Article Text

Abstract
Background Complex tumor and immune microenvironment render pancreatic ductal adenocarcinoma (PDAC) resistant to immune checkpoint inhibitors (ICIs). Therefore, a strategy to convert the immune hostile into an immunopermissive tumor is required. Recent studies showed that intratumoral injection of Toll-like receptor 9 agonist IMO-2125 primes the adaptive immune response. Phase I and II trials with intratumoral IMO-2125 demonstrated its safety and antitumoral activity.
Methods We generated an array of preclinical models by orthotopically engrafting PDAC-derived cell lines in syngeneic mice and categorized them as high, low and no immunogenic potential, based on the ability of tumor to evoke T lymphocyte or NK cell response. To test the antitumor efficacy of IMO-2125 on locally treated and distant sites, we engrafted cancer cells on both flanks of syngeneic mice and treated them with intratumoral IMO-2125 or vehicle, alone or in combination with anti-PD1 ICI. Tumor tissues and systemic immunity were analyzed by transcriptomic, cytofluorimetric and immunohistochemistry analysis.
Results We demonstrated that intratumoral IMO-2125 as single agent triggers immune system response to kill local and distant tumors in a selected high immunogenic subtype affecting tumor growth and mice survival. Remarkably, intratumoral IMO-2125 in combination with systemic anti-PD1 causes a potent antitumor effect on primary injected and distant sites also in pancreatic cancer models with low immunogenic potential, preceded by a transition toward an immunopermissive microenvironment, with increase in tumor-infiltrating dendritic and T cells in tumor and lymph nodes.
Conclusion We demonstrated a potent antitumor activity of IMO-2125 and anti-PD1 combination in immunotherapy-resistant PDAC models through the modulation of immune microenvironment, providing the rationale to translate this strategy into a clinical setting.
- gastrointestinal neoplasms
- tumor microenvironment
- immunotherapy
Data availability statement
All data relevant to the study are included in the article or uploaded as supplemental information. All methodologies, analyses, and results are included in the article.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is the fourth leading cause of cancer-related mortality among adults in developed countries.1 In 2030, it is expected to become the second cause of death among all tumors.2 Indeed, most of patients are diagnosed with metastatic disease, and only 15%–20% of them are eligible for upfront surgical resection.3 In addition, disease relapse is common in patients undergoing surgery and postoperative therapy, with 80% of resected patients developing metastases within 2 years.4 The poor prognosis of PDAC is mainly due to metastasis development and limited efficacy of available systemic treatments.
In the last decades, immunotherapy has revolutionized cancer treatment offering an effective strategy for the management of solid tumors.5 Recent studies demonstrated that the response to immune checkpoint inhibitors (ICIs), such as anti-PD1 therapy, was improved in patients with high tumor-infiltrating lymphocytes (TILs) while poor T cell priming or immunological ignorance was responsible of ICI inefficacy.6 7 In PDAC, the presence of a pronounced desmoplastic reaction, accumulation of stroma and formation of an immunosuppressive environment with limited capacity to evoke a sustained CD8+ T-cell inflammatory infiltrate contributed to resistance and inefficacy to systemic treatments. Therefore, agents that increase TILs might be expected to improve anti-PD1 response rate.8 9
Toll-like receptors (TLRs) are key molecules of the innate immune system playing a role in regulating adaptive immune responses.10 11 IMO-2125, a synthetic oligonucleotide-based agonist of TLR9 with immune-stimulating activity, binds to and activates TLR9 expressed by plasmacytoid dendritic cells (DCs) and B cells leading to a signaling cascade activation, which ultimately induces CD8+ T-cell mediated response against tumor.12 13
We have previously shown that IMO-2125 interferes with epidermal growth factor receptor (EGFR)-related signaling pathway and is able to inhibit tumor growth in mice models following intraperitoneal or oral administration. Moreover, we showed that IMO-2125 cooperates with monoclonal antibodies anti-EGFR cetuximab14 15 and anti-VEGF bevacizumab16 in several cetuximab-sensitive and cetuximab-resistant colorectal cancer models and in pancreatic cancer models harboring KRAS mutation.
More recently, the discovery that intratumoral injection of TLR9 agonist could convert an ‘immune cold’ into an ‘immune hot’ tumor fostered the interest for new combination strategies for the treatment of cold and immunotherapy refractory tumors.17 Mechanistically, the intratumoral immunotherapy induced a specific in situ immunization using the tumor as its own vaccine, thus bypassing major limits of systemic treatments such as safety, bioavailability and immune cell priming, achieving local effective concentration while using low drug doses. Such local immune stimulation can be translated into systemic antitumor effects, thanks to the migration of primed immune cells at distant untreated sites.18 19
Results from the phase I/II ILLUMINATE-204 trial demonstrated the safety and activity of intratumoral IMO-2125, also named tilsotolimod, in combination with systemic ICI ipilimumab in patients with anti-PD1 refractory advanced melanoma.20 Interestingly, the antitumor activity was observed in both injected (enestic) and non-injected (anenestic) lesions (NCT02644967).
To date, no information is available about the potential effect of TLR agonists combined with ICI in the complex context of poorer TIL infiltrated and ICI refractory tumors, such as that of PDAC. Recently, different PDAC subtypes have been described and characterized for their tumor microenvironment (TME) and immune context,21–23 which can be translated in a better stratification of patients with PDAC.
We took advantage of the above-described new findings, and here, we investigated the effects of IMO-2125, alone or in combination with ICI, in preclinical in vivo mouse models, which recapitulate the different human molecular PDAC subtypes.
Materials and methods
Cell lines and materials
Cell lines were kindly provided by Dr D Tuveson’s laboratory, at Cold Spring Harbor Laboratory (New York, USA) and Dr P Cappello’s laboratory, at CeRMS laboratory (Turin, Italy), and were maintained in their original culturing conditions. Cells were daily checked by morphology and routinely tested to be mycoplasma free by PCR assay. IMO-2125 was provided by Idera Pharmaceuticals (Cambridge, Massachusetts, USA); anti-PD1 checkpoint inhibitor and the relative control were purchased from Leinco Tech (Fenton, Missouri, USA).
Cell proliferation and migration assay
In 96-well plates, 1.0×103 cells/well were seeded. At the indicated hours, sulforhodamine B (Sigma-Aldrich, St Louis, Missouri, USA) assay was used to obtain relative estimates of viable cell number according to manufacturer instruction. For migration assay, cells were scratched, washed gently with cold phosphate buffered saline (PBS) solution 1× and rinsed with fresh medium. Photographs at five different points at least were taken immediately and after 24 and 32 hours of culture.
Protein extraction and Western blotting
Lysates was separated by SDS-PAGE and probed with antibodies against, TLR-9 (ab12121) from Abcam (Cambridge, UK) and beta-actin (4970s), E-cadherin (3195s), LC3B (2775s) and vimentin (5741s) from Cell Signaling Technology (CST, Danvers, Massachusetts, USA). Immunoreactive proteins were detected using an enhanced chemiluminescence reagent (ECL, Millipore, Burlington, Massachusetts, USA) according to the manufacturer’s instructions. Images were captured by UVITEC Digital Image Scanning System (UVITEC, Cambridge, UK).
