, engineered to express firefly luciferase as imaging reporter to allow monitoring of tumor growth in vivo, were engrafted into NSG mice. Five days later, activated human PBMCs were injected intravenously into the animals, followed by SINV-CAR with or without tFab 30 min later. By day 24, following SINV-CAR injection, mice treated with SINV-CAR/tFab displayed significantly reduced tumor bioluminescence (BLI) compared with control mice infused with SINV-CAR alone (figure 3B,C; online supplemental figure S3A). Control mice began developing hind-limb paralysis due to tumor localization in the spine, which necessitated sacrificing all animals at a much earlier time point (10 days earlier according to median survival times) than mice treated with SINV-CAR/tFab (figure 3D). We attempted to quantify CAR+ and CD3+ human T cells circulating in the peripheral blood. While only very small numbers of CAR+ and CD3+ human T cells were detected at early time points (online supplemental figure S3B,C), we found a substantial quantity of CAR+CD3+ human T cells in the peripheral blood of all mice treated with SINV-CAR/tFab at the time of sacrifice, while the CD3− cells did not express the CAR (figure 3E; online supplemental figure S3D). These greater levels of CAR+CD3+ human T cells were attributed to greater T cell transduction by SINV-CAR/tFab versus SINV-CAR and not attributed to simply greater total number of T cells in the peripheral blood, as total T cell counts were similar between both treatment groups (online supplemental figure S3E).

SINV-CAR targeted with tFab generates functional CAR-T cells directly in vivo. (A) Experimental schema of the mouse model. Following a lose dose of irradiation (100 rad), mice were injected with FFLuc BV-173 (5×105 cells) intravenously. Five days later, mice were injected intravenously with 5×106 activated PBMCs followed by either SINV-CAR alone or SINV-CAR plus tFab 30 min later. (B) Representative tumor bioluminescence (BLI) (color scale: min=1×106; max=5×107) for mice treated according to scheme from panel A. (C) BLI kinetics for all mice treated according to scheme from panel A. Lighter lines represent individual mice, while bolded lines represent the means for the treatment groups. Summary of two independent experiments (n=10 mice for each condition). ***P=0.0002; ****p<0.0001, two-way ANOVA with Bonferroni correction. (D) Kaplan-Meier survival curve for all mice (n=10 mice per condition) treated according to scheme from panel A. *P=0.0242, log-rank