Mouse models
For the generation of pancreatic cancer orthotopic models, pancreatic cancer cells (2.5×105 cells/mouse) in a 1:1 solution of Matrigel and cold PBS were injected into the pancreas parenchyma of recipient C57BL/6J strain male mouse models at 6 weeks of age. Briefly, an incision was made in the left abdominal side at the level of the spleen. DT4313, FC1245, FC1242, RC416, CR705, DT6606 cells were injected into the tail region of the pancreas using insulin syringes (BD micro-fine 25 Gauge). The injection was considered successful by the development of bubbles without signs of leakage. The peritoneum was sutured with short-term absorbable suture (Vetsuture), and the skin was closed with wound clips. Mice were euthanized at the indicated time points. Following weekly manual palpation starting 10 days following transplantation, tumor-bearing mice were subjected to high-contrast ultrasound imaging using the Vevo 2100 System with a MS250, 13–24 MHz scan-head (VisualSonics, Amsterdam, The Netherlands). For the generation of heterotopic mice models, pancreatic cancer cell lines (DT4313, FC1245, FC1242, RC416) in a solution of 1:1 Matrigel and cold PBS were injected in both flanks of individual C57BL/6J male mouse strain models at 6 weeks of age (right flank 1.5×105 cells; left flank 0.7×105 cells).
Treatment was initiated when tumor volume reached ~100 mm3, which is the inclusion criterion. Tumor-bearing mice received PBS or IMO-2125 (2.5 mg/kg, twice a week for 2 weeks) into the tumor implanted in the right flank. After 2 weeks of IMO-2125 or PBS treatment, three mice per group were euthanized and biological materials collected for downstream analysis (RNA-seq, Immunohistochemestry (IHC), cytokines analysis). No significant body weight differences were detected on treatments. The remaining mice (10 mice per group) were randomly assigned to receive intraperitoneal injection of anti-PD1 or IgG as control (10 mg/kg, twice a week for 2 weeks). At the end of the treatment, three mice per group were sacrificed and tumors collected for further analyses. Tumor growth was determined by measuring the long and short diameters of the tumor using calipers. Tumor volume was calculated using the formula 0.52×length×width.2 Age-matched treatment naive C57BL/6J mice served as controls. Mouse were euthanized using carbon dioxide inhalation when evidence of advanced bulky disease developed or at cutoff of 2 cm3, which was considered the day of death for the purpose of survival evaluation. To test TME differences in anti-PD1 response: syngeneic orthotopic mouse models of three different cell line-derived graft (CDG) model (FG1245, FC1242 and DT4313) were randomly assigned (n=3 per group) to receive anti-PD1 or IgG as control (intraperitoneal 10 mg/kg twice a week for 2 weeks). Tumor size was measured with VEVO 3100 ultrasound device weekly.
The methods for animal study followed the ARRIVE Guidelines 2.0.24
RNA isolation and quantitative RT-PCR assay
RNA was obtained using TRizol reagent (Invitrogen Corporation, Carlsbad, California, USA) according to manufacturer’s instructions. The cDNA was evaluated for real-time PCR with QuantStudio 3 (Thermo Fisher Scientific, Waltham, Massachusetts, USA) using specific primer and SYBR Green. QuantiTect Primer Assays (Qiagen, Hilden, Germany) were used to quantify cDNA levels of CDH1, VIM and TLR9. Gene expression was calculated using 2−DDCT method and normalized to β-actin expression.
Histology
Whole 3 µm tissue sections were dewaxed and rehydrated. Antigen retrieval was performed using Bond Epitope Retrieval Solution 2 (AR9640, Leica Microsystems). A pathologist who was blinded to treatment allocation of the mice reviewed the H&E-stained slides of cancer tissues.
The following antibodies were used for immunohistochemical staining with established procedures: Ly6G (ab25377), CD11b (ab133357), CD3 (ab5690), CD68 (ab213363), FAP-a (ab218164), IFNγ (ab216644), MLH1 (ab92312), PMS2 (PA5-86491), MSH2 (ab212188) and MSH6 (ab92471) from Abcam and Ki-67 (D3B5, 9129s) from CST.
Multiplex cytokines profiling
Plasma specimens were analyzed for TNFα, MIP1α, MIP1-β, MCP1, IL-1b, IL-6, IL-10, IL-17A, KC and G-CSF using Luminex XMAP multiplexing technology according to manufacturer’s instructions (Bioplex 200, Bio-Rad, Hercules, California, USA). Cytokines concentrations in samples were determined from the standard curve using a five-point regression.
Flow cytometry analysis of peripheral blood and tumor-infiltrating immune cells
Flow cytometry-based immunophenotype of tumors was performed according to already published protocols.25 One million cells were incubated with CD16/CD32 (14-0161-82) (BioLegend, San Diego, California, USA) antibody and subsequently stained with the appropriate antibodies. Spleen and lymph node cell components were stained with antibody staining panels: a PBMC subset panel and a T cell subset panel. The Peripheral Blood Mononucletae Cells (PBMC) subset panel antibody cocktail of CD3 (FITC, 100204), CD8-PerCP (100734), CD45 (BV421,103134), CD19 (115530), CD86 (PE, 105106), CD11b (PERCP,101228), CD80 (APC, 104714), Ly6G (APC-CY7, 127624), from BioLegend, and CD4 (25-0041-82), CD25 (APC, 17-0251-82), LY-6C (48-5932-82), CD11C (FITC, 11-0114-82) from eBioscience (Thermo Fisher Scientific). Samples were acquired on an FACS Canto II (BD Biosciences, San Jose, California, USA) and analyzed with FlowJo software (FlowJo LLC, Ashland, Oregon, USA).
Lymph nodes analysis
Inguinal and axillary lymph nodes from each flank were measured. To retrieve immune cells, lymph nodes were dissociated with 200 µg/mL liberase and 40 µg/mL DNAse I (Sigma-Aldrich) in Roswell Park Memorial Institute (RPMI) culture medium with 2% Fetal Bovine Serum (FBS) for 20 min at 37°C. To retrieve neoplastic cells, lymph nodes were dissociated with 200 µg/mL Type IV Collagenase (Gibco, Thermo Fisher Scientific) and 100 µg/mL DNAse I (Sigma) in RPMI‐2% FBS for 30 min at 37°C.
RNA-sequencing (RNA-seq)
RNA integrity number (RIN) was measured on an Agilent 2100 Bioanalyzer system. Only RNA samples with an RIN > 7 were used for cDNA library construction. All cDNA libraries were sequenced using paired-end strategy (read length 150 bp) on an Illumina HiSeq 2000 platform.
Quality of raw reads was checked with FASTQC.26 Transcripts were quantified with the alignment-free method implemented in Salmon 0.11.3.27 Mouse genome and transcriptome from Gencode Release M18 (GRCm38.p6) were used. Quantified transcripts were imported to the statistical software R with the tximport package28 and aggregated to the gene level using the option tx2gene. The matrix of gene counts was then converted to a DESeq data set with the function DESeqDataSetFromTximport function implemented in DEseq2 package V.1.22.2 and normalized using the rlog function. Differentially expressed genes were identified with the same package. Gene set variation analysis was performed on log2 normalized counts with the GSVA package 1.30.029 using custom and MSigDB gene sets. Gene set enrichment analysis was performed with the fgsea package applying 10,000 permutations, Benjamini–Hochberg procedure for correcting p values and a p value cutoff of 0.05. Heat-maps were generated either with the Bioconductor packages ComplexHeatmaps V.1.20.0 or heat-map.
Statistical analysis
All results, when applicable, were expressed as the means±SD. Statistical analysis was performed using the Prism 5 software program. Unpaired Student’s two-tailed t-tests were used to compare two groups when appropriate. For multiple groups, one-way analysis of variance with Tukey’s post-test was used. P values <0.05 were considered as statistically significant. Kaplan–Meier curves were generated using the Prism 5 software program and analyzed by log rank tests.
Results
In vitro and ex vivo characterization of orthotopic pancreatic cancer isograft mouse models
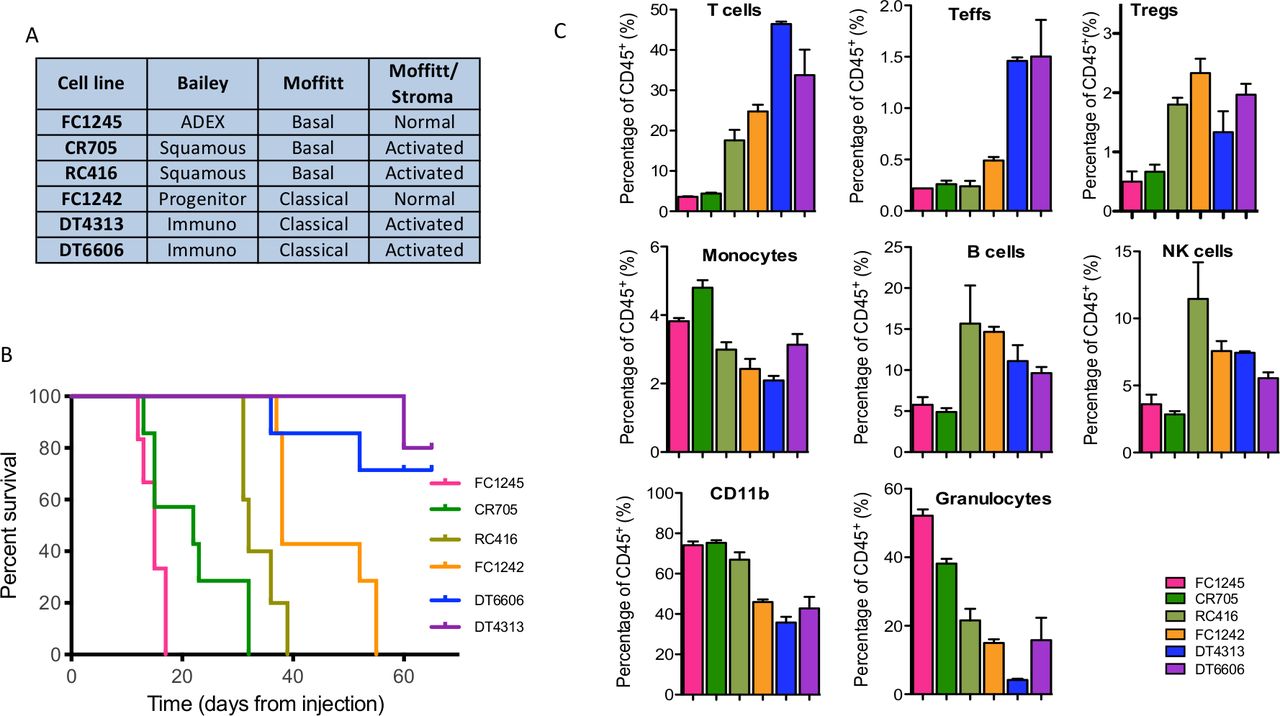
To study the effect of IMO-2125 on the immunological landscape of PDAC and, in parallel, on tumor growth, we generated a set of preclinical models by engrafting six mouse PDAC-derived cell lines into the pancreas of recipient C57BL/6J immune-competent mice. At first, cells were tested in vitro for TLR9 and epithelial to mesenchymal transition (EMT) markers expression (online supplemental figure S1A). Although TLR9 is expressed, IMO-2125 treatment did not affect cell proliferation, migration and EMT in vitro (online supplemental figure S1B–D) leading to the hypothesis that it could affect tumor growth independently on the baseline tumor cell-intrinsic characteristics. Thereafter, ex vivo RNA-seq analysis of each mouse pancreatic cancer model (n=3) was performed and showed that murine cancer cell lines were representative of the human pancreatic cancer molecular subtypes22 23 30 (figure 1A). In particular, the two murine CDGs DT4314 and DT6606 resembled the immunogenic subtype according to Bailey, suggesting that they were infiltrated by immune cells (figure 1A). In keeping with human long-term survivors showing increased level of immune infiltration,6 immunogenic CDGs showed longer survivals (figure 1B). CDGs from FC1242 cell line were consistently classified as classical/pancreatic progenitors, while CDGs from CR705 and RC416 aligned with the more aggressive basal-like/squamous subtype and had shorter survival (figure 1B). CDGs from FC1245 aligned with the ADEX and the basal-like subtype according to Bailey and Moffitt, respectively, and displayed the worst survival of all preclinical models here analyzed (figure 1B). When looking at Moffitt classification based on stromal gene expression, two CDGs were classified as ‘normal’ and four were classified as ‘activated’ (figure 1A). IHC analysis of MLH1, MSH2, MSH6 and PMS2 MicroSatellite Instability markers demonstrated that all models were microsatellite stable, with DT4313 PDAC model stable and yet immunogenic (online supplemental figure S2). Stratification according to stromal gene expression was not informative of mouse survival (figure 1B).22 To validate inference from RNA-seq analysis about differences in the quality of stromal infiltration in the CDG cohort, we subjected individual CDG to immune characterization by FACS analysis (figure 1C). We observed that classical and stroma-activated cancer subtypes (DT4313 and DT6606) were characterized by high percentage of infiltrating T lymphocytes and by a low percentage of CD11b-positive cells and macrophages compared with basal and normal-stroma subtypes (FC1245). We infer that stroma-activated subtype could generate a more pronounced immune response than the other subtypes.30 These analyses showed the subtype collocation of each pancreatic cancer cell line, allowing us to test the intriguing hypothesis that TLR9 agonist IMO-2125 could inhibit tumor growth of pancreatic cancer subtypes that retain the ability to evoke immune response.
Supplemental material
Supplemental material
Supplemental material

Characterization of orthotopic pancreatic cancer isograft mouse models. (A) RNA-seq of pancreatic cancer cell-derived graft (CDG) recapitulates the Moffitt and Bailey human pathology; (B) Kaplen-Meier survival analysis of C57BL/6J mice bearing the indicated pancreatic cancer subtypes; (C) comparative flow cytometry analysis of immune infiltrating components of cancer tissues from CDG bearing mouse models. T cells, CD45+CD3+; Teffs, CD45+CD8+CD3+; Tregs, CD45+CD3+CD4+CD25+FOXP3+; monocytes, CD45+CD11b+Ly6C+; B cells, CD45+CD19+; NK cells, CD45+CD49b+; granulocytes, CD45+CD11b+Ly6G+. NK, natural killer.