test. (E) Representative flow plots (left panel) and quantification (right panel) of CAR-T cells in the CD3− (open symbols) and CD3+ (filled symbols) populations (gated on live cells) in the peripheral blood at the time of euthanasia (n=10 each condition, mean shown). *P=0.0100 and *p=0.0202, unpaired t-test. ANOVA, analysis of variance; CAR, chimeric antigen receptor; PBMCs, peripheral blood mononuclear cells.
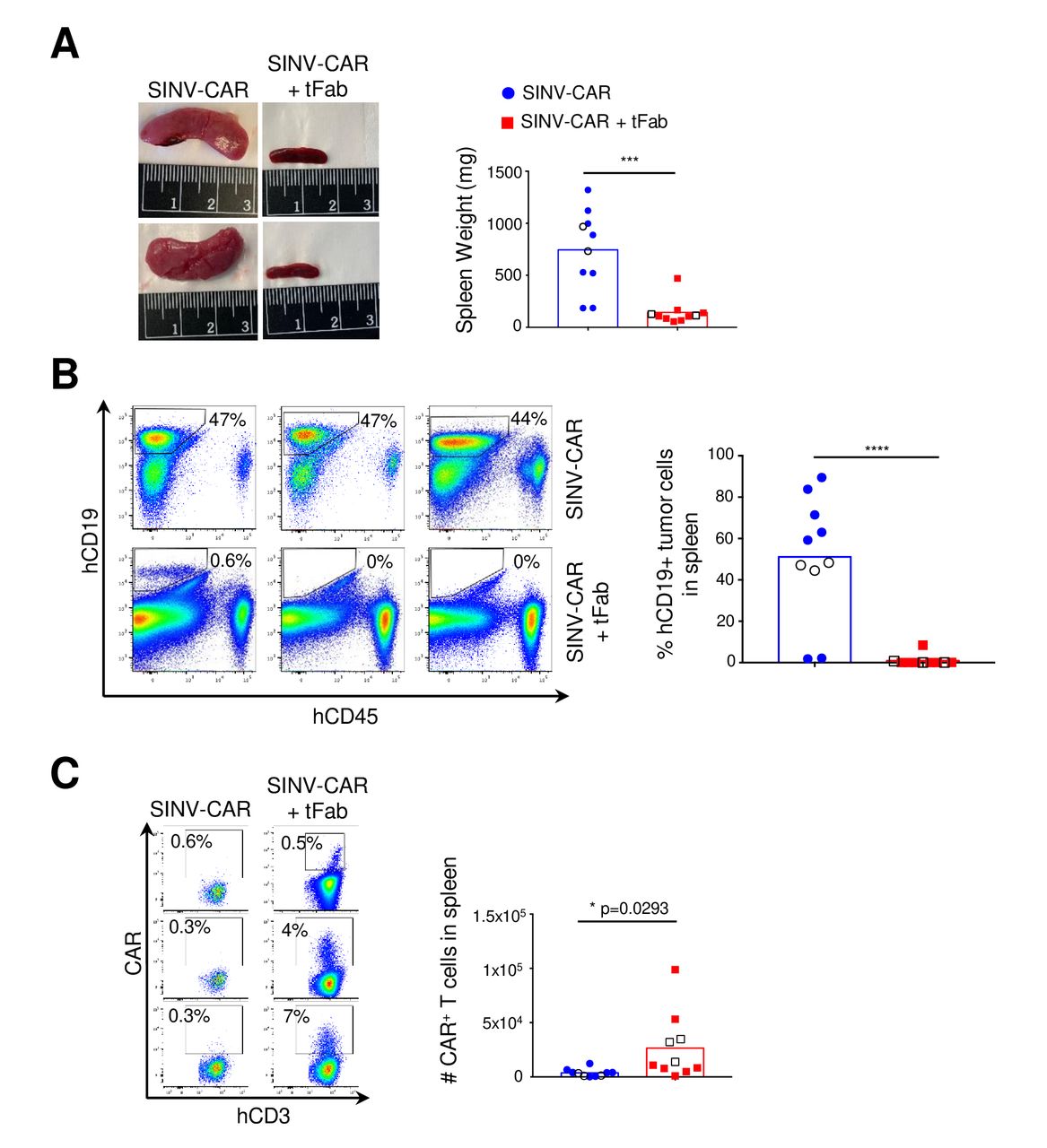
The aggressive BV-173 B cell lymphoma model appears to result in accumulation and spread of tumor cells in the spleen: at the time of sacrifice, we discovered very enlarged spleens in mice treated with SINV-CAR alone (figure 4A; online supplemental figure S4A), with a very high proportion of BV-173 tumor cells in the enlarged spleens (>50% of the total cell populations on average) (figure 4B). In contrast, the overall size and weight of spleens from mice treated with SINV-CAR/tFab appeared comparable with those from normal, healthy mice. Analysis of the cellular composition of the spleens revealed higher infiltration of CAR-T cells in mice treated with SINV-CAR/tFab (figure 4C; online supplemental figure S4C,D), which correlated with much lower numbers of CD19+ BV-173 tumor cells (<1% on average) (figure 4B). Taken together, these data suggest that generating even a relatively small number of CAR-T cells directly in vivo is sufficient to enable tumor suppression in lymphoid organs and significantly prolong the median survival time of tumor-bearing mice.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
SINV-CAR targeted with tFab suppresses tumor growth in spleen. (A) Mice engrafted with FFLuc BV-173 tumor cells and treated with either SINV-CAR alone or SINV-CAR plus tFab were euthanized, and spleens were weighed (right, n=10, mean shown). Representative images of the spleens (left panel). ***P=0.0002, unpaired t-test. (B) Representative flow plots (left panel) and summary (right panel) of the percentage of human CD19+ tumor B cells infiltrating the spleen of mice engrafted with FFLuc BV-173 and treated with either SINV-CAR alone or SINV-CAR plus tFab at time of sacrifice (n=10, mean shown). ****P<0.0001, unpaired t-test. (C) Representative flow plots (left panel) and quantification (right panel) of human CAR-T cells (gated on CD3+CD45+) in the spleen at the time of euthanasia (n=10 each condition, mean shown). Empty symbols denote the flow plots shown to the left. *P=0.0293, unpaired t-test. CAR, chimeric antigen receptor; tFab, tandem Fab format.