To test this hypothesis, we initially selected cancer cell lines with high immunogenic potential (DT4313 and RC416), low immunogenic potential (FC1242) and no immunogenic potential (FC1245), based on the ability to evoke T lymphocyte or natural killer (NK) cell immune response in syngeneic mouse models.
The combination therapy of locally IMO-2125 and systemic anti-PD1 antibody affected pancreatic tumors with high immunogenic potential at both local and distant sites
To evaluate whether local stimulation with IMO-2125 could generate systemic antitumor response in uninjected (anenestic) lesions (abscopal effect), C57BL/6J mice were injected subcutaneously on both animal flanks with DT4313, FC1242, FC1245 or RC416 cancer cell lines (n=38 for each cell line) and subsequently were randomly assigned to receive IMO-2125 (2.5 mg/kg intratumorally twice a week for 2 weeks) or vehicle on tumor tissue in the right flank (hereinafter referred to as T1) and anti-PD1 (10 mg/kg or IgG, intraperitoneally twice a week for 2 weeks) alone or in combination (hereinafter referred to as T2) (figure 2A). At the end of T1 and T2 treatment protocol, three mice for each group were euthanized and tumors, spleen and lymph nodes were collected and analyzed as reported below.
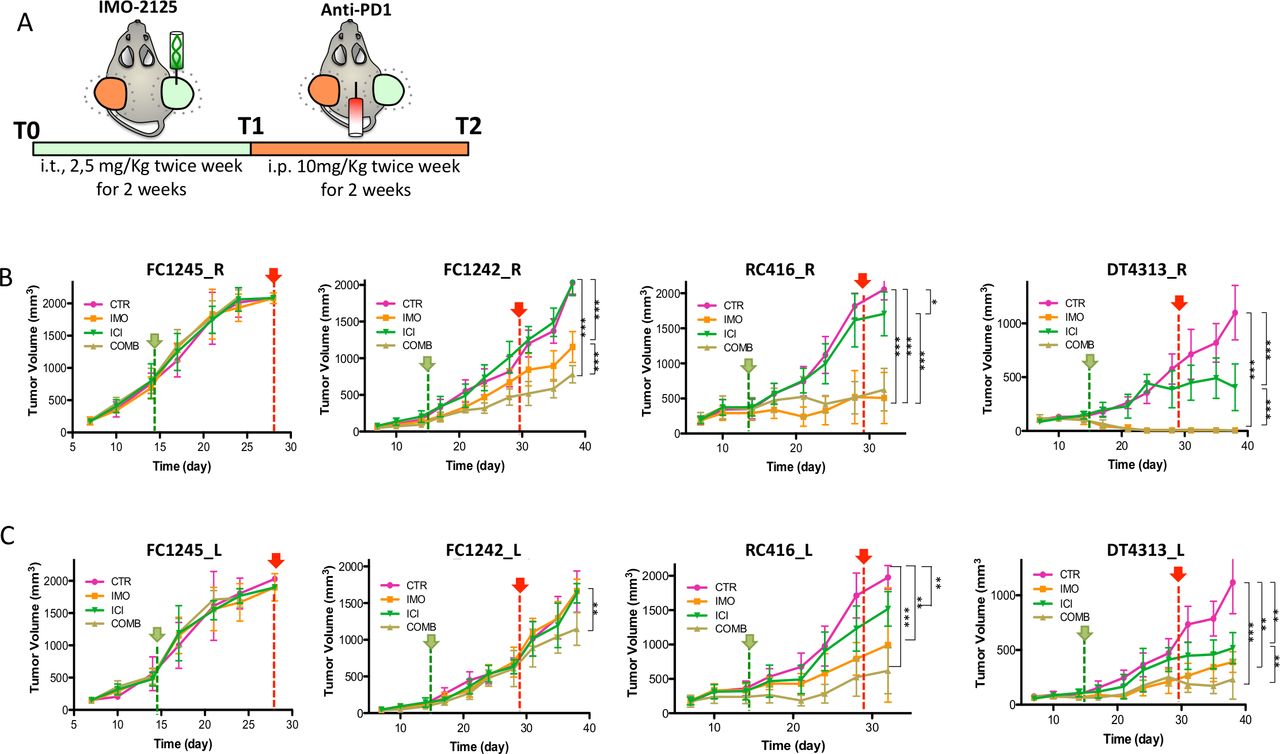
The combination therapy of locally IMO-2125 and systemic anti-PD1 antibody affects pancreatic tumors with high immunogenic potential at both local and distant sites. (A) Schematic representation of the mouse models treatment. Syngeneic mouse models bearing subcutaneous pancreatic tumors on both flanks were randomly assigned (n=10 per group) to receive IMO-2125 (intratumorally 2.5 mg/kg twice a week for 2 weeks, green line) and anti-PD1 (intraperitoneally 10 mg/kg twice a week for 2 weeks, red line) or combination. Tumor size was measured with a caliper system of both IMO-2125 locally treated (R) and untreated (L) tumors. Mean tumor volume and SD of locally treated (B) and opposite locally untreated (C) tumor in mm3 are shown (*p<0.05, **p<0.01, ***p<0.001 of the last point, green pointer, T1; red pointer, T2.
IMO-2125 showed no efficacy in terms of tumor shrinkage in CDG FC1245 tumors that have no immunological potential (figure 2B). Further, the analysis of FC1242 cancer cell line bearing mice demonstrated that IMO-2125 as single agent had a weak efficacy on tumor with low immunological potential (figure 2B,C). Moreover, the combination with ICI significantly increased the efficacy of IMO-2125 on tumor growth. Notably, IMO-2125 showed high efficacy and curative effect on RC416 and DT4313 high immunological CDG models (figure 2B). Both tumor models were sensitive to ICI as single agent, suggesting that the immunological classification correctly identifies tumors that might show some degree of sensitivity to immunotherapy.
We tested three different subtypes of CDG models to establish whether the different TME of the orthotopic and heterotopic model could influence the response to ICI-based therapy. ICI treatment did not exert any effect either in orthotopic or in the heterotopic models of both the FC1245 and the FC1242 CDG models, whereas DT4313-bearing mice confirmed to be the most immunotherapy sensitive model. Thus, although TME may be different in different location it does not affect the response rate to ICI therapy in our models (online supplemental figure S3).
Supplemental material
Next, we analyzed the IMO-2125 abscopal effect on the same tumor-bearing models, demonstrating that IMO-2125 was able to increase the efficacy of ICI in the anenestic lesions only in tumors with immunological potential (figure 2C).
Effect of IMO-2125, ICI and combination treatment on survival rate was also examined (online supplemental figure S4). According to tumor growth delay, IMO-2125 treatment failed to prolong mouse survival when injected with no immunogenic potential CDGs while showing significant effect when injected with the high immunogenic ones and, in a lesser extent, with the low immunogenic CDG, both alone and in combination with ICI.
Supplemental material
IMO-2125-based immune reprogramming fosters a suppressive to permissive immune transition
We investigated transcriptional changes induced by IMO-2125 in tumors with different immunogenic profiles. Tumor tissue from DT4313, FC1245 and FC1242 CDG models treated (intratumorally) with IMO-2125 or vehicle (T1) was analyzed by RNA-seq. CDG models clustered separately according to the cell identity and regardless of the treatment, with a better separation between clusters of DT4313 IMO-2125 treated or untreated models (figure 3A). Accordingly, in this latter group a larger proportion of genes was significantly regulated following treatment (figure 3B). Transcriptional signatures indicative of biological processes such as activation of inflammation, immune response activating signaling and adaptive immunity, among others, were enriched in DT4313 tumors treated with IMO-2125 (figure 3C). The five most significant upregulated biological processes identified by Gene Set Enrichment Analysis (GSEA) demonstrated that IMO-2125 dramatically increased the expression of proinflammatory signaling pathway, including the expression of cytokines (figure 3D).

IMO-2125-based immune reprogramming favors a suppressive to permissive immune transition. Tumor tissue from FC1245, FC1242 and DT4313 tumor-bearing-mouse models treated with IMO-2125 or vehicle control (T1) analyzed by RNA-sequencing. (A) PCA demonstrates that FC1245, FC1242 and DT4313 CDG tumor tissue cluster separately according to the cell identity and regardless of the treatment, with a major variance in the clusters of DT4313 (high immunological potential) and FC1242 (low immunological potential) compared with FC1245 (no immunological potential) on IMO-2125 treatments. (B) Volcano plots of genes differentially expressed between locally treated (n = 3) and untreated (n = 3) FC1245, FC1242 and DT4313 tumors. (C) Biological processes activated and suppressed in DT4313 tumor treated with IMO-2125 (intratumorally) compared with untreated DT4313. Top 40 gene sets with an adjusted p value <0.05 are displayed; (D) five most significant upregulated (fold >1.5, p value <0.05) biological processes identified by GSEA in DT4313 tumor treated with IMO-2125 (intratumorally). (E) Multiplex analysis of proinflammatory circulating factors. Plasma from peripheral blood was collected ffrom each group of mice (n=3) treated or untreated with IMO-2125. Concentrations of proinflammatory cytokines were analyzed using Luminex technology. Concentrations of cytokines (pg/mL) were calculated. The mean values and SD are shown.
To confirm the ability of IMO-2125 to modulate the immune system, we evaluated the expression of proinflammatory circulating cytokines in tumor-bearing mice. We demonstrated that IMO-2125 stimulated a release of IFNγ and chemokines with distinct immune properties (including CCL2, CCL4, CCL5, CXCL10 and IL-10) (figure 3E). IMO-2125 (intratumorally) increased proinflammatory blood circulating cytokines that control the production, differentiation and function of granulocytes and macrophages (GMCSF, MIP2), as well as inflammatory and immune regulatory cytokines (IL1a, IL10, CCL2, CCL4) and cytokine produced by activated T cell (IL17A) (figure 3E and online supplemental figure S5). Altogether, our data show that IMO-2125 activated a strong immune response in pancreatic tumor subtype with intrinsic immunogenic potential by increasing the release of cytokines and chemokines able to attract and activate antitumor immune cells.
Supplemental material
To confirm the putative molecular features that contribute to IMO-2125 efficacy, we mined our RNA-seq dataset to determine basal transcriptional differences between high immunogenic potential DT4313, low immunogenic potential FC1242 and no immunogenic potential FC1245 cancer models in an unbiased manner.
As expected, DT4313 and FC1242 tumor tissues showed several differentially expressed genes (online supplemental figure S6A). Gene Ontology (GO) Biological Processes analysis confirmed the high immunological potential behavior of DT4313 compared with FC1242 (online supplemental figure S6B,C) with an increase of genes for immunological response and for the expression of secreted proinflammatory factors (online supplemental figure S6D). Volcano Plot and GSEA on differential expressed genes in DT4313 compared with FC1245 (online supplemental figure S7A,B) also showed, in a greater extent, a substantial activation of immune pathways (online supplemental figure S7C), with an accentuated positive regulation of lymphocyte differentiation and activation (online supplemental figure S7D).
Supplemental material
Supplemental material
Since reduction of immunogenic molecules on cell surface might contribute to hide tumor cells from immune recognition, reducing IMO-2125 effect, we investigated the basal autophagy flux in our models (online supplemental figure S8). Following lysosomal inhibition with chloroquine, FC1245 and FC1242 cells showed a basal increase of LC3-II autophagy marker compared with RC416 and DT4313 cells. These results stimulate the hypothesis that basal autophagy flux might influence IMO-2125 response of our pancreatic cancer cell lines through the regulation of immunogenicity.
Supplemental material
In order to investigate the effect of IMO-2125 treatment on anenestic sites, gene expression profile of the DT4313 left implant tumors was analyzed. The RNA-seq-based Principal Component Analysis (PCA) showed that while anti-PD1 as single agent did not alter gene expression of DT4313 left implant tumors, IMO-2125 and combination treatments were able to induce an immune-activated genetic program switch that could be responsible for shrinkage of tumor volume (online supplemental figure S9A). Volcano scatter-plot showed that treatments slightly affected the gene expression of anenestic DT4313 compared with the effect seen in locally IMO-2125-treated cancer models (online supplemental figure S9B). Ridge plot of the GSEA (online supplemental figure S9C) and GO (online supplemental figure S9D) analyses of differentially expressed genes confirmed that combination treatment was able to evoke immune response in DT4313 anenestic tumor.
Supplemental material
Intratumoral injection of IMO-2125 increased tumor-infiltrating dendritic and T cells
To validate RNA-seq data and investigate how IMO-2125 elicits an immune response reducing tumor volume, we analyzed major immune cell population subsets in the available matched tissues.