Conclusion
In conclusion, we have developed a modular gene delivery platform, using a mutant Sindbis LV vector redirected to target cells via bispecific tFab binders. This LV platform enabled highly specific and effective gene delivery to target cells in vivo even at modest MOIs. Compared with recent studies of in vivo generated CAR-T cells from the Buchholz team using Nipah pseudotyped LVs, we injected similar particle doses per mouse between our studies (5×1010 SINV-LV vs 1–2.5×1011 Niv-LV).28 30 31 Although particle titers were measured by the same method of nanoparticle tracking analysis using similar Nanosight instruments, Buchholz et al21 established a different efficacy mouse model engrafting Nalm-6 tumor B cells, so a direct comparison of our in vivo results is not feasible. Similarly, direct comparisons of transduction efficiency in vitro are difficult: we determine MOI based on a qPCR titer, which provide an absolute count of viruses carrying the transgene without inferring details on viral particle functionality or infectivity. In contrast, many groups report functional titers established from transduction assays on particular cell lines, which produce different titer values without specific details of viral transgene or particle content.23 25 27 29 30 32 40 41 43–45 53 Although retargeted measles and Nipah pseudotyped LVs seem to achieve significantly greater in vitro transduction rates compared with our SINV-LV/tFab in primary PBMCs, it is worth noting that our transduction protocol did not include spinoculation or enhancers such as Vectofusin-1.23 27 29–31 Nevertheless, measles and Nipah pseudotyped LVs retargeted to T cells were able to transduce unstimulated cells in vitro and in vivo, which we are currently unable to achieve with our SINV-LV/tFab. This difference might be explained by the divergent mechanisms used by alphaviruses and paramyxoviruses for infection: alphaviruses, such as Sindbis, require pH-dependent membrane fusion following endocytosis to deliver the cargo DNA, whereas paramyxoviruses, such as measles and Nipah, are pH independent and do not rely on endocytosis to deliver their cargo DNA into cells.22
The differences between Sindbis and other paramyxoviruses notwithstanding, we showed here that a single dose of our targeted Sindbis-based LV vector was able to generate CAR-T cells from circulating T lymphocytes in a humanized tumor mouse model of B cell leukemia. The in vivo engineered CAR-T cells from this proof-of-concept study greatly suppressed CD19+ tumor cell growth and prolonged the overall survival time of mice, despite the highly aggressive nature of the tumor model. The efficient transduction was critically dependent on the bispecific tFab binders redirecting mutant Sindbis pseudotyped LVs to circulating T-lymphocytes. While the other previously discussed LV surface engineering approaches employ mutations and addition of either adapter molecules for non-covalent targeting or directly incorporated targeting ligands, our SINV-LV only requires mutations without the need for incorporating additional residues that might compromise particle integrity and titer. Our system does not require empirical testing of new targeting ligands swapped onto the particle surface as covalent attachments but only engineering of the bispecific binder molecule. Furthermore, antibody engineering with directed evolution will allow us to screen for a library of bispecific binder molecules enabling fully tunable affinity toward both LV surface glycoproteins and cell receptors. By exchanging our current anti-CD3 cell receptor binding Fab arm for specificity toward a different receptor, we could target CD4+ or CD8+ T cells without modifications to our LV vector, even enabling us to multiplex with trispecific binders for enhanced cell specificity. This modular nature provides numerous possibilities for a wide variety of cell and gene therapy applications beyond just CAR-T cell immunotherapy, but it would require separate development of both bispecific binder and LV vector if a different cell type were to be targeted with a different genetic cargo, such as a different CAR molecule or therapeutic transgene.