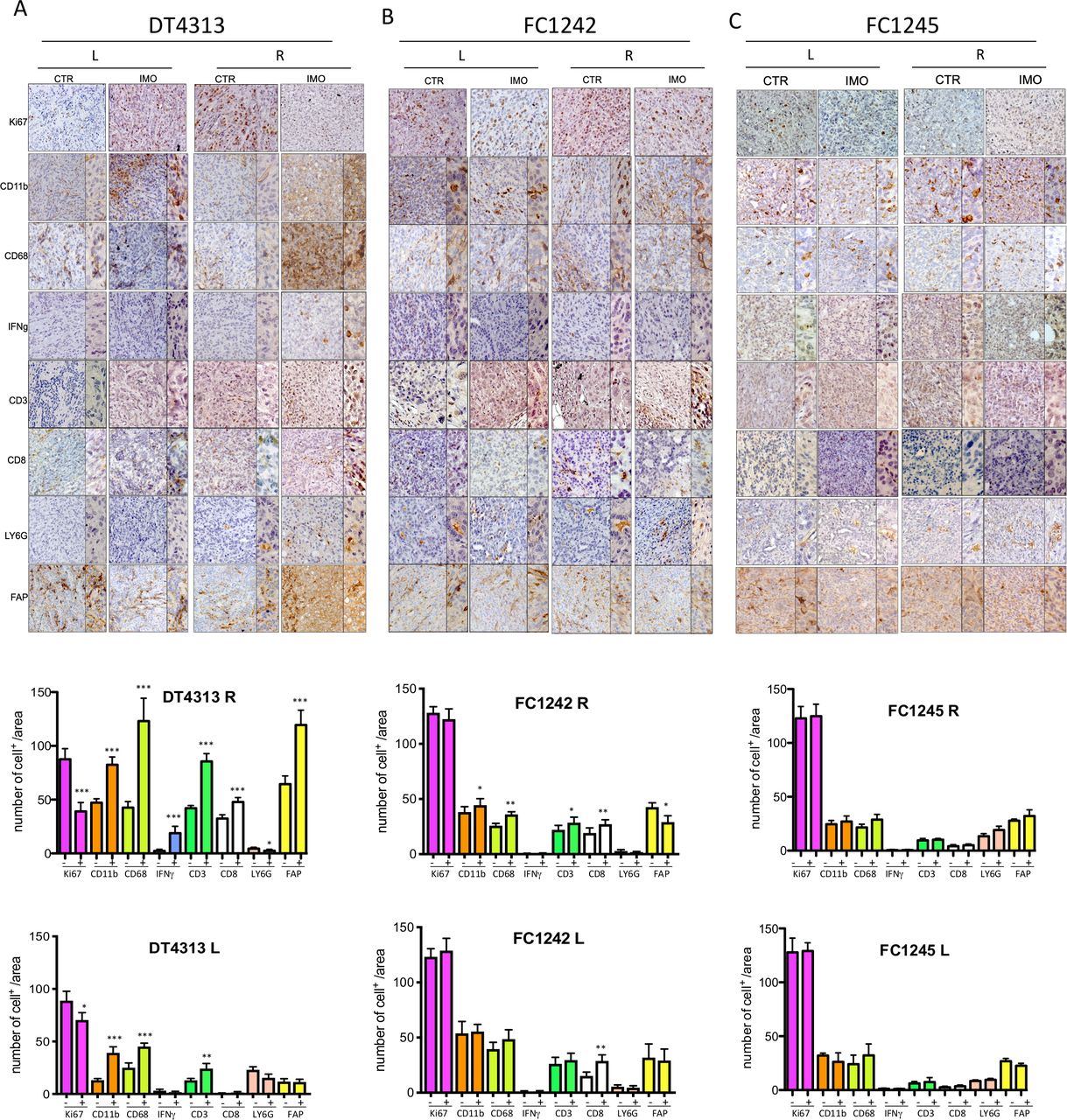
According to GSEA results and the subtype class, IHC analysis of tumor tissue of mice bearing DT4313 showed an immunoreactive FAP+ stroma and increased amounts of total T cells (CD3+ cells), CD8+ T cells, CD68-expressing macrophages and to a statistically significant reduction of Ki67 proliferation marker (figure 4A) compared with FC1242 and FC1245 models (figure 4B,C). Of note, IMO-2125 (intratumoral) treatment led to an increase of IFNγ release more evident for the tumor with high immunological potential (figure 4A) compared with FC1242 and FC1245 tumors (figure 4B,C).

Intratumoral injection of IMO-2125 increases infiltrating dendritic and T cells. Immunohistochemical staining for the reported markers in tumor tissue from mice bearing (A) DT4313, (B) FC1242 and (C) FC1245 subcutaneously injected cell line-derived graft cells. Anenestic (left panel) and anestic (right panel) IMO-2125 effects are reported. Quantification of each marker is provided as the average number of indicated marker-positive cells per mm2. From five to eight individual areas per case were examined. Statistical associations were calculated by Student’s t-test. *p<0.05; **p<0.01; ***p<0.001 (L, left flank tumor; R, right flank tumor).
In summary, IHC analysis demonstrated an increase of IFNγ, compatible with the activation of adaptive immunity and inflammation. Analysis of tumor immune components revealed that IMO-2125 (intratumoral) therapy might mobilize CD3+CD8+ T cells both locally and in the anenestic distant tumor, thus contributing to anti-PD1-mediated suppression of immunotolerance. IMO-2125 as single agent significantly increased the percentage of T cells both in locally IMO-2125 treated (right flank) and on the opposite anenestic tumor (left flank) of DT4313 (high immunological potential) and FC1242 (low immunological potential) CDGs. No effects on the immunomodulation of T cells were evident in the FC1245 tumor models (figure 4C).
Since IMO-2125 increased the expression of IFNγ and, in turn, the adaptive immunity signaling, we analyzed whether systemic anti-PD1 intraperitoneal treatment could ameliorate IMO-2125 effect (online supplemental figure S10). Combination of IMO-2125 (intratumoral) and anti-PD1 (intraperitoneal) treatments affected the frequency of CD8+ TILs in DT4313 and FC1242 models.
Supplemental material
Intratumoral injection of IMO-2125 promotes immune system activation and lymph nodes volume increase
To gain insight into the mechanism of IMO-2125-enhanced immune response, we evaluated the effect of intratumoral injection on the systemic immunity by analyzing the immune cell population of the lymph nodes and the splenic mononuclear isolated cells.
We first evaluated the volume of both IMO-2125-treated and untreated tumor-draining lymph nodes in each mouse model (figure 5A). Compared with control mice, a statistically significant increase of lymph node volume (DT4313, p<0.0001; FC1242, p<0.0001; RC416, p<0.0003; FC1245, p<0.0407) was detected in all IMO-2125-treated groups (figure 5A). The increment of lymph node size, after IMO-2125 treatment, correlated with the number of immune cells in the lymph nodes (online supplemental figure S11A).
Supplemental material

IMO-2125 (intratumorally) promotes immune system activation and lymph nodes volume increase. (A) Lymph node volume of Tumor Draining Lymph Nodes (TDLNs, (inguinal and axillary) of the both animal flanks. Lymph nodes were isolated from the both animal flanks of the indicated tumor-bearing mice models. Lymph nodes (inguinal and axillary) average size and Standard Deviation (DS) are plotted. Statistical difference is calculated with a paired two-tailed t-test; Flow cytometry analysis of the indicated immune cell populations from smashed lymph nodes of (B) locally treated (R) and (C) untreated (L) FC1242 and DT34313 tumor-bearing mouse models; dendritic cells were gated as CD11c+MHCII+ cells. Statistical differences were analyzed with two-way analysis of variance. All data represent average ±SEM (n=4 per group) (*p<0.05, **p<0.01, ***p<0.001).
Since the hallmark of IMO-2125 treatment is the activation of immune cells, including macrophages, DCs and T cells, we analyzed the frequency of immune cell populations from lymph nodes of each flank (figure 5B,C). We observed an increase of percentage of CD11b+ myeloid cells and CD19+ B cells in both the lymph nodes draining the IMO-2125-treated (intratumorally) (figure 5B) and untreated tumors (figure 5C). Moreover, we identified a significant upregulation of CD80 costimulatory molecule in CD11c+ MHCII+ DCs isolated from lymph nodes draining the DT4313 tumors treated with IMO-2125 (figure 5B). In agreement, a statistically positive reduction of the myeloid suppressor (CD11b+, Ly6G+) cells of the same lymph nodes was observed (figure 5B,C). Interestingly, this effect was not exclusive to lymph nodes of high immunological potential tumor mouse models, indicating an immune system activation-specific response by IMO-2125 independent from tumor subtype.
The effect of IMO-2125 treatment was less evident on systemic immunity, as demonstrated by flow cytometry analysis of immune cell populations from the spleen of each mouse model (online supplemental figure S11B).