The antitumor activity that we observed in vivo, considering the low number of CAR-expressing T cells, is consistent with the superior performance and self-renewal capacity of CAR-T cells directly generated in vivo compared with that of CAR-T cells stimulated and expanded more extensively ex vivo. However, this theory has yet to be thoroughly explored by us or any other research group generating CAR-T cells directly in vivo. The only study to date to directly compare efficacy of in vivo versus ex vivo generated CAR-T cells was led by Matthias and colleagues, who utilized a non-viral polymeric nanoparticle platform for systemic delivery of anti-CD19 CAR to a fully mouse model of leukemia.54 Their results suggest that CAR-T cells, whether engineered directly in vivo with their novel integrating nanoparticle platform or ex vivo from traditional LV transduction, could achieve similar levels of tumor remission and prolonged animal survival in a preclinical mouse model of leukemia. However, the comparison in this study was made from different gene delivery vectors at high doses for both nanoparticles (five sequential injections of 3×1011 particles) and ex vivo generated CAR-T cells (5 million CAR-T per mouse). Future studies comparing in vivo and ex vivo generated CAR-T cells should apply identical gene delivery vectors in both treatment groups and contextualize the efficacious dose range of ex vivo generated CAR-T cells by injecting at the lower dosage limit for therapy. Simultaneously, the dosage range of systemically delivered gene delivery vector should be optimized so as to correlate an in vivo vector dose with similar efficacy observed from a particular dosage of ex vivo generated CAR-T cells. Simply measuring the number of CAR+ T cells generated in a tumor-bearing mouse model following different doses of gene vector may not represent the complex activity of CAR engagement and cell stimulation occurring in vivo upon tumor antigen recognition. As the new field of in vivo CAR-T engineering continues to develop, carefully designed studies comparing performance and relevant doses in context to more traditional ex vivo engineered CAR-T are imperative to understand the feasibility of this novel approach and essential in understanding the limitations that must be circumvented for such a therapy to reach the clinic.
Supplemental material
Data availability statement
Data are available on reasonable request. All data relevant to the study are included in the article or uploaded as supplementary information.
Ethics statements
Patient consent for publication
Ethics approval
All tumor mouse model experiments were performed in accordance with UNC Animal Husbandry and Institutional Animal Care and Use Committee (IACUC) guidelines and were approved by UNC IACUC (Protocol #: 18–251).
Acknowledgments
Animal studies were performed within the UNC Lineberger Animal Studies Core Facility at the University of North Carolina at Chapel Hill. The UNC Lineberger Animal Studies Core is supported in part by an NCI Center Core Support Grant (CA16086) to the UNC Lineberger Comprehensive Cancer Center. Special thanks is given to both the Biomedical Research Imaging Center (BRIC) and Flow Cytometry Core Facility at UNC for use of their instruments, which are supported in part by a Cancer Center Core Support Grant (P30 CA016086) to the UNC Lineberger Comprehensive Cancer Center. Transmission electron microscopy and grid preparation were performed by Paul Risteff at the UNC Hooker Imaging Core Facility. Special thanks to Boris Kantor and Logan Brown of the Duke University Viral Vector Core for their production of concentrated, in vivo grade lentiviruses used in these studies.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors JTH and EL contributed equally to this work. All authors conceptualized the project framework and designed experiments. JTH and TMJ designed and developed the bispecific binder molecule and lentiviral vector. JTH performed in vitro transductions. EL isolated and cultured primary cells. JTH and EL conducted tumor coculture cytotoxicity studies. EL handled mice and performed bioluminescent imaging. JTH and EL processed mouse tissue samples and performed flow cytometry phenotypic analyses. JTH and EL analyzed the data and performed statistical analysis. All authors contributed in writing and editing the manuscript.
Funding This work was supported in part by the National Science Foundation Graduate Research Fellowship Program (DGE-1650116, JTH), NIDCR of the National Institutes of Health (F32DE026683, TMJ), the National Institutes of Health (5-R01-CA193140, EL, BS, GD), The David and Lucile Packard Foundation (2013-39274, SKL), Eshelman Institute for Innovation (SKL), a Dissertation Completion Fellowship from The Graduate School at UNC-Chapel Hill (JTH), and startup funds from the Eshelman School of Pharmacy and Lineberger Comprehensive Cancer Center at UNC-Chapel Hill (SKL).
Competing interests TMJ has an equity interest in Dualogics LLC, a company aimed at commercializing bispecific antibodies.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.