Discussion
TLR9 is a member of the TLR family, key receptors of the innate immune system playing a crucial role in regulating adaptive immune response by activation of Th1-type signaling. The expression of TLR9 in several components of TME is pointing out TLR9 as potential pharmacological target in cancer therapy. We previously showed that IMO-2125 has a strong in vivo activity in colorectal and pancreatic cancer models by interfering with EGFR-related signaling and synergizing with cetuximab14 15 and with anti-VEGF monoclonal antibody bevacizumab.16 Different studies report TLR9 expression in both cancer and immune cells31–34 and its potential ability to enhance the effect of current immunotherapy.35 36
Immunotherapy revolutionized treatment of several cancers with high degree of T-cell infiltration (hot tumors), whereas low efficacy was reported in tumors with low levels of T-cell infiltration (cold tumors). The chance to couple immunostimulating drugs with an ICI opens up the possibility to potentiate the therapeutic efficacy in refractory tumors. In this line, results from the phase I/II ILLUMINATE-204 trial demonstrated the safety and activity of intratumoral injection of the TLR9 agonist IMO-2125 in combination with systemic ICI ipilimumab in patients with anti-PD1 refractory advanced melanoma. Interestingly, the antitumor activity was observed in both injected (enestic) and non-injected (anenestic) lesions (NCT02644967). Other clinical studies are investigating the activity of intratumoral IMO-2125 in colorectal cancer.
To date, PDAC is unfortunately an example of immunotherapy refractory tumor.37 Tumor cell-intrinsic aberrations of PDAC can lead to a ‘cold’ tumor by excluding cells that can mount an effective antitumor response or by attracting immunosuppressive population to the TME.38 In fact, PDAC shows a low-moderate mutational burden with low immunogenic potential and high infiltration of MDSCs, Treg cells and, in addition, carcinoma-associated fibroblasts, resulting in massive collagen deposition and fibrotic response and, ultimately, a high immunosuppressive microenvironment.39 40 Of note, it has been recently reported that PDAC subtypes with a TME that is not only rich in CD4+ and CD8+ effectors T cells but also poor in immunosuppressive cell populations were associated with a better patient outcome.41–43 The molecular subtyping of PDAC21–23 30 44 in several subgroups with specific characteristics opened a new scenario in the treatment of this pathology.
In this study, we demonstrated that TLR-9 agonist, IMO-2125, affects the growth of a specific pancreatic tumor subtype with immunogenic potential, triggering immune system response to kill local and distant tumors. Our results candidate pancreatic cancer subtyping as a criterion to select patients who could benefit from TLR9-immune-based therapy.
To prove that tumors with particular immune characteristics are more susceptible to a therapy based on local activation of the immune system, we characterized and established a panel of mouse pancreatic tumors in syngeneic models reproducing the molecular subtypes of PDAC.
In order to test the strength of our models and their capability to summarize the human pathology, we analyzed each PDAC subtype for gene expression markers and immune cell infiltration, demonstrating that our PDAC mouse subtype models well recapitulate human pathology including the prognostic significance (as reviewed in the study by Moffitt et al21).
Although the identification of PDAC subtypes has opened new possibilities for treatment, to date little has yet translated into more effective therapies.
In this context, we propose TLR9-based immunotherapy specifically in some subtypes of tumors potentially capable of initiating a cellular immune response. Remarkably, it has been shown that TLR9 agonists stimulate a cell-mediated inflammatory response in particularly hot tumor types, such as colon and lymphoma, and there are clinical trials in patients with refractory solid tumors currently in advanced clinical trial phases (NCT02644967, NCT03445533, NCT03052205).
In this study, we demonstrated that the intratumoral injection of IMO-2125 in pancreatic cancer subtype with high immunogenic potential (DT4313 and RC416) cured locally treated tumors inducing both a cytokine storm and an immune cellular response. In addition to the local effect, IMO-2125 leads to an antitumor response also on locally untreated distant tumor site. The IMO-2125-mediated immune cell-based response is further increased by anti-PD1 systemic treatment. Accordingly, it has been demonstrated that intratumoral injection of a TLR9 agonist in colorectal cancer reverts resistance to PD-1 blockade by expanding multifunctional CD8+ T cells.45
The intratumoral injection of IMO-2125 in the pancreatic cancer subtype with low immunogenic potential (FC1242) had less efficacy on tumor growth compared with subtypes with high immunogenic potential. On distant site, IMO-2125 was completely ineffective while a slight not statistically significant effect resulted when used in combination with anti-PD1.
No tumor growth inhibition was obtained on locally IMO-2125-treated or on distant tumor site of the pancreatic cancer subtype without immunogenic potential (FC1245).
TLR9-based immunotherapy primes host immune system to recognize and kill tumor cells. It has already been shown that IMO-2125 intratumorally injected in models of lymphoma and colon cancer elicits infiltration of T lymphocytes into tumors, resulting in the regression of both locally treated and distant tumors, and that this effect is attributable to active T lymphocytes (CD8+).45 46
It has been reported that the lack of effect of immunotherapy-based strategies on the growth of PDAC depends on multiple factors including its aggressive biology and poor immunogenicity. The latter is due to the abundant desmoplastic stroma, which can impede effector T cell infiltration and the ability of tumors to escape from immune system by masking the pattern of antigenic epitopes.47 48
It has been suggested that PD1 immunotherapy could activate T cells in the tumor-draining lymph nodes, which might contribute to the antitumor response.49 To this purpose, we resected inguinal and axillary tumor-draining lymph nodes after each treatment in order to evaluate the actual contribution of lymph nodes in therapeutic efficacy. IMO-2125-treated tumor-draining lymph nodes from all our models (with or without immunogenic potential) showed a statistically significant volume increase as well as an expansion of immune cell content, including myeloid cells, CD8+ T cells, B cells. Moreover, higher expression of CD80 costimulatory molecule was identified in CD11c+ MHCII+ DCs isolated from DT4313 tumor-draining lymph nodes treated with IMO-2125. According to these data, Joncker and colleagues50 showed that tumor elimination correlates with rapid recruitment of DCs presenting the tumor antigen to both CD4+ and CD8+ T cells in the tumor-draining lymph nodes, a key process for the development of protective antitumor response. Since the IMO-2125 effect on lymph nodes was clearly evident in all our models, we hypothesize that a common immune cell-mediated mechanism occurs, but that the efficacy is determined by intrinsic characteristic of cancer cells.
Recent study reported that autophagy is an escape mechanism from the immune system also in pancreatic tumors through an increase in MHC degradation.47 51 We tested in vitro the basal autophagic flux of our cancer models, demonstrating that the IMO-2125 unresponsive cells expressed an increased autophagy compared with the sensitive ones. Thus, we speculate that the high basal autophagic flux of some tumors might be a crucial strategy to escape from T lymphocytes surveillance, rendering tumor cells refractory to immunomodulatory drugs such as IMO-2125. However, further studies combining autophagy inhibition with TLR9 agonists and immunotherapy in PDAC tumors with low or no immunogenic potential are needed to confirm this hypothesis.
Overall, we might infer that antigen presenting cells (APCs), such as DCs, were recruited by IMO-2125 local treatment into the tumor and then present the captured antigens, including tumor antigens, on the MHC molecules to T cells, leading to lymph node priming and activation of effector T-cell responses.
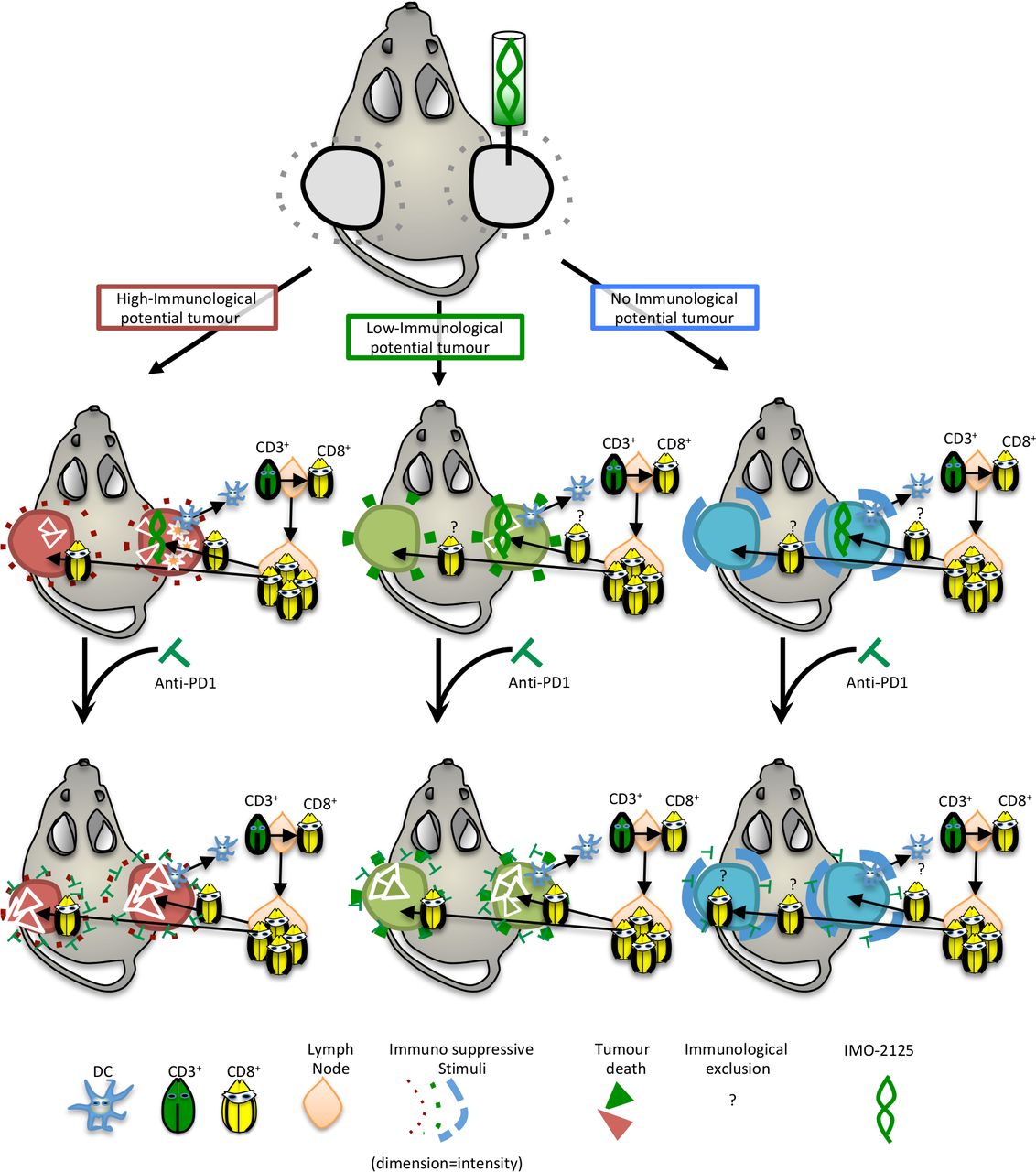
Alongside, a chemokine gradient exists to guide the activated T-cell infiltration to the tumor site. As a result of T-cell receptor and neoantigen–MHC complex interaction, the killing of cancer cells occurs (figure 6). Finally, we propose intratumoral injection of IMO-2125 as a promising therapeutic approach for immune sensitization of specific PDAC subtypes with immunogenic potential.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Schematic representation of IMO-2125 mechanism of action. IMO-2125 (intratumoral) treatment leads to dendritic cell migration in draining lymph nodes of all pancreatic ductal adenocarcinoma, whereas only in the high immunological potential tumors the expression of IFNγ determines a cytokines storm that exacerbates the IMO-2125 antitumor effect. The expansion and activation of T cells in tumor-draining lymph nodes occur for all IMO-2125-treated tumors. Whereas in tumor with high immunological potential, T cells are able to efficiently kill cancer cells, tumors with low or no immunological potential are able to escape to T lymphocytes through an increase of autophagy flux and expression of immune suppressive cytokines that limited efficacy of IMO-2125. Anti-PD1 treatment is able to increase the efficacy of IMO-2125, potentiating the infiltration of T cells, only in tumor with high and low immunological potential, while no effect in the extremely refractory tumors is evident.
In conclusion, in our study we have identified tumors with high, low and no immunological potential. Intratumoral injection of TLR-9 agonist IMO-2125 in a selected high immunogenic subtype triggers immune system response to kill local and distant tumors. Importantly, intratumoral injection of IMO-2125 favors a transition toward an immune-permissive microenvironment sensitizing PDAC to anti-PD1 therapy in tumor subtypes usually refractory to immune therapy. Altogether, our results support a new approach to treat PDAC that may be translated into a clinical setting.
Data availability statement
All data relevant to the study are included in the article or uploaded as supplemental information. All methodologies, analyses, and results are included in the article.
Ethics statements
Patient consent for publication
Ethics approval
In vivo experiments were conducted in accordance with guidelines of the Institutional OPBA and Italian Ministry of Health Ethic Committee.
Acknowledgments
We would like to thank the Italian Pancreatic Cancer Community (IPCC, www.i-pcc.org) and ‘Fondazione Nadia Valsecchi Onlus’ for the precious networking in pancreatic cancer field. We would like to thank Granitto A and Esposito AC for help in the histopathological examination of the tumors and for technical assistance. We would like to thank the members of the Cen.Ri.S. animal facility, Lupoi L, D’Aquino M, Caristo ME for their valuable assistance during in vivo experiments.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Twitter @nasca_vincenzo, @paolacappello07, @emilio.bria
Contributors CC, GP, PC, VC and GT conceived and designed the study; CC and GP designed and performed the majority of the experiments; PC provided cancer cells; FS, VN, FDS and SU performed experiments; MM performed IHC analyses; PD and AA performed bioinformatics analyses. CC, GP, AS, CS, EB and GT wrote the manuscript.
Funding This work was supported by the AIRC IG grant number 18599 to GT; AIRC5x1000 grant number 12182 and grant from Italian Ministry of Health FIMCUP_J38D19000690001 to AS; AIRC StartUp Grant No. 18178 to VC; AIRC IG grant number 20583 to EB; AIRC IG grant number 23416 to CS; My First AIRC Grant "Luigi Bonatti e Anna Maria Bonatti Rocca”, grant number 23681 to CC.